Introduction
Autistic spectrum disorder (ASD) (MIM 209850) is a
complex neurodevelopmental disorder that results in social and
communication impairment, as well as repetitive and stereotyped
patterns (1,2). Other non-psychiatric symptoms of ASD
include intellectual disability, motor delay, hypotonia and
seizures (2). Individuals affected
with ASD have also been reported to exhibit neuroanatomical
structure alterations (3).
Epidemiologically, the frequency of ASD is estimated to be ~60
cases per 10,000 children, increasing from early epidemiological
studies that originally reported 2–5 cases per 10,000 children
worldwide (2,4).
Genetically, ASD has been described as a
multifactorial genetic disorder, which in certain cases is
associated with a specific mutation or syndrome, such as fragile X
syndrome (Xq27.3) (5), tuberous
sclerosis (9q or 16p) (6) and
Angelman/Prader-Willi syndrome (15q11-q13 chromosomal abnormality)
(7,8). Mutations in genes that encode
proteins associated with synaptic transmission and activity,
including NLGN3/NLGN4 and SHANK3, have been
reported in patients with ASD (2,9–11).
Other reported changes include protein synthesis-associated genes
(FMR1 and PTEN), transcription factors (MEF2)
(11), and neurotransmitter
proteins and receptors [glutamate and gamma-aminobutyric acid
(GABA) receptors] (10). Previous
reports have linked ASD to dysfunctional energy metabolism in the
central nervous system (CNS), which may describe changes in
mitochondrial DNA, or nuclear DNA associated with mitochondrial
function (10). In addition, there
are numerous cases where ASD appears to arise from a de novo
genetic alteration (9).
In the effort to characterize novel genetic
alterations associated with ASD, the aim of the present study was
to investigate potential susceptibility loci of ASD, utilizing the
highly consanguineous and inbred nature of numerous families within
the population of Saudi Arabia. Families with ASD-affected
individuals were recruited in the present study, and loss of
heterozygosity analyses were performed. The present study presents
possible regions that may be linked to ASD, as obtained from a loss
of heterozygosity analysis.
Materials and methods
Patients and samples
The present study was performed under the approval
of the tertiary care center King Faisal Specialist Hospital and
Research Center (KFSHRC RAC no. 2080001; Riyadh, Saudi Arabia). The
