Introduction
There is compelling evidence that apolipoprotein E
deficiency (ApoE−/−) combined with a high-fat diet
regulates toll-like receptor 4 (TLR4) expression and promotes
atherosclerosis development. Massaro and Massaro (1) reported that ApoE−/− mice
displayed reduced alveologenesis as compared with wild-type strain
controls, and that ApoE−/− had an effect on lung
pathological changes. Similarly, Naura et al (2) demonstrated that ApoE−/−
mice on a high-fat diet displayed lung inflammation. Goldklang
et al (3) indicated that
ApoE−/− mice on a high-fat Western-type diet (WD) showed
emphysema due to TLR4 activation (3). Samokhin et al (4) suggested that ApoE−/− mice
on a high-fat diet developed granulomas similar to those observed
in human sarcoidosis. Accordingly, reports of the effects of HFD on
lung pathological changes in ApoE−/− mice differ
greatly.
Macrophages are important inflammatory cells
implicated in the initiation of inflammation, and they have
critical roles in the pathogenesis of foam cell formation (5). TLR4 is a key initiator of innate
immunity that is able to promote an adaptive immune response. TLR4
recognizes lipopolysaccharide (LPS), resulting in the activation of
the myeloid differential factor 88 (MyD88)- and toll-interleukin-1
receptor domain-containing adapter inducing interferon-β
(TRIF)-dependent downstream signaling pathways. TLR4 signalling has
a critical role in the progression of atherosclerosis and lung
inflammation (6,7). A previous study by our group revealed
that ApoE−/− mice developed pulmonary capillaritis via
up-regulation of TLR4 and nuclear factor (NF)-κB (8). However, to date, the role of TLR4
signalling in the pathogenesis of lung lipidosis have not been
studied, to the best of our knowledge.
The aim of the present study was to determine
whether ApoE deletion combined with hypercholesterolemia induces
lung inflammation and lipidosis. Furthermore, TLR4 knockdown was
employed to investigate whether TLR4 signalling is implicated in
those pathological changes.
Materials and methods
Animals and experimental design
All of the procedures and protocols were approved by
the Animal Care Committee of Fujian Medical University (Quanzhou,
China) and followed the guidelines of the Animal Management Rules
of the Chinese Ministry of Health. Eighty eight-week-old male
ApoE−/− mice and sixty age- and gender-matched wild-type
mice with a C57BL/6 genetic background (B6) were obtained from the
Peking University Animal Centre (Beijing, China). In the first
group, thirty ApoE−/− and B6 mice were fed a WD
(containing 0.25% cholesterol and 15% cocoa butter; MD12032) or a
normal chow diet (ND; MD12031; Yangzhou Medicience Ltd, Yangzhou,
China) for 4, 12 or 24 weeks, respectively (n=10 in each group). In
the second group, the ApoE−/− and wild-type mice were
injected with short hairpin TLR4 interference lentivirus
(Lv-shTLR4) or empty vector (both from Invitrogen Life
Technologies, Paisley, UK) at 1×108 transducing units
for each mouse through the caudal vein (n=10). They were fed the WD
for 12 weeks. All the animals were under standardized lighting
conditions (12-h light/dark cycle) and temperature (21±1°C).
Mineral water was administered ad libitum. At the end of the
experiment the mice were sacrificed by overdose of pentobarbital
(90 mg/kg; intraperitoneal injection; Huayehuanyu Ltd, Beijing,
China). The bronchoalveolar lavage fluid (BALF) was collected and
the left lung lobe tissue was collected for histomorphological
examination, whereas the right lung lobe tissue was collected for
RNA and protein analysis.
Hematoxylin and eosin (HE) staining for
lung pathomorphological changes
The lung tissue was stained with hematoxylin and
eosin (HE; Sigma-Aldrich, St. Louis, MO, USA). The mean linear
intercept and septal thickness were quantified as described by
Wendel et al (9). This
assessment was repeated for 10 terminal respiratory units in one
random tissue section per mouse. All the images were acquired using
a BX51 microscope (Olympus, Center Valley, PA, USA) and analyzed
using Image-Pro Plus 6.0 (Media Cybernetics, Inc., Bethesda, MD,
USA). The evaluation was performed by two experienced pathologists
who were blinded to the treatments that the mice had received,
according to methods previously described (10).
Oil red O staining for lipidosis in the
lung and quantitation of pulmonary cholesterol content
For assessment of lipidosis, the frozen lung
sections were stained with Oil Red O (Sigma-Aldrich). The
cholesterol content of the lung tissue was quantified according to
Bates et al (11). The free
and total cholesterol contents were calculated using a cholesterol
standard (Sigma-Aldrich). The cholesteryl ester content was
calculated by subtracting the free cholesterol from the total
cholesterol for each sample. The cholesterol content was expressed
as ‘micrograms of lipid per gram of animal’.
Double immunofluorescent staining for
assessment of TLR4 in macrophages
For the localization of TLR4 expression in
macrophages, double immunofluorescence staining using the CD68
macrophage marker and anti-TLR4 (all diluted at 1:100; Abcam,
Cambridge, UK) was performed on the lung sections. CD68 and TLR4
double-labelled cells were quantified as a fraction of the total
cell nuclei in each lung section.
Western blot analysis of TLR4, MyD88,
NF-κB p65, TRIF and interferon regulatory factor 3 (IRF3)
expression in the lung tissues
To determine the TLR4, MyD88 phosphorylated
(p)-NF-κB, TRIF3 and IRF3 protein levels in the lung tissue,
western blot analysis was performed as described by Zhang et
al (12). Primary antibodies
(all from Abcam, Cambridge, UK) were used at the indicated
dilutions as follows: Rabbit polyclonal to TLR4 (cat. no. ab13556;
1:500); rabbit polyclonal to MyD88 (cat. no. ab2064; 1:1,000);
rabbit polyclonal to NF-κB p65 (cat. no. ab7970; 1:1,000); rabbit
polyclonal to p65-NF-κB (cat. no. ab7970; 1:500); rabbit polyclonal
to TRIF (cat. no. ab13810; 1:500); rabbit monoclonal to IRF3 (cat.
no. ab68481; 1:1000). Proteins (30 µg) were run on a 10%
SDS-PAGE gel and transferred to polyvinylidene fluoride membranes.
Following incubation with 10% non-fat milk for 1 h, the membranes
were probed with primary antibodies overnight at 4°C and then
incubated with HRP-labeled goat anti-rabbit secondary antibodies
(1:2,000; Santa Cruz Biotechnology, Inc., Dallas, TX, USA). The
protein levels were normalized using β-actin as a loading control.
The relative optical density of the protein bands was measured
using a Zeineh Laser Densitometer (Biomed Instruments, Inc.,
Fullerton, CA, USA) after subtracting the film background.
ELISA of cytokine profiles in BALF
The cytokine concentrations were determined using
sandwich ELISA for interferon-γ (IFN-γ), tumor necrosis factor
alpha (TNF-α), interleukin-4 (IL-4), IL-6 and IL-17 (IFN-γ, TNF-α,
IL-4, IL-6 and IL-17 ELISA kits all from eBioscience, San Diego,
CA, USA) in BALF according to the manufacturer’s instructions. All
the measurements were performed in duplicate.
Serum lipid analysis
The fasting serum samples were collected in
20-week-old mice of different genotypes following fasting for 8 h.
