Introduction
Traumatic brain injury (TBI), a major public health
problem globally, is the leading cause of mortality and morbidity
in adults and children (1). TBI is
caused by primary as well as secondary injury mechanisms. Primary
damage is as a result of mechanical factors immediately following
trauma, while secondary injury is produced by complicating
processes which are initiated at the moment of impact but do not
present clinically for a period of time. Moreover, TBI induces an
amount of inflammatory responses that are believed to participate
in the pathogenesis of secondary injury (2). In particular, these inflammatory
responses incorporate the upregulation of adhesion cytokines,
permeation of neutro-phils and macrophages as well as activation of
glia and neurons (3). Tumor
necrosis factor α (TNF-α), interleukin-1β (IL-1β) and IL-6 are
crucial pro-inflammatory cytokines involved in the in flammatory
responses after TBI (4–6). In the serum and cerebral spinal fluid
of TBI patients and in brain parenchyma of animals with
experimental brain injuries, elevated levels of these cytokines
have been detected (7–9). In spite of the potential
pathophysiological function of these cytokines in TBI, their role
has remained controversial. Evidence from animal experiments has
implied that in the initial post-trauma period, elevated levels of
TNF-α, IL-1β and IL-6 are harmful, and suppressing the expression
of these cytokines may reduce tissue damage and brain edema, and
improve the functional outcome (3,10).
Furthermore, multiple previous studies have shown
that glutamate is the major excitatory neurotransmitter in the
brain (2,11). Accumulation of additional
extracellular glutamate and succeeding overstimulation of
glutamatergic receptors increases the production of excitotoxic
oxygen/nitrogen species, which induce oxidative stress resulting in
neuronal death (12). Moreover,
high-affinity glutamate transporters are able to clear the majority
of the glutamate from the extracellular space under physiological
conditions (13). There are five
different proteins within the glutamate transporter family in the
mammalian central nervous system (13). Excitatory amino acid transporter
(EAAT)1 and EAAT2, known as glutamate/aspartate transporter (GLAST)
and glutamate transporter-1 (GLT-1), respectively, are
predominantly expressed in astrocytes and account for 80% of the
total glutamate uptake in the brain (14). The potential of GLAST and GLT-1 to
limit extracellular glutamate levels makes them a potential target
in diseases associated with glutamate excitotoxicity (11,15).
Therefore, a pharmacological approach aimed at increasing glutamate
transporter protein levels may be an effective strategy for TBI
treatment.
Dextromethorphan (DM) is a non-narcotic anti-tussive
drug that initially attracted attention due to its anti-convulsant
and neuroprotective properties (16). Since then, multiple studies have
demonstrated that DM has a high safety profle in humans and is
neuroprotective in a variety of experimental injury models,
including cerebral ischemia, epilepsy, neurodegenerative disorders
and acute brain injury (17–19).
However, few experiments have explored the underlying mechanism of
the neuroprotective effect of DM in animals in the setting of
TBI.
To determine the potential mechanism of the
neuroprotective effect of DM following TBI, the present study aimed
to investigate the hypothesis that DM exerts neuroprotective
effects via attenuation of pro-inflammatory cytokines and
upregulation of glutamate transporter proteins following TBI in
rats.
Materials and methods
Animals
A total of 150 male Sprague-Dawley rats [obtained
from Shanghai Jiaotong University Experimental Animal Center
(Shanghai, China)], weighing 280–320 g, were allowed free access to
food and water under optimal keeping conditions (12-h light/dark
cycle; 22°C) prior to the operation. The study was performed in
accordance with the Institutional Guidelines for the Care and Use
of Laboratory Animals and was approved by the Shanghai Jiaotong
Univeristy School of Medicine (Shanghai, China).
Model of TBI
A previously described controlled cortical impact
(CCI) injury procedure was utilized. Rats were anesthetized with
sodium pentobarbital (i.p; 50 mg/kg; Solar Biotechology, Beijing,
China) and placed in a stereotaxic frame. A 5-mm craniotomy was
performed over the left parietal cortex, centred on the coronal
suture and 3 mm lateral to the sagittal suture. Considerable care
was taken to avoid injury to the underlying dura. Injury was
performed using a pneumatic piston with a rounded metal tip (2.5 mm
diameter) that was angled 22.5° vertically so that the tip was
perpendicular to the brain surface at the centre of the craniotomy.