present study was a collaboration between the departments of
Genetics and Neuroscience, and the Autism Research Center at
KFSHRC. Families with >1 affected individual, regarded as
multiplex families, were identified and recruited. The majority of
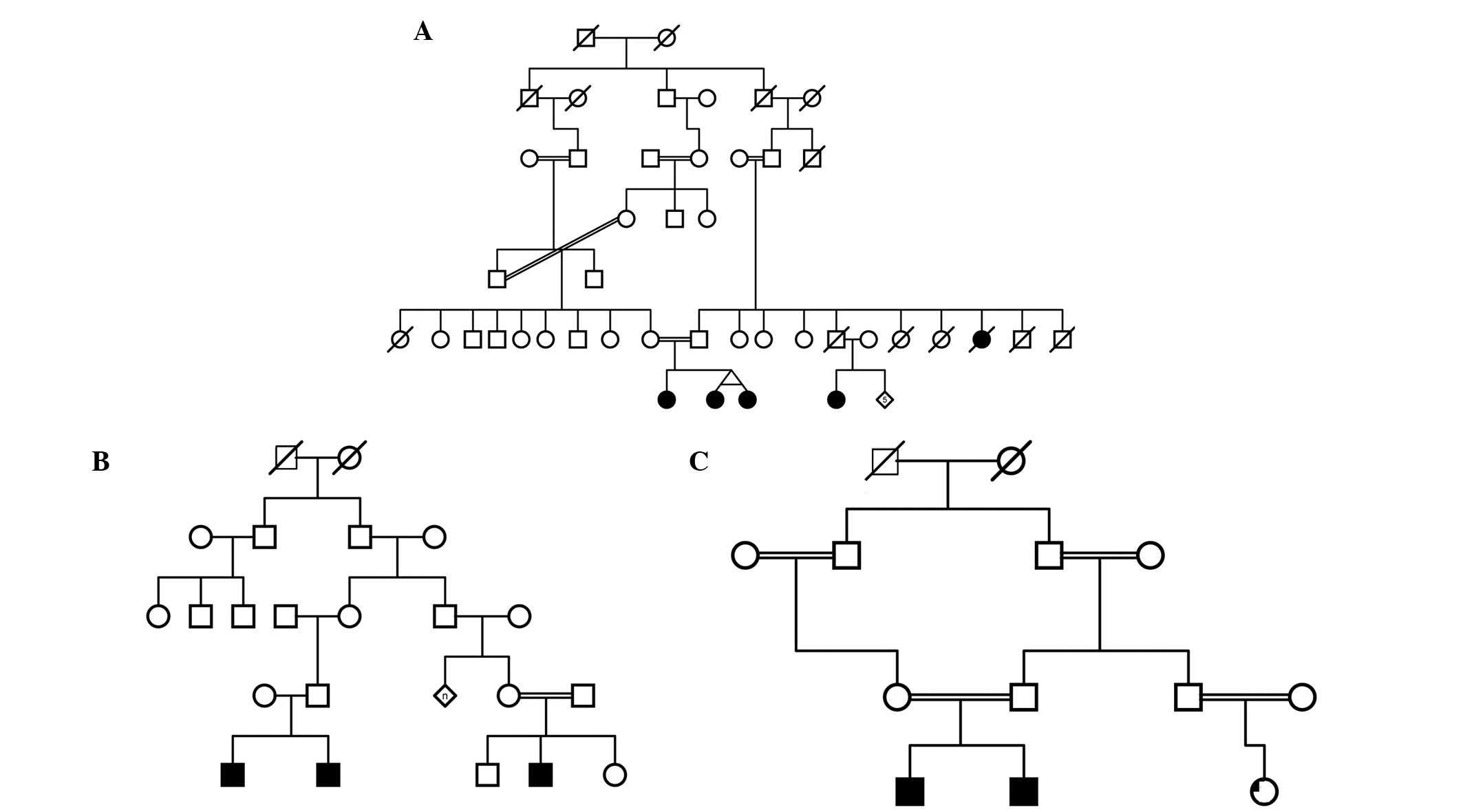
the families recruited contained consanguineous marriages. Examples
of family pedigrees from a selection of the recruited families are
shown in Fig. 1. Diagnosis of ASD
was made at KFSHRC, according to the ADI-R and DSM-VI criteria
(1,2). Families with individuals diagnosed
with ASD secondary to known genetic or metabolic disorders were
excluded. Blood samples (3–5 ml) were obtained in EDTA tubes from
family members for subsequent genetic analysis. The family members
provided written informed consent in adherence to institutional and
international guidelines (RAC#2080001).
DNA extraction
DNA was extracted from the peripheral blood samples
using the Gentra Systems Puregene DNA Isolation kit (Qiagen,
Valencia, CA, USA), according to the manufacturer’s instructions.
DNA concentration was determined spectrophotometrically using a
Nanodrop (Thermo Fisher Scientific, Waltham, MA, USA).
Loss of heterozygosity analysis
All DNA samples were genotyped using
GeneChip® Mapping 250K and 6.0 arrays, and the Axiom
Genome-Wide CEU 1 Human Mapping Array (Affymetrix, Santa Clara, CA,
USA), according to the manufacturer’s instructions. Briefly, 25–250
ng of high-quality genomic DNA was digested using the restriction
endonuclease StyI for the 250K array, and StyI and
NspI restriction endonucleases for the 6.0 array. The
products were then ligated with an adaptor. Generic primers that
recognize the adaptor sequence were used to amplify the
adaptor-ligated DNA fragments in the recommended size range. The
amplified polymerase chain reaction products were then fragmented
with the Affymetrix fragmentation reagent to 50–200 bp, and
end-labeled with biotinylated dideoxyadenosine triphosphate using
terminal deoxynucleotidyl transferase. The end-labeled fragments
were then hybridized to GeneChip® Human Mapping 250K or
6.0 arrays. For the Axiom Set, the target DNA was amplified using
the Axiom 2.0 Amp kit and then fragmented according to the
manufacturer’s recommendations (Affymetrix). The pellets were then
re-suspended and hybridized, and genotyping was performed using the
GeneTitan® Multi-Channel Instrument (Affymetrix).