The total cholesterol (TC), triglycerides (TG), high-density
lipoprotein cholesterol (HDL-C) and non-HDL-C were measured as
previously described (13) using
reagents from Nanjing KeyGen Biotech Co., Ltd. (Nanjing,
China).
Lentiviral short hairpin RNA
(Lv-shRNA)-mediated TLR4 gene silencing
The shRNA targeting of the TLR4 gene (GenBank
accession no., NM021297.2) was screened and tested according to the
protocol of according to Zhu et al (14). The target sequence was designed and
chemically synthesized by the United Gene Company (Shanghai,
China). This shRNA comprises an RNA duplex containing a sense
strand: 5′-GATCCGCACTCTTGATTGC AGTTTCATTCAAGAGATGAAACTGCAATCAAGAGTG
CTTTTTTG-3′ and an antisense strand: 5′-AATTCAAA
AAAGCACTCTTGATTGCAGTTTCATCTCTTGAATGA AACTGCAATCAAGAGTGCG-3′. The
inserted TLR4 cDNA sequence was confirmed by DNA sequencing.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
qPCR was performed to test the efficacy of TLR4
knockdown. Total RNA was isolated from the lung tissue using the
RNAiso Plus kit (Takara Biotechnology Co., Ltd., Dalian, China). A
total of 500 ng RNA was used as the template for cDNA generation
with the RNA RT kit (Takara Bio, Inc., Shiga, Japan). cDNA was
immediately reverse-transcribed from the isolated RNA and
subsequently qPCR was performed using Power SYBR Green PCR Master
Mix (Takara Bio, Inc.) on the Master Mix System (Roche, Basel,
Switzerland). The primer sequences (5′ to 3′) were TLR4 forward,
ATGGCATGGCTTACACCACC and reverse, GAGGCCAATTTTGTCTCCACA; GADPH
forward, AGGTCGGTGTGAACGGATTTG and reverse,
TGTAGACCATGTAGTTGAGGTCA. The PCR conditions were as follows:
Initial denaturation at 95°C for 5 min, denaturation at 94°C for 45
sec, annealing at 50°C for 1 min and extension at 72°C for 1 min.
The PCR was performed for 35 cycles followed by a final extension
step at 72°C for 10 min. The PCR product was quantitatively
analyzed with LabWorks 4.5 analysis software (UVP LLC, Upland, CA,
USA). Relative quantification of the target gene mRNA was performed
using the comparative ΔΔCT-method, normalized to (GADPH) and a
relevant ApoE−/− empty vector control to obtain
2−ΔΔCT.
Statistical analysis
All values are expressed as the mean ± standard
error unless otherwise indicated. The group comparisons were
performed using Student’s t test (2-sample test) or analysis
of variance. A P-value of 0.05 was regarded to indicate a
statistically significant difference between values. The
statistical analysis was performed using SPSS 17.0 software (SPSS,
Chicago, IL, USA).
Results
Age-dependent inflammation and lipidosis
in the lungs of ApoE−/− WD mice
In the ApoE−/− mice receiving the WD for
four weeks, inflammatory cell infiltration was noted around the
capillaries and venules (Fig. 1A).
Following WD for 12 weeks, the ApoE−/− mice showed
thickened alveolar septa, exudate-filled alveolar spaces, ruptured
septa and bullae formation (Fig.
1B). Widely distributed granulomas were observed in the
ApoE−/− mice following WD for 24 weeks (Fig. 1C). By contrast, among the animals
receiving ND treatment for 4 or 12 weeks, the ApoE−/−
mice displayed a minimal number of inflammatory cells in the
peribronchiolar and perivascular sites (Fig. 1D and 1E), and very few granulomas developed at
24 weeks of ND (Fig. 1F). Those
manifestations were absent in the ApoE−/− mice fed an ND
for four weeks or in the wild-type mice fed a WD for 24 weeks
(Fig. 1G–L). Collectively, these
data suggested that pulmonary inflammation developed more
extensively and earlier in the ApoE−/− WD mice than in
the littermates on ND. In the wild-type mice on WD, no signs of
inflammation were observed, as indicated by normal bronchioles and
alveoli.
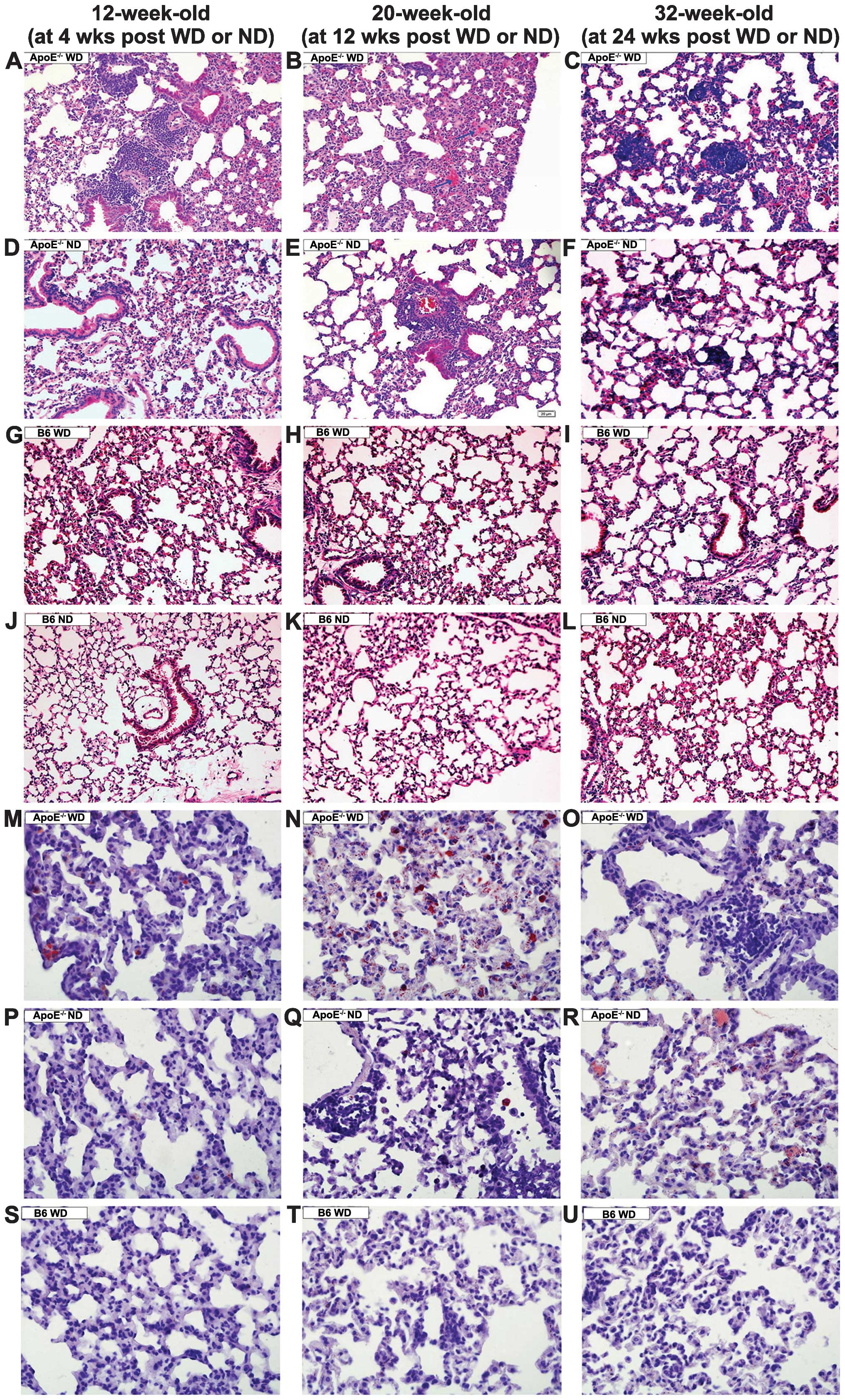 | Figure 1Lung inflammation and lipidosis in
mice of different genotypes fed an ND or WD. Micrography
demonstrating (A) increased inflammatory cell infiltration, (B and
E) exudation and thickened alveolar septa, and (C and F) granuloma
development in the ApoE−/− mice. (D, G–L, S–U) Normal
lung structure was present. (M–R) Oil red O staining indicated that
foam cells (red) were present in the septa and alveolar lumina.