A velocity of 4 m/s and a deformation depth 2 mm below the dura
were used. The bone flap was immediately replaced and sealed, and
the scalp was closed with sutures. The body temperature was
monitored throughout the surgery by a rectal probe, and the
temperature was maintained at 37.0±0.5°C using a heated pad. Rats
were placed in a heated cage to maintain body temperature while
recovering from anesthesia. Rats, which revealed no symptoms
following TBI were excluded from further experiments.
Group and drug administration
Rats were randomly assigned to a sham-operated group
(sham; n=30), a group which received TBI only and which was treated
with equal volumes of 0.9% saline solution (vehicle; n=60), and a
TBI group treated with DM (DM; n=60). DM was dissolved in 0.9%
saline and stored at 4°C. After brain injury, DM was immediately
administered by intraperitoneal injection in the DM group following
TBI (30 mg/kg body weight). All tests were run in a blinded manner,
and the animal codes were revealed only at the end of the
behavioral and histological analyses.
Evaluation of brain edema
Brain edema were evaluated by analysis of the brain
water content as described previously (2). Rat brains were separated and weighed
immediately with a chemical balance to determine the wet weight
(WW). Following drying in a desiccating oven for 24 h at 100°C, dry
tissues were weighed again to determine the constant dry weight
(DW). The percentage of water in the tissues was calculated
according to the formula: % Brain water = (WW-DW)/WW) x100.
Recovery of motoric function
The neurobehavioral status of the rats was evaluated
using a set of 10 tasks, collectively termed Neurologic Severity
Score (NSS), which tests reflexes, alertness, coordination and
motoric abilities (20). One point
is awarded for failure to perform a particular task; thus, a score
of 10 reflects maximal impairment, whereas a normal rat scores 0.
Post-injury, the NSS was evaluated at 1, 3 and 5 days. Each animal
was assessed by an observer who was blinded to the animal
treatment. The difference between the initial NSS and that at any
later time was calculated for each rat, and this value (ΔNSS)
reflected the spontaneous or treatment-induced recovery of motoric
function.
Immunofluorescence
Brain tissues were fixed in 4% para-formaldehyde
(Solar Biotechology) for 24 h and immersed in 30% sucrose solution
(Solar Biotechology) with 0.1 mol/l phosphate-buffered saline (PBS;
pH 7.4; Solar Biotechnology) until sinking to the bottom. Tissue
samples 200 µm apart from each section from the anterior to
the posterior cortex (bregma -1.90 to -3.00 mm) obtained from the
TBI rats and embedded in optimal cutting temperature resin. 15
µm frozen sections were sliced with a frozen slicer
microtome (Leika CM1950; Leika, Mannheim, Germany), treated with
0.4% Triton-100 for 10 min and blocked in normal donkey serum for 1
h. The frozen sections were incubated with mouse
anti-neuron-specific nuclear protein (NeuN) polyclonal antibody
(Santa Cruz Biotechnology, Dallas, TX, USA; diluted 1:100)
overnight at 4°C. The next day, sections were incubated with an
anti-mouse immunoglobulin (Ig)G (Santa Cruz Biotechnology; diluted
1:1,000) for 2 h at 37°C in the dark. Images were captured using a
laser scanning confocal microscope (Olympus FV1000; Olympus, Tokyo,
Japan). Primary antibodies were replaced with PBS in the negative
control group.