GeneChip® Operating software (version
1.4) and Genotyping Console™ (GTC; version 3.01) software
(Affymetrix) were used for primary data analysis, normalization
against internal control features on the chip, genotype calling and
quality control (overall single nucleotide polymorphism call rate,
95–99%). Data generated from the arrays were then analyzed using
Affymetrix Genotyping Console (version 3.01) and/or a homozygosity
mapper program (http://www.homozygositymapper.org/) (12). Regions of shared homozygosity
between affected individuals within a family, but not shared with
unaffected individuals, were identified. Also, regions of shared
homozygosity between affected individuals from different families
were identified. The cutoff characteristic was that the region had
to be shared between all of the affected individuals from at least
four different families. Unaffected individuals from three random
families were used as controls. Regions <2 Mb were excluded from
the analysis, and gender-specific chromosomes were not
analyzed.
Identified regions of homozygosity were entered into
the Suspects Candidate Gene Search v.28.3 (http://www.cgem.ed.ac.uk/resources/suspects/)
from the University of Edinburgh (Edinburgh, UK), in order to
prioritize genes in an order of their reported relevance to the
development of ASD (12). The top
three candidate genes were reported in the present study.
Results
A total of 13 multiplex families were recruited and
analyzed using GeneChip® Mapping 250K and 6.0 arrays,
and the Axiom Set. There were >27 affected individuals within
the recruited families. A selection of the pedigrees is presented
in Fig. 1.
Regions of homozygosity identified from the
independent analysis of each family were spread across all
chromosomes. The chromosome with the most regions of homozygosity
was identified as chromosome 5 (Table
I). Shared regions of homozygosity between the affected
individuals from all of the families were also determined. There
were seven regions that were shared between all individuals, with
two regions on chromosome 11 (Table
II). The number of genes in the regions identified ranged
between 3 genes to several hundreds. Notably, no regions of
homozygosity (above 2 Mb in size) were observed on chromosomes 13,
14 and 22 as per the analysis criteria used.
 | Table IRegions of homozygosity identified in
the analysis of multiplex families (n=13). |
Table I
Regions of homozygosity identified in
the analysis of multiplex families (n=13).
| Chromosome | Coordinates | Top 3 genes | Family ID |
|---|
| 1 | 17, 606, 235-20, 525,
209 | TAS1R2, HTR6,
PLA2G2F | F14 |
| 66, 592, 949-70, 541,
226 | NP_065999,
NP_060238, SLC35D1 | F14 |
| 90, 600, 674-10, 477,
047 | ATP6V0B, SLC1A7,
ATPIF1 | F20 |
| 208, 362, 435-211,
304, 878 | NP_061134, ATF3,
RPS6KC1 | F8 |
| 2 | 16, 280, 027-18, 724,
599 | KCNS3, SMC6L1,
VSNL1 | F14 |
| 111, 883, 724-114,
024, 469 | SLC20A1, MERTK,
NP_116213 | F23 |
| 162, 797, 863-165,
675, 258 | KCNH7, FAP,
NP_775783 | F23 |
| 129, 417, 474-133,
043, 938 | NP_079305,
PLEKHB2, CFC1 | F6 |
| 180, 338, 192-192,
340, 990 | ITGAV, NAB1,
COL3A1 | F7 |
| 3 | 49, 088, 836-51, 462,
363 | BSN, DOCK3,
LAMB2 | F20 |
| 155, 790, 787-160,
134, 049 | Q8NGV9, SSR3,
GMPS | F14 |
| 4 | 3, 691, 191-5, 842,
856 | MSX1, CYTL1,
STK32B | F14 |
| 37, 930, 813-44,
149, 564 | CHRNA9, APBB2,
SLC30A9 | F14 |
| 75, 534, 780-81,
987, 898 | FRAS1, ANTXR2,
GK2 | F7 |
| 91, 479, 821-96,