A–L: hematoxylin and eosin staining; magnification, ×200; M–U: Oil
red O staining, magnification, ×400. Bars represent the mean ±
standard error of seven mice. *P<0.05 compared with
mice of the same genotype on a ND. #P<0.05 compared
with the B6 mice fed on the same diet. B6, C57BL/6J; ND, normal
chow diet; WD, high-fat Western-type diet; ApoE,
apolipoprotein. |
Lipid-laden cells were observed by oil red O
staining in the septa and alveolar lumina in the ApoE−/−
WD mice at 12 weeks (Fig. 1N),
whereas scattered lipid-filled cells were observed in the
ApoE−/− WD mice at 4 and 24 weeks (Fig. 1M and O) or the ApoE−/−
ND littermates at 24 weeks. No foam cells were present in the
wild-type mice on the WD even for 24 weeks (Fig. 1S–U).
Additionally, at 12 weeks, the alveolar septal
thickness and mean linear intercept were obviously greater in the
ApoE−/− WD mice when compared with that in the
ApoE−/− ND or B6 WD mice. The ApoE−/− ND mice
showed a marginal increase in alveolar septal thickness and mean
linear intercept; however, with no statistical significance. The B6
WD mice exhibited normal alveoli and septal thickness.
The lung cholesteryl content quantitation showed
that at 12 weeks, the total cholesterol content in the lungs of
ApoE−/− WD mice was elevated 5.69-fold relative to that
in the B6 WD mice and 4.08-fold compared to that in the
ApoE−/− ND mice, predominantly due to marked elevation
in cholesteryl ester levels. In ApoE−/− WD mice,
cholesteryl ester was 29.48-fold higher than that in the B6 WD mice
and 6.95-fold higher than that in the ApoE−/− ND mice.
In the wild-type B6 mice, the levels of lung cholesterol were
altered insignificantly, irrespective of the diet.
Increased TLR4 expression in pulmonary
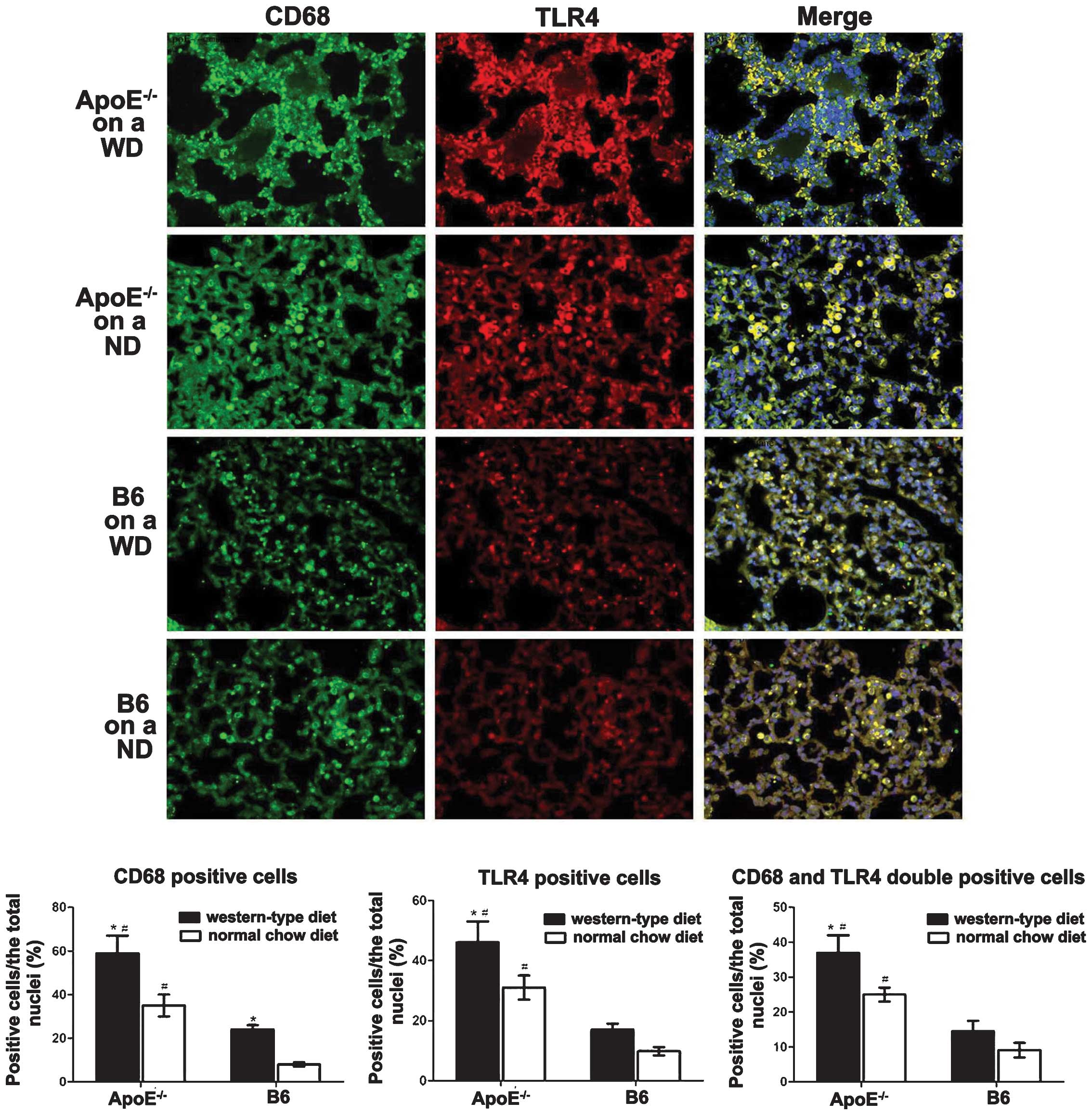
macrophages of ApoE−/− WD mice
TLR4 in macrophages is critical in pulmonary
inflammation and lipidosis. To investigate the implications of TLR4
in macrophages in the pathogenesis of lung injury, TLR4 expression
and lung macrophages were co-localised with double
immunofluorescent staining. The CD68+ macrophages
(green) in the lung were enriched in TLR4 (red). The
ApoE−/− mice fed a WD for 12 weeks exhibited markedly
greater macrophage infiltration and TLR4 expression in the alveolar
septum compared with that in the B6 WD mice (Fig. 2). The ApoE−/− ND mice
displayed a moderately increased number of CD68+
TLR4+ cells (yellow) in the lung (Fig. 2).