Western blot analysis
Briefly, rats were anesthetized and underwent
intracardiac perfusion with 0.1 mol/l phosphate-buffered saline
(PBS; pH 7.4). The cortex region of the brain was rapidly isolated,
total protein was extracted and the protein concentration was
determined using the bicin-choninic acid method (Solarbio, Beijing,
China). Samples were subjected to 30:0.8% (w/v)
acrylamide/bisacrylamide SDS-PAGE. Separated proteins on the gel
were transferred onto polyvinylidene difluoride membranes (Roche
Diagnostics, Mannheim, Germany). Blots were blocked with 5%
fat-free dry milk for 1 h at room temperature. Following blocking,
the membrane was incubated with indicated primary antibodies
overnight at 4°C, including rabbit anti-IL-1β polyclonal antibody
(Santa Cruz Biotechnology; diluted 1:500), rabbit anti-IL-6
polyclonal antibodies (Santa Cruz Biotechnology; diluted 1:500),
rabbit anti-TNFα polyclonal antibody (Santa Cruz Biotechnology;
diluted 1:500), rabbit anti-GLAST polyclonal antibody (Santa Cruz
Biotechnology; diluted 1:500), rabbit anti-GLT-1 polyclonal
antibody (Santa Cruz Biotechnology; diluted 1:500) and mouse
anti-β-actin monoclonal antibody (Santa Cruz Biotechnology; diluted
1:500) overnight at 4°C. Samples were then incubated with
horseradish peroxidase-conjugated anti-rabbit IgG and anti-mouse
IgG (Cell Signaling Technology, Inc., Danvers, MA, USA; diluted
1:5,000) for 2 h at room temperature. Following incubation with the
properly titrated secondary antibody, the immunoblot on the
membrane was visible after development with an enhanced
chemiluminescence detection system (Aoboxing Biotechnology,
Beijing, China) and densitometric signals were quantified using an
imaging program. Immunoreactive bands of all proteins expressed
were normalized to intensity of corresponding bands for β-actin.
The western blot results were analyzed using ImageJ 1.41 software
(National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
All values are expressed as the mean ± standard
deviation. SPSS 16.0 (SPSS, Inc., Chicago, IL, USA) was used for
statistical analysis of the data. Statistical analysis was
performed using analysis of variance followed by the
Student-Newman-Keuls post-hoc tests. P<0.05 was considered to
indicate a statistically significant difference between values.
Results
Treatment with DM attenuates TBI-induced
cerebral edema
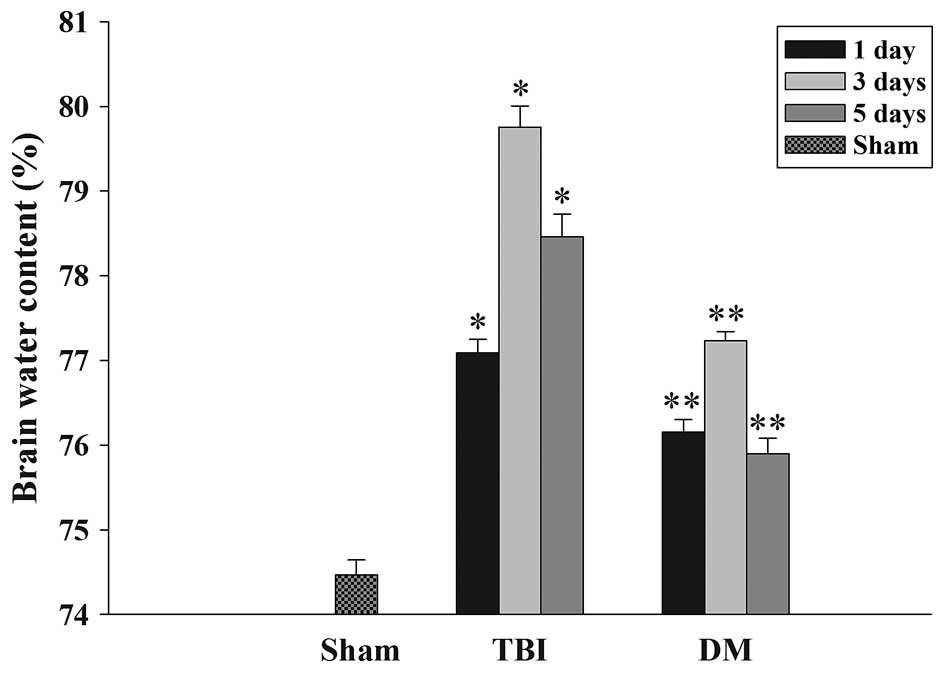
The wet-dry weight method was used to evaluate brain
edema. As shown in Fig. 1, the
brain water content was significantly increased in the TBI group
compared with that in the sham group at 1, 3 and 5 days after TBI.
Of note, the tissue water content in the DM treatment group was
significantly reduced at 1, 3 and 5 days compared with that in the
TBI group at the same time-point.
Treatment with DM attenuates TBI-induced
motoric deficits