553, 748 | PDLIM5, GRID2,
BMPR1B | F3 |
| 127, 483, 173-131,
017, 058 | SLC25A31, PHF17,
PDZK6 | F6 |
| 5 | 10, 671, 102-12,
018, 348 | CTNND2,
DAP | F14 |
| 13, 115, 454-15,
292, 186 | DNAH5, TRIO,
ANKH | F23 |
| 43, 721, 462-46,
644, 204 | HCN1, FGF10,
MRPS30 | F14 |
| 63, 181, 011-65,
623, 491 | TRIM23, ADAMTS6,
HTR1A | F14-F7 |
| 70, 580, 012-78,
664, 650 | F2RL1, F2RL2,
F2R | F14 |
| 91, 871, 294-95,
831, 186 | KIAA0372, PCSK1,
NR2F1 | F14 |
| 105, 398, 456-108,
334, 980 | EFNA5,
FBXL17 | F6 |
| 110, 309, 108-113,
085, 059 | SRP19, APC,
WDR36 | F23 |
| 154, 784, 618-157,
912, 731 | ADAM19, CRSP9,
SGCD | F23 |
| 6 | 43, 852, 052-46,
694, 741 | DSCR1L1, ENPP4,
SLC35B2 | F20 |
| 51, 472, 664-58,
554, 555 | PKHD1, MCM3,
ICK | F6-F14 |
| 61, 784, 585-65,
361, 195 | PTP4A1, KHDRBS2,
GLULD1 | F14 |
| 89, 467, 426-91,
642, 040 | GABRR2, GABRR1,
MDN1 | F14 |
| 117, 801, 917-120,
016, 046 | MAN1A1, DCBLD1,
C6orf60 | F14 |
| 128, 741, 476-132,
437, 814 | AKAP7, CTGF,
LAMA2 | F14 |
| 7 | 44, 167-3, 935,
550 | GPR146, LFNG,
CHST12 | F23 |
| 33, 956, 386-38,
816, 634 | SEPT7, AMPH,
GPR154 | F14 |
| 39, 251, 002-42,
234, 071 | GLI3, INHBA,
CDC2L5 | F14 |
| 78, 299, 408-93,
347, 267 | GRM3, SRI,
CACNA2D1 | F14 |
| 88, 881, 477-91,
474, 214 | PFTK1, CYP51A1,
CLDN12 | F14 |
| 118, 385, 860-122,
485, 086 | SLC13A1, KCND2,
WNT16 | F1 |
| 132, 226, 627-137,
446, 264 | SLC13A4, PTN,
NUP205 | F7 |
| 143, 159, 897-147,
967, 548 | CNTNAP2, TPK1,
O60393 | F14 |
| 149, 811, 568-153,
488, 778 | MLL3, ACCN3,
KCNH2 | F14 |
| 8 | 95, 013, 089-99,
313, 599 | UQCRB, GDF6,
NP_057218 | F14 |
| 113, 038, 464-118,
887, 549 | CSMD3, THRAP6,
NP_115710 | F14 |
| 8, 966, 544-10,
388, 436 | MSRA, TNKS,
PPP1R3B | F20 |
| 29, 493, 059-31,
510, 288 | GSR, WRN,
LEPROTL1 | F23 |
| 9 | 77, 676, 620-85,
812, 95 | SLC28A3, NTRK2,
TLE1 | F14 |
| 125, 138, 695-137,
750, 235 | GRIN1, KCNT1,
SLC25A25 | F23 |
| 10 | 24, 259, 097-26,
357, 328 | GPR158, THNSL1,
PRTFDC1 | F6 |
| 34, 935, 425-36,
373, 753 | CREM, NP_699199,
CUL2 | F23 |
| 56, 919, 560-59,
481, 992 | ZWINT,
ZWINTAS | F7 |
| 79, 202, 450-85,
447, 212 | SFTPD, ANXA11,
PPIF | F23 |
| 11 | 43, 773, 141-51,
682, 438 | RAPSN, PSMC3,
SPI1 | F3 |
| 110, 025, 057-112,
216, 763 | PTS, TIMM8B,
DLAT | F23 |
| 12 | 4, 042, 363-10,
283, 711 | A2M, CHD4,
TNFRSF1A | F7 |
| 37, 345, 650-39,
627, 393 | KIF21A, SLC2A13,
ABCD2 | F23 |
| 15 | 80, 676, 184-82,
887, 137 | AP3B2, BTBD1,
RPS17 | F23 |
| 95, 798, 343-101,
692, 828 | ADAMTS17, CHSY1,
IGF1R | F17 |
| 16 | 65, 000, 489-70,
848, 261 | SLC12A4, CTCF,
ATP6V0D1 | F1-F14 |
| 6, 392, 511-8, 255,
216 | A2BP_HUMAN,
Q8WZ91, Q14229 | F14 |
| 63, 133, 419-77,
264, 064 | SLC12A4, CTCF,
ATP6V0D1 | F14 |
| 17 | 32, 587, 614-35,
196, 133 | GPR158L1, ERBB2,
NEUROD2 | F3 |
| 21, 890, 916-27,
461, 374 | SLC6A4, SLC13A2,
UNC119 | F14 |
| 76, 007, 579-78,
771, 650 | SLC25A10, FOXK2,
SLC16A3 | F23 |
| 18 | 36, 589, 650-38,
808, 690 | PIK3C3 | F23 |
| 19 | 4, 463, 233-7, 013,
199 | SLC25A23,
NP_775908, PTPRS | F8 |
| 20 | 13, 821, 260-16,
084, 725 | FLRT3,
C20orf133, Q9NQI9 | F14 |
| 21 | 23, 309, 707-39,
155, 954 | ATP5J, ATP5O,
GRIK1 | F23 |
| Table IIRegions of shared homozygosity
obtained from analyzing all affected individuals from multiplex
families using GeneChip® Mapping 250k and 6.0
arrays. |
Table II
Regions of shared homozygosity
obtained from analyzing all affected individuals from multiplex
families using GeneChip® Mapping 250k and 6.0
arrays.