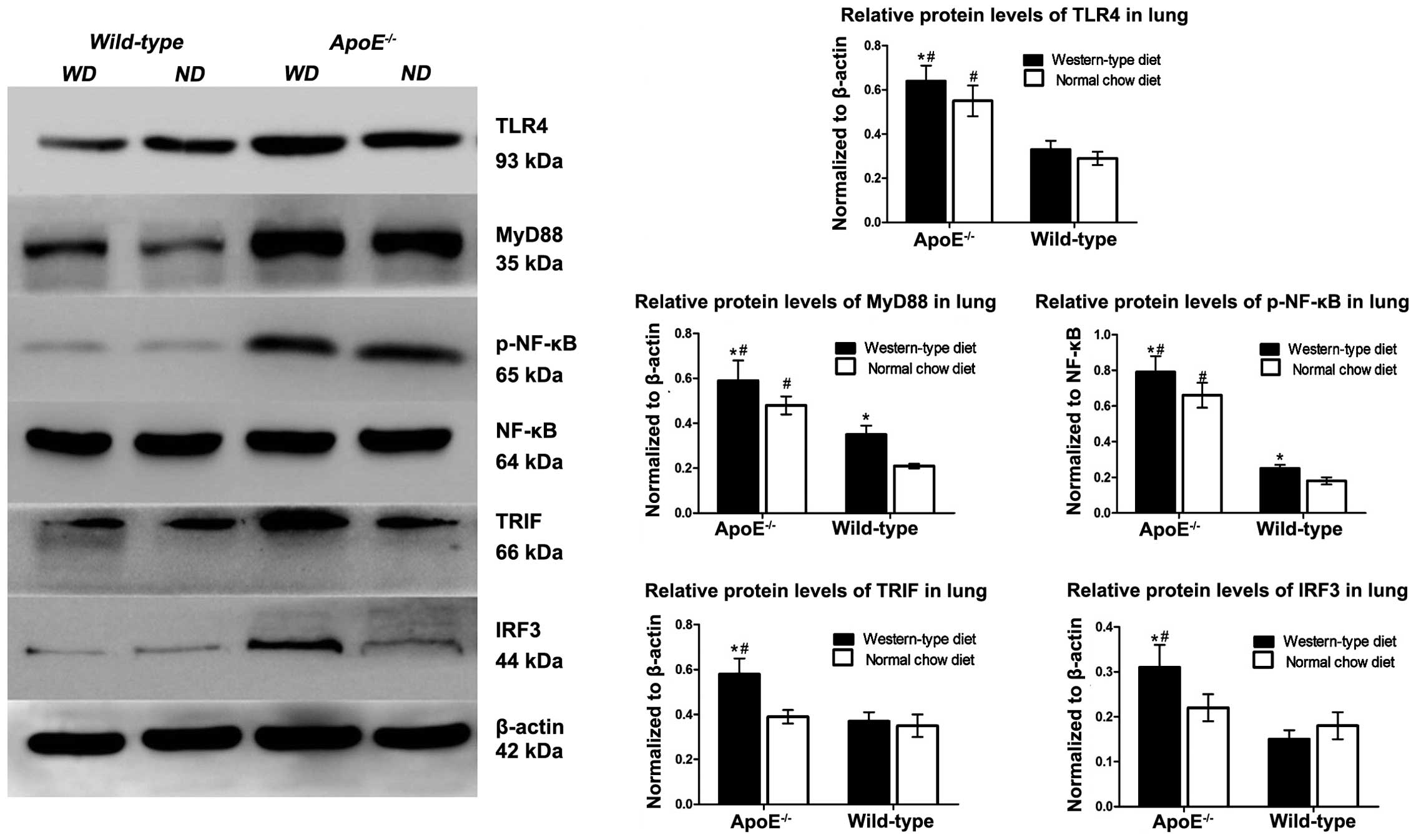
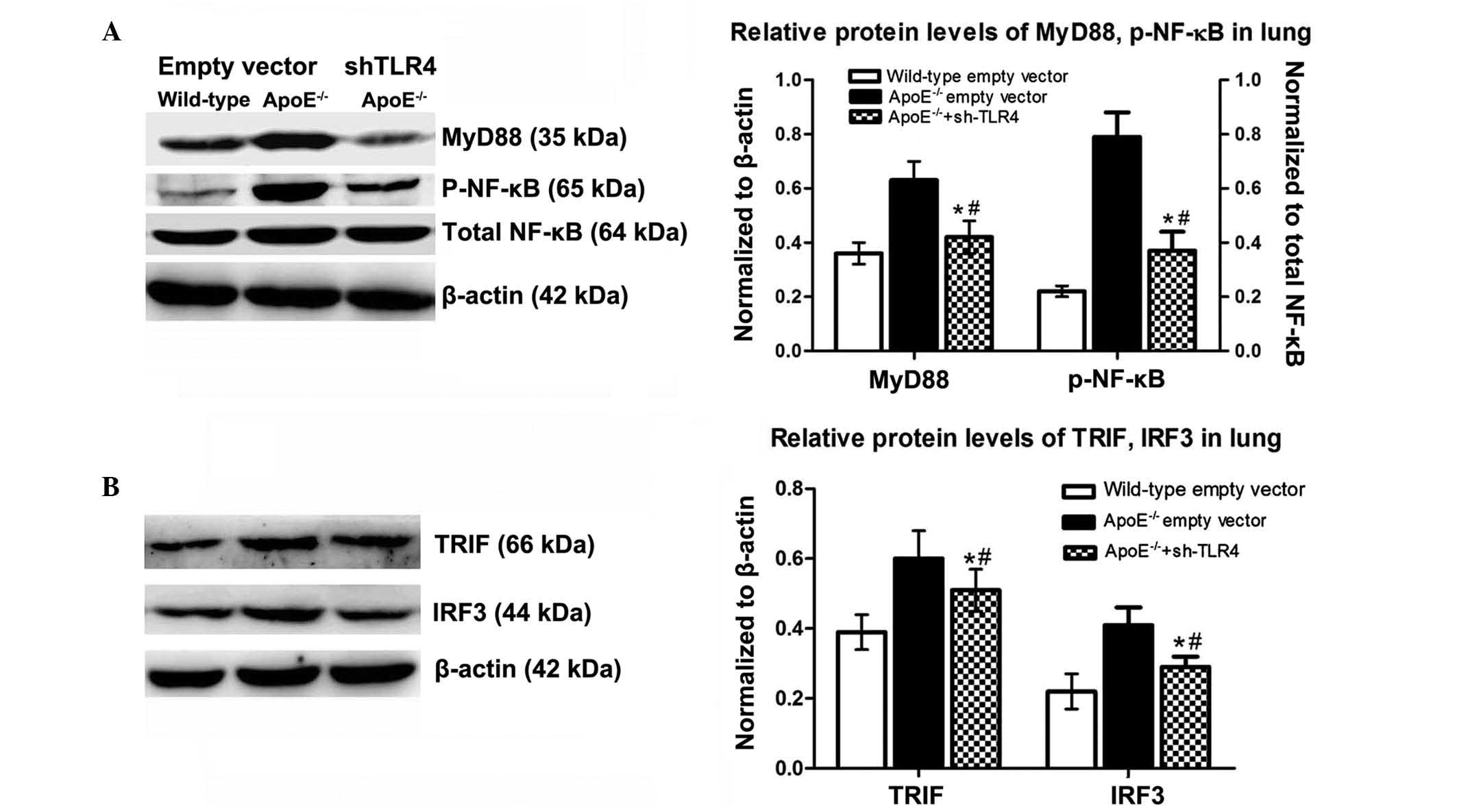
TLR4 and its downstream activation in
ApoE−/− WD mice
MyD88/NF-κB and TRIF/IRF3 are key downstream
molecules in the TLR4 signalling pathway. To determine whether
these molecules were involved in pulmonary inflammation and
lipidosis, the protein levels of TLR4, as well as its downstream
MyD88-dependent (MyD88, NF-κB) and -independent (TRIF and IRF3)
molecules in the lungs were detected by western blot analysis.
Following WD for 12 weeks, the levels of TLR4, MyD88 and p-NF-κB
were markedly upregulated in the ApoE−/− mice compared
with those in the B6 mice. The TRIF and IRF3 levels were
significantly increased (Fig. 3).
Among the mice receiving the ND, the levels of MyD88 and NF-κB in
the ApoE−/− mice were moderately elevated relative to
those in the corresponding B6 controls; however, the expression
levels of TRIF and IRF3 were marginally altered in the
ApoE−/− mice in comparison with those in the B6
animals.
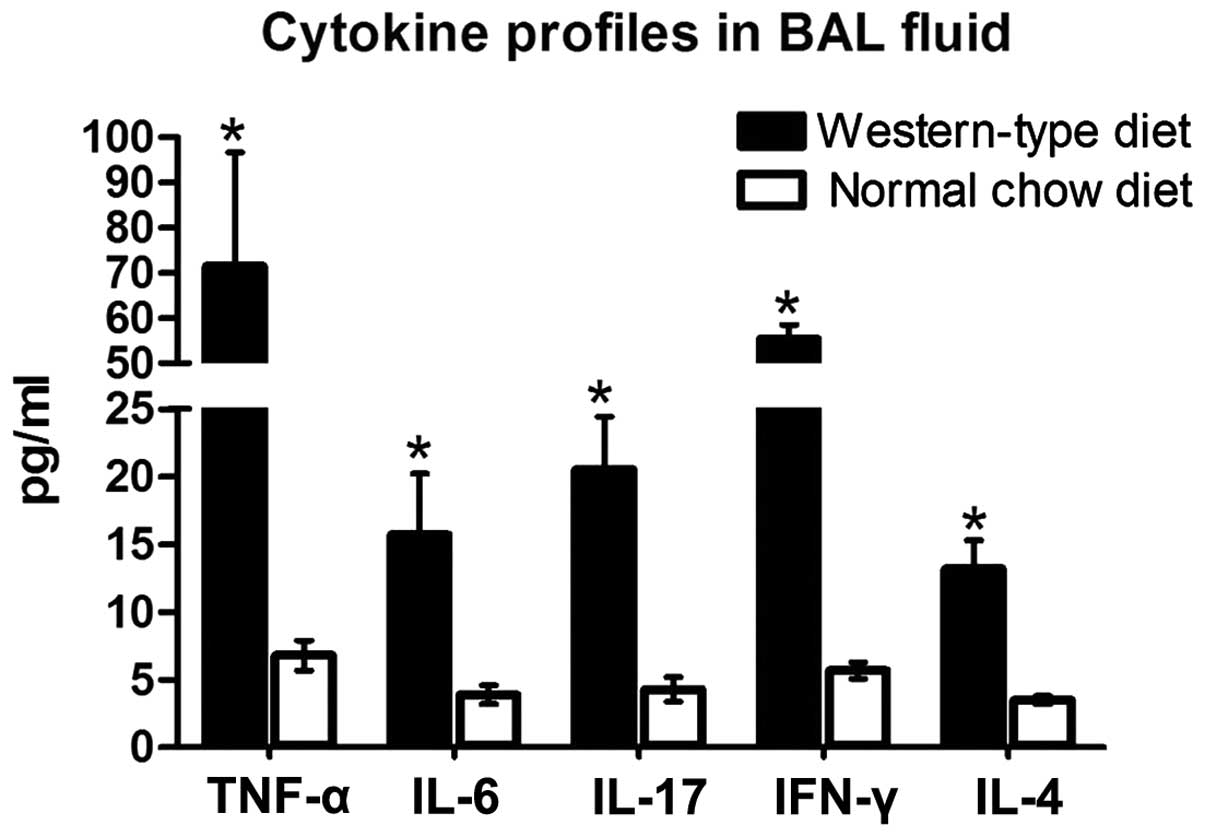
Increased BALF levels of IFN-γ, TNF-α,
IL-4, IL-6 and IL-17 in ApoE−/− WD mice
An inflammatory response involves the recruitment of
immune cells and changes in cytokines. The levels of IFN-γ, TNF-α,
IL-4. IL-6 and IL-17 in BALF were detected in the mice following
12-weeks of reciving their respective diet. In the
ApoE−/− mice, the levels of pro-inflammatory cytokines
IFN-γ, TNF-α, IL-6 and IL-17 in BALF were markedly elevated by
8.7-fold, 9.5-fold, 3.0-fold and 3.8-fold, respectively, compared
with those in the littermates on an ND. The anti-inflammatory
cytokine IL-4 was significantly increased by 2.7-fold (Fig. 4). The levels of these cytokines
were undetectable in the B6 mice receiving ND or WD.
Hyperlipidemia in the ApoE−/−
WD mice
The serum lipids were detected to evaluate the
effects of the high-fat Western-type diet on the lipid profiles of
the experimental mice. Compared with those of their corresponding
genotype mice receiving the ND, the serum TC, TG and non-HDL-C
levels were markedly elevated in the ApoE−/− mice
receiving the WD, and, to a lesser extent, in the wild-type mice
following 12 weeks of WD. The HDL-C levels in the
ApoE−/− mice decreased, whereas they remained comparable
in the wild-type mice (Table
I).
 | Table ILipid profile of the
ApoE−/− and wild-type mice fed on the high-fat
Western-type or normal chow diet for 12 weeks. |
Table I
Lipid profile of the
ApoE−/− and wild-type mice fed on the high-fat
Western-type or normal chow diet for 12 weeks.