| Chromosome | Coordinates and
Axiom | Top 3 genes |
|---|
| 1 | 102, 688, 721-105,
073, 165 | COL11A1, AMY1A,
AMY1A |
| 2 | 134, 545, 725-136,
584, 137 | ACMSD, LCT,
MGAT5 |
| 3 | 49, 856, 601-51,
968, 474 | GRM2, DOCK3,
SLC38A3 |
| 7 | 77, 339, 194-80,
531, 718 | SEMA3C, CD36,
GNAI1 |
| 11 | 47, 240, 293-49,
279, 489 | PSMC3, RAPSN,
SPI1 |
| 48, 549, 401-50,
779, 621 | FOLH1, TYRL,
OR4C13 |
| 15 | 25, 581, 119-27,
659, 946 | APBA2, HERC2,
OCA2 |
Discussion
The regions of homozygosity identified in the
present study contained numerous genes that have previously been
reported as possible causes of ASD (13). The results included the following
genes: GRIN1, GRIK1, GRID2, GPR158L1,
GLULD1 and GRM3, which are associated with glutamate
receptor and proteins, RAPSN, which is associated with
acetylcholine receptors and transmission, and GABRR1 and
GABRR2, which are associated with GABA receptors (14). The results of the present study
support the numerous reports that suggest ASD may be a
synaptopathic disease (15).
Furthermore, numerous genes in these regions are associated with
mitochondria and are energy-associated, including ATP5J,
SLC25A25, SLC25A31 and MRPS30, which have also
been implicated in previous studies (16,17).
A third group of genes identified in these regions were consistent
with embryonic/development-associated genes, including, WNT
16 and NEUROD2, suggesting a possible early insult to
the central nervous system in the developing embryo (18).
A number of the regions reported to be associated
with the development of ASD include: i) Duplication or deletion of
16p11.2, which has been implicated in patients with ASD as well as
in animal models, which may cause alterations in brain anatomy and
behavior (19,20); ii) a 1.5-Mb microdeletion in region
14q23.2–23.3, which is associated with ASD, as well as
spherocytosis (21); iii) a
deletion at 2p15 (22); iv) an
alteration at 8p23.2 (23); and v)
a 535-Kb deletion at 3p26.3 (24).
However, the results of the present study did not indicate any
alterations to these regions, suggesting that ASD-associated gene
alterations differ across various populations.
As shown in Table
II, the number of regions identified when analyzing all of the
affected individuals from the different families was lower, as
compared with the regions obtained from analyzing the affected
individuals within their respective families. In addition, there
were no regions that were universally present in all of the
affected individuals from the different families. However, 7 shared
regions were observed between certain affected individuals. This
difference may be attributed to the fact that ASD is a genetically
complex and multifactorial disorder, which concurs with numerous
previous studies and reports regarding the genetics of ASD
(3,4,9).
There may be more than one genetic change per family, and different
genetic changes between families. Biallelic mutations have been
reported in other disorders with complex phenotypes (25–27).