| Lipid profile | ApoE−/−
| Wild-type
|
|---|
| High-fat
Western-type | Normal chow | High-fat
Western-type | Normal chow |
|---|
| TC (mmol/l) | 20.01±3.26a,b | 11.45±0.93b | 3.25±0.48a | 2.09±0.47 |
| TG (mmol/l) | 2.84±0.35a,b | 1.65±0.54b | 1.03±0.24a | 0.81±0.12 |
| Non-HDL
(mmol/l) | 18.83±0.41a,b | 10.8±2.84b | 1.71±0.37a | 0.38±0.55 |
| HDL-C (mmol/l) | 0.32±0.23a,b | 0.47±0.16b | 1.47±0.54 | 1.59±0.28 |
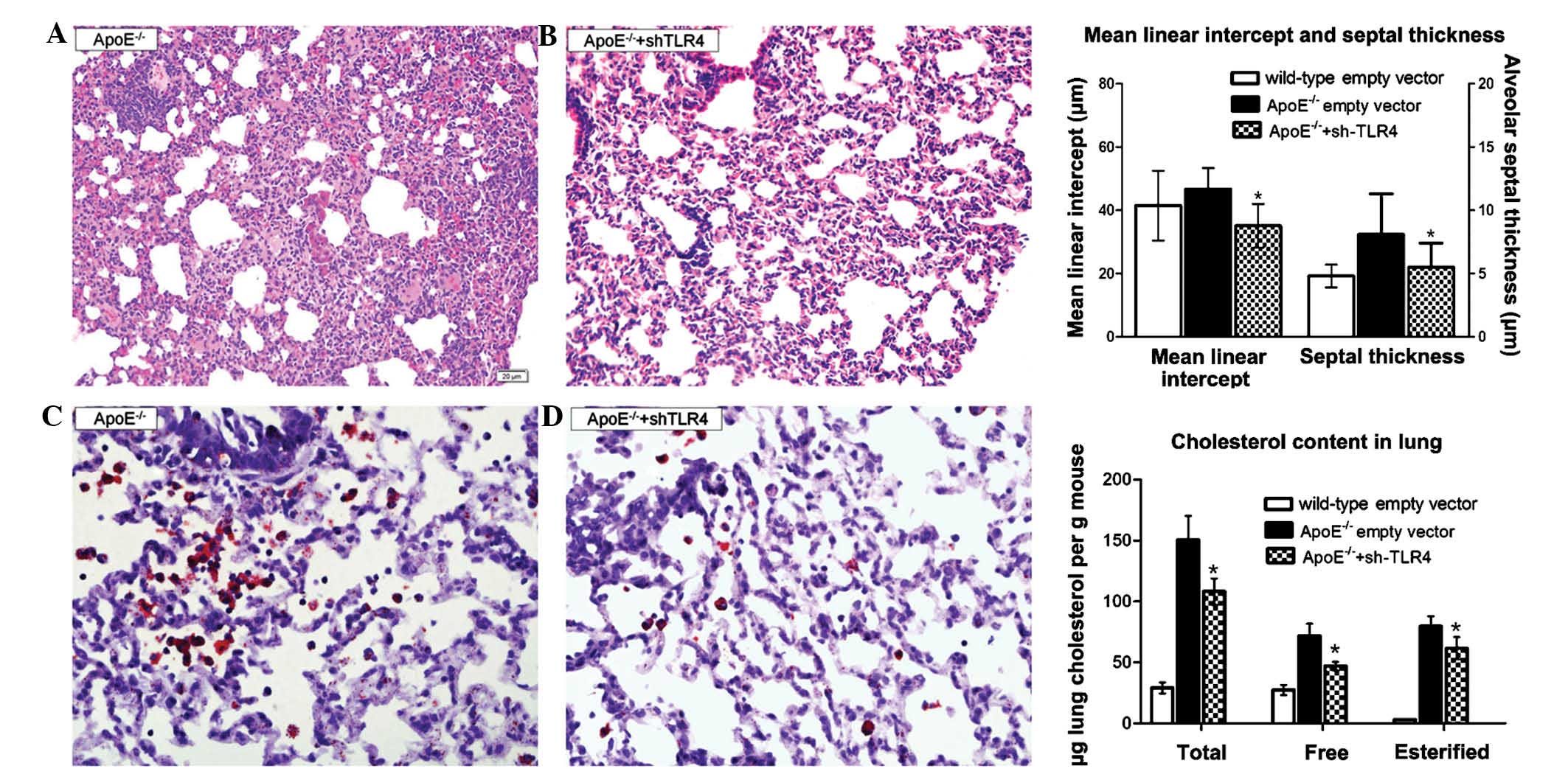
TLR4-targeted gene silencing ameliorates
inflammation and lipidosis in the ApoE−/− mice
To further validate whether the TLR4 molecule
contributed to lung injury in the ApoE−/− mice, the TLR4
pathway was blocked using Lv-shRNA-TLR4. Treatment with Lv-TLR4
shRNA for 12 weeks significantly attenuated the septal thickness
(5.5±1.9 µm vs. 8.1±3.2 µm) and the mean linear
intercept (35.1±6.9 µm vs. 46.7±11.6 µm). The lung
cholesterol content, including the esterified and free cholesterol,
was diminished (Fig 5). The serum
lipid profiles changed insignificantly with the TLR4-shRNA
lentivirus treatment (data not shown).
Inactivating the MyD88-dependent NF-κB
downstream pathways by TLR4 interference
Following TLR4-targeted gene silencing, all the
signalling molecules (MyD88, p-NF-κB, TRIF, IRF3) were
downregulated by 46, 53, 15, and 29%, respectively, with a
predominant inhibitory effect on the MyD88-dependent pathway. In
the ApoE−/− mice, levels of these signaling molecules in
the lung remained significantly higher than those of the B6
counterparts fed the WD (Fig.
6).
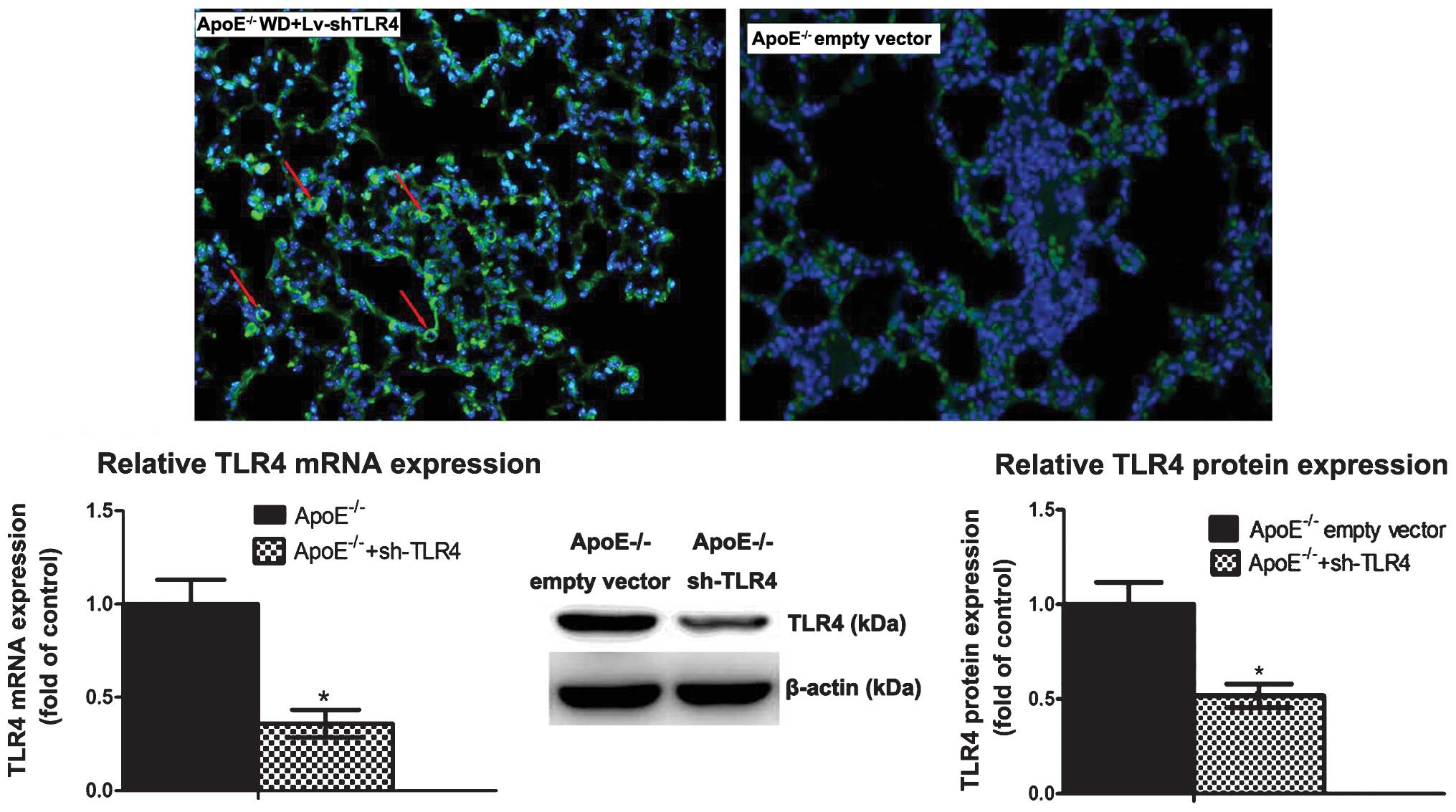
Efficiency and safety of lentivirus
transfection in vivo
GFP fluorescence in the lung was still observed at
12 weeks following transfection, which suggested a successful
transfection of shRNA lentivirus (Fig.