One of the challenges in studying the genetics of
ASD in Saudi Arabia is that the Saudi population, as evident in the
families that were recruited in the present study, is highly
consanguineous and inbred. This represents a challenge in
identifying genetic regions that are associated with ASD, as
opposed to regions that may be normally shared as a result of
identity by descent. In order to overcome this challenge, it has
been proposed for future studies that appropriate tests and
statistics are conducted prior to genetic analysis that allow for
discrimination of these two types of regions, such as the
coefficient of inbreeding (28).
Another challenge facing the genetic study of ASD is
that ASD represents a variable spectrum, in which the milder forms
may not be detected with conventional clinical methods. This is
relevant to the results of the present study in that when the
results of affected individuals are compared with normal controls,
there may be missing/excluded regions. This may possibly be due to
the controls having a mild form of ASD that the clinic was unable
to detect. Further study of ASD genetics is therefore dependent on
the development of more specific clinical tools. Another way to
overcome this issue in future studies is to include information on
clinically normal individuals, including their intelligence
quotient, social interaction status and their school performance.
As has been reported previously, the possibility of the presence of
multiple phenotypes within the same family suggests involvement of
multiple interacting loci, which adds to the complexity of the
inheritance of ASD (28).
As mentioned previously, all of the regions
identified in the present study were >2 Mb. It is possible that
more regions and possible genes may be identified for analysis by
examining smaller regions. This is the aim for future studies by
our group, as well as the inclusion of gender-associated
chromosomes in the analyses.
Candidate autism genes usually have distinct
biological roles and interactions. In order to attain an improved
understanding regarding the pathogenesis of ASD, it is important to
discover the location and functional role of autism-associated
proteins in biological pathways, and hence in neuronal function and
resulting behavior (29). An ASD
interactome was generated to meet the obstacles caused by this
hereditary neurological disorder, and to identify proteins that
interact with ASD-associated proteins (30). Of note, when the ASD interactome
was created, a high connectivity was identified between two
ASD-associated proteins that initially appeared functionally
unrelated, SHANK3 and TSC1. SHANK3 is an adapter protein in the
post-synaptic regions that may have a role in the organization of
the dendritic spine and synaptic junction, and TSC1 is a tuberous
sclerosis 1 protein that regulates mechanistic (serine/threonine
kinase) target of rapamycin, a promoter of protein synthesis.
However, these two proteins share ≥21 protein associates and were
found to interact in a complex scaffold at the post-synaptic region
(30), thus suggesting that
various ASD-associated proteins may share a common key pathway
associated with disease development. Similarly, the genes
identified in the present study involve various categories of
proteins that may also share a common molecular pathway, and may be
further investigated through a protein-protein interaction assay.
Furthermore, since ASD genes differ between populations, creating
an interactome for these Saudi ASD proteins would be beneficial not
only in the development of treatment, but also in developing
powerful therapies for a specific ethnic group.
Acknowledgments
The present study was supported by the King Faisal
Specialist Hospital and Research Center (KFSHRC) (RAC# 2080001) and
the Center for Autism Research. The authors of the present study
would also like to acknowledge the genotyping core facility at
KFSHRC.
References
|
1
|
American Psychiatric Association:
Diagnostic and statistical manual of mental disorders. 4th. text
rev.2000
|
|
2
|
Levy SE, Mandell DS and Schultz RT:
Autism. Lancet. 374:1627–1638. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bailey A, Le Couteur A, Gottesman I, et
al: Autism as a strongly genetic disorder: evidence from a British
twin study. Psychol Med. 25:63–77. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bryson S: Epidemiology of autism: overview
and issues outstanding. Handbook of Autism and Pervasive
Developmental Disorders. 2nd. Cohen D and Volkmar F: John Wiley and
Sons; New York: pp. 41–46. 1997
|
|
5
|
Rogers SJ, Wehner DE and Hagerman R: The
behavioral phenotype in fragile X: symptoms of autism in very young
children with fragile X syndrome, idiopathic autism, and other
developmental disorders. J Dev Behav Pediatr. 22:409–417. 2001.