7). To further confirm the efficacy of lentivirus-mediated TLR4
gene silencing, the levels of TLR4 mRNA and protein in the lung
were determined. Compared with ApoE−/− WD mice, TLR4
mRNA expression in the Lv-sh-TLR4 subgroup was reduced by 64.1%
paralleled by a reduction of TLR4 protein by 49.3%. No adverse
effects occurred during the trial, indicating that lentivirus
transfection was safe (data not shown). Collectively, the results
demonstrated an efficient and safe lentivirus-mediated transfection
of shRNA in vivo.
Discussion
The present study reported that in genetically
susceptible ApoE−/− mice, a 12-week high-fat diet
induced pulmonary lipidosis, as illustrated by an elevated lung
cholesterol content and increased alveolar macrophage foam cell
formation. It was discovered that, dependent on the time period of
receiving the diet, the ApoE−/− WD mice exhibited
inflammatory injury that was characterized by initial leukocyte
recruitment (week 4), increased alveolar septal thickness, a mean
linear intercept (week 12) and granuloma formation (week 24). The
ApoE−/− ND mice or wild-type WD mice manifested a
low-grade or no inflammation. The expression of TLR4 and its
downstream molecules MyD88, p-NF-κB, TRIF and IRF3 were markedly
upregulated in the 12-week-old ApoE−/− WD mice, whereas
their expression was slightly changed in the ApoE−/− ND
and wild-type WD mice. Blocking the TLR4 pathway was able to
ameliorate lipidosis and inflammation in the ApoE−/− WD
mice. To the best of our knowledge, the present study was the first
to reveal that an ApoE deficiency combined with a high-fat diet
caused lung lipidosis and inflammation via the TLR4 signaling
pathway. Of note, it was found that the blocking of TLR4 could not
fully ameliorate lipidosis and inflammation, suggesting that other
signaling pathway(s) may be involved in those pathomorphological
changes.
Inflammatory response in
ApoE−/− WD mice
Evidence has indicated that the respiratory system
and cardiovascular system are intricately intertwined (15). It has been well documented that the
ApoE−/− mice on a high-fat diet developed pulmonary
arterial hypertension (16); those
on a Paigen diet exhibited more severe pulmonary hypertension
(17). An ApoE mimetic peptide was
able to prevent airway inflammation and goblet cell hyperplasia in
ApoE−/− mice challenged by house dust mites (18). The present study indicated that
ApoE−/− WD mice developed lung inflammation, which was
characterized by initial inflammatory cell infiltration, resultant
lipid phagocytosis and exudation, and ultimately, proliferation.
The findings of the present study are partially substantiated by a
study reporting that ApoE−/− mice on a WD for 10 weeks
developed inflammation and emphysema (3). However, the results of the present
study contradicted those reported by Samokhin et al
(4), who claimed that
ApoE−/− mice on a high-fat diet developed granulomas.
These conflicting results may be partly explained by the difference
in the lipid content in the diet and the duration of the high-fat
diet. The wild-type mice on the WD exhibited hypercholesterolemia
and hypertriglyceridemia with no evidence of lung inflammation and
lipidosis. Gene-diet interaction effects were possibly involved in
this outcome (19). In the
wild-type mice, lipotoxicity induced by the WD resulted in
microinflammation only. However, the ApoE-deficient mice treated
with the same diet exhibited obvious inflammation injury,
suggesting that lipotoxicity or ApoE deletion alone is not
sufficient to induce inflammation injury.
Pulmonary lipidosis in the
ApoE−/− WD mice
The primary function of ApoE is to facilitate lipid
transport into cells by receptor-mediated endocytosis mediated by
the low-density lipoprotein receptor. Adenosine
triphosphatase-binding cassette transporter A1 (ABCA1) mediates the
efflux of cholesterol to lipid-poor apolipoproteins (ApoA1 and
ApoE) (20). It was reported that
ABCA1−/− mice displayed lung cholesterol accumulation
and inflammation (21). The
results of the present study revealed that lung lipidosis occurred
in the ApoE−/− mice receiving the WD for 12 weeks.
Lipid-laden macrophages were scarce in the ApoE−/− WD
mice at 24 weeks, which indicated that the interaction between the
genetic and nongenetic factors occurred only at the critical
periods.
Several pulmonary diseases are associated with a
disruption of lipid homeostasis and inflammation, including
endogenous lipid pneumonia, alveolar proteinosis, respiratory
distress syndrome and Niemann Pick disease (22). The disease type of the lung
abnormity in ApoE−/− WD mice in the present study
remains inconclusive.
TLR4 and activation of downstream
molecules in ApoE−/− WD mice
TLR4 downstream signalling comprises at least two
distinct pathways as follows: The MyD88-dependent activation of the
NF-κB pathway that leads to the production of inflammatory
cytokines and a MyD88-independent pathway associated with the
production of interferon-beta and the maturation of dendritic
cells. The results of the present study showed that TLR4 signalling
activated the MyD88/NF-κB and the TRIF/IRF3 pathway to elicit lung
inflammation and lipidosis in the ApoE−/− WD mice. It
was indicated that MyD88 and p-NF-κB were elevated in the wild-type
mice fed a WD, which was partially consistent with a study
reporting that a high-fat diet led to the upregulation of TLR4 and
NF-κB expression in the intestines of wild-type mice (23).
The present study demonstrated that ApoE deficiency
in combination with a WD induces lipidosis and chronic inflammation
in lungs through the TLR4 pathway. There is evidence for the
correlation between respiratory and cardiovascular diseases
(24); however, the precise
mechanisms underlying this co-morbidity have remained elusive, and
research on the correlation of the two disorders is in its early
stages. It is well-documented that TLR4 signalling has an important
role in atherosclerosis (25). The
findings of the present study illustrated that TLR4 signalling may
be a possible common pathway that contributes to lung injury and
atherosclerosis, which may provide valuable information for
elucidating the lung-heart cross-talk. However, evidence provided
in the present study is limited, and future studies on the gene
silencing of MyD88 may be required to validate the involvement of
TLR4 downstream signaling in the WD-induced lung pathology in the
absence of ApoE more convincingly. Due to the complexity of the
mechanism of the gene-environment interaction (26), the neuroendocrine system, the
changes in the gene methylation pattern and the roles of other TLRs
in the animal model used in the present study, further
investigation is warranted.