View Article : Google Scholar
|
|
6
|
Baker P, Piven J and Sato Y: Autism and
tuberous sclerosis complex: prevalence and clinical features. J
Autism Dev Disord. 28:279–285. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Steffenburg S, Gillberg CL, Steffenburg U
and Kyllerman M: Autism in Angelman syndrome: a population-based
study. Pediatr Neurol. 14:131–136. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sutcliffe JS, Nurmi EL and Lombroso PJ:
Genetics of childhood disorders: XLVII. Autism, part 6: duplication
and inherited susceptibility of chromosome 15q11-q13 genes in
autism. J Am Acad Child Adolesc Psychiatry. 42:253–256. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Geschwind DH: Autism: many genes, common
pathways? Cell. 135:391–395. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Smith M, Spence MA and Flodman P: Nuclear
and mitochondrial genome defects in autisms. Ann N Y Acad Sci.
1151:102–132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Walsh CA, Morrow EM and Rubenstein JL:
Autism and brain development. Cell. 135:396–400. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Adie EA, Adams RR, Evans KL, Porteous DJ
and Pickard BS: Speeding disease gene discovery by sequence based
candidate prioritization. BMC Bioinformatics. 6:552005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Freitag CM: The genetics of autistic
disorders and its clinical relevance: A review of the literature.
Mol Psychiatry. 12:2–22. 2007. View Article : Google Scholar
|
|
14
|
Tarabeux J, Kebir O, Gauthier J, et al S2D
team: Rare mutations in N-methyl-D-aspartate glutamate receptors in
autism spectrum disorders and schizophrenia. Transl Psychiatry.
1:e552011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bourgeron T: A synaptic trek to autism.
Curr Opin Neurobiol. 19:231–234. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Anitha A, Nakamura K, Thanseem I, et al:
Brain region-specific altered expression and association of
mitochondria-related genes in autism. Mol Autism. 3:122012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tang G, Gutierrez Rios P, Kuo SH, et al:
Mitochondrial abnormalities in temporal lobe of autistic brain.
Neurobiol Dis. 54:349–361. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Olson JM, Asakura A, Snider L, Hawkes R,
Strand A, Stoeck J, Hallahan A, Pritchard J and Tapscott SJ:
NeuroD2 is necessary for development and survival of central
nervous system neurons. Dev Biol. 234:174–187. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Horev G, Ellegood J, Lerch JP, et al:
Dosage-dependent phenotypes in models of 16p11.2 lesions found in
autism. Proc Natl Acad Sci USA. 108:17076–17081. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kumar RA, Marshall CR, Badner JA, et al:
Association and mutation analyses of 16p11.2 autism candidate
genes. PLoS One. 4:e45822009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Griswold AJ, Ma D, Sacharow SJ, et al: A
de novo 1.5 Mb microdeletion on chromosome 14q23.2–23.3 in a
patient with autism and spherocytosis. Autism Res. 4:221–227. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu X, Malenfant P, Reesor C, et al:
2p15-p16.1 microdeletion syndrome: molecular characterization and
association of the OTX1 and XPO1 genes with autism spectrum
disorders. Eur J Hum Genet. 19:1264–1270. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nucaro A, Pisano T, Chillotti I, Montaldo
C and Pruna D: Chromosome 8p23.2-pter: a critical region for mental
retardation, autism and epilepsy? Clin Genet. 79:394–395; author
reply 396. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cottrell CE, Bir N, Varga E, et al:
Contactin 4 as an autism susceptibility locus. Autism Res.
4:189–199. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cole LW, Sidis Y, Zhang C, et al:
Mutations in prokineticin 2 and prokineticin receptor 2 genes in
human gonadotrophin-releasing hormone deficiency: molecular
genetics and clinical spectrum. J Clin Endocrinol Metab.
93:3551–3559. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shaw ND, Seminara SB, Welt CK, et al:
Expanding the phenotype and genotype of female GnRH deficiency. J
Clin Endocrinol Metab. 96:E566–E576. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Katsanis N, Eichers ER, Ansley SJ, et al:
BBS4 is a minor contributor to Bardet-Biedl syndrome and may also
participate in triallelic inheritance. Am J Hum Genet. 71:22–29.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Risch N, Spiker D, Lotspeich L, et al: A
genomic screen of autism: evidence for a multilocus etiology. Am J
Hum Genet. 65:493–507. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kalkman HO: A review of the evidence for
the canonical Wnt pathway in autism spectrum disorders. Mol Autism.
3:102012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sakai Y, Shaw CA, Dawson BC, et al:
Protein interactome reveals converging molecular pathways among
autism disorders. Sci Transl Med. 3:86ra492011. View Article : Google Scholar : PubMed/NCBI
|