Acknowledgments
The present study was supported by the Major
Scientific Research Project Foundation of Fujian Medical University
(grant no. 09ZD019), the Construct Foundation of the Key Clinical
Discipline of Fujian Medical University (grant no. XKPY201102), the
High-level Talent Development Foundation of Fujian Province (grant
no. 2109901) and Fujian Provincial Natural Science Foundation
(grant no. 2013J0101).
References
|
1
|
Massaro D and Massaro GD: Apoetm1Unc mice
have impaired alveologenesis, low lung function and rapid loss of
lung function. Am J Physiol Lung Cell Mol Physiol. 294:L991–L997.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Naura AS, Hans CP, Zerfaoui M, Errami Y,
Ju J, Kim H, Matrougui K, Kim JG and Boulares AH: High-fat diet
induces lung remodeling in ApoE-deficient mice: an association with
an increase in circulatory and lung inflammatory factors. Lab
Invest. 89:1243–1251. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Goldklang M, Golovatch P, Zelonina T,
Trischler J, Rabinowitz D, Lemaître V and D’Armiento J: Activation
of the TLR4 signaling pathway and abnormal cholesterol efflux lead
to emphysema in ApoE-deficient mice. Am J Physiol Lung Cell Mol
Physiol. 302:L1200–L1208. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Samokhin AO, Bühling F, Theissig F and
Brömme D: ApoE-deficient mice on cholate-containing high-fat diet
reveal a pathology similar to lung sarcoidosis. Am J Pathol.
176:1148–1156. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Byers DE and Holtzman MJ: Alternatively
activated macrophages and airway disease. Chest. 140:768–774. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Michelsen KS, Wong MH, Shah PK, Zhang W,
Yano J, Doherty TM, Akira S, Rajavashisth TB and Arditi M: Lack of
Toll-like receptor 4 or myeloid differentiation factor 88 reduces
atherosclerosis and alters plaque phenotype in mice deficient in
apolipoprotein E. Proc Natl Acad Sci USA. 101:10679–10684. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chaudhuri N, Whyte MK and Sabroe I:
Reducing the toll of inflammatory lung disease. Chest.
131:1550–1556. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ni JQ, Ouyang Q, Lin L, Huang Z, Lu H,
Chen X, Lin H, Wang Z, Xu D and Zhang Y: Role of toll-Like receptor
4 on lupus lung injury and atherosclerosis in LPS-challenge
ApoE(−/−) mice. Clin Dev Immunol. 2013:4768562013.
View Article : Google Scholar
|
|
9
|
Wendel DP, Taylor DG, Albertine KH,
Keating MT and Li DY: Impaired distal airway development in mice
lacking elastin. Am J Respir Cell Mol Biol. 23:320–326. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang X, Ren Q, Wu T, Guo Y, Liang Y and
Liu S: Ezetimibe prevents the development of non-alcoholic fatty
liver disease induced by high-fat diet in C57BL/6J mice. Mol Med
Rep. 10:2917–2923. 2014.PubMed/NCBI
|
|
11
|
Bates SR, Tao JQ, Collins HL, Francone OL
and Rothblat GH: Pulmonary abnormalities due to ABCA1 deficiency in
mice. Am J Physiol Lung Cell Mol Physiol. 289:L980–L989. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang Z, Chen N, Liu JB, Wu JB, Zhang J,
Zhang Y and Jiang X: Protective effect of resveratrol against acute
lung injury induced by lipopolysaccharide via inhibiting the
myd88-dependent Toll-like receptor 4 signaling pathway. Mol Med
Rep. 10:101–106. 2014.PubMed/NCBI
|
|
13
|
Moghadasian MH, McManus BM, Nguyen LB,
Shefer S, Nadji M, Godin DV, Green TJ, Hill J, Yang Y, Scudamore CH
and Frohlich JJ: Pathophysiology of apolipoprotein E deficiency in
mice: relevance to apo E-related disorders in humans. FASEB J.
15:2623–6230. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhu C, Chen YL, Wang XJ, Hu XS, Yu ZB and
Han SP: ShRNA-mediated gene silencing of AHR promotes the
differentiation of P19 mouse embryonic carcinoma cells into
cardiomyocytes. Mol Med Rep. 6:513–518. 2012.PubMed/NCBI
|
|
15
|
Dick TE, Hsieh YH, Dhingra RR, Baekey DM,
Galán RF, Wehrwein E and Morris KF: Cardiorespiratory coupling:
common rhythms in cardiac, sympathetic, and respiratory activities.
Prog Brain Res. 209:191–205. 2014.PubMed/NCBI
|
|
16
|
Hansmann G, Wagner RA, Schellong S, Perez
VA, Urashima T, Wang L, Sheikh AY, Suen RS, Stewart DJ and
Rabinovitch M: Pulmonary arterial hypertension is linked to insulin
resistance and reversed by peroxisome proliferator-activated
receptor-gamma activation. Circulation. 115:1275–1284.
2007.PubMed/NCBI
|
|
17
|
Lawrie A, Hameed AG, Chamberlain J, Arnold
N, Kennerley A, Hopkinson K, Pickworth J, Kiely DG, Crossman DC and
Francis SE: Paigen diet-fed apolipoprotein E knockout mice develop
severe pulmonary hypertension in an interleukin-1-dependent manner.
Am J Pathol. 179:1693–1705. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yao X, Remaley AT and Levine SJ: New kids
on the block: the emerging role of apolipoproteins in the
pathogenesis and treatment of asthma. Chest. 140:1048–1054. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Qi L: Gene-diet interactions in complex
disease: Current findings and relevance for public health. Curr
Nutr Rep. 1:222–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shang W, Yu X, Wang H, et al: Fibroblast
growth factor 21 enhances cholesterol efflux in THP-1
macrophage-derived foam cells. Mol Med Rep. 11:503–508. 2015.
|
|
21
|
Delvecchio CJ, Bilan P, Nair P and Capone
JP: LXR-induced reverse cholesterol transport in human airway
smooth muscle is mediated exclusively by ABCA1. Am J Physiol Lung
Cell Mol Physiol. 295:L949–L957. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Goldstein LS, Kavuru MS, Curtis-McCarthy
P, Christie HA, Farver C and Stoller JK: Pulmonary alveolar
proteinosis: clinical features and outcomes. Chest. 114:1357–1362.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang N, Wang H, Yao H, Wei Q, Mao XM,
Jiang T, Xiang J and Dila N: Expression and activity of the
TLR4/NF-κB signaling pathway in mouse intestine following
administration of a short-term high-fat diet. Exp Ther Med.
6:635–640. 2013.PubMed/NCBI
|
|
24
|
Nemmar A, Al-Maskari S, Ali BH and Al-Amri
IS: Cardiovascular and lung inflammatory effects induced by
systemically administered diesel exhaust particles in rats. Am J
Physiol Lung Cell Mol Physiol. 292:L664–L670. 2007. View Article : Google Scholar
|
|
25
|
Chansrichavala P, Chantharaksri U, Sritara
P and Chaiyaroj SC: Atorvastatin attenuates TLR4-mediated NF-kappaB
activation in a MyD88-dependent pathway. Asian Pac J Allergy
Immunol. 27:49–57. 2009.PubMed/NCBI
|
|
26
|
Ayala E, Kudelko KT, Haddad F, Zamanian RT
and de Jesus Perez V: The intersection of genes and environment:
development of pulmonary arterial hypertension in a patient with
hereditary hemorrhagic telangiectasia and stimulant exposure.
Chest. 141:1598–1600. 2012. View Article : Google Scholar : PubMed/NCBI
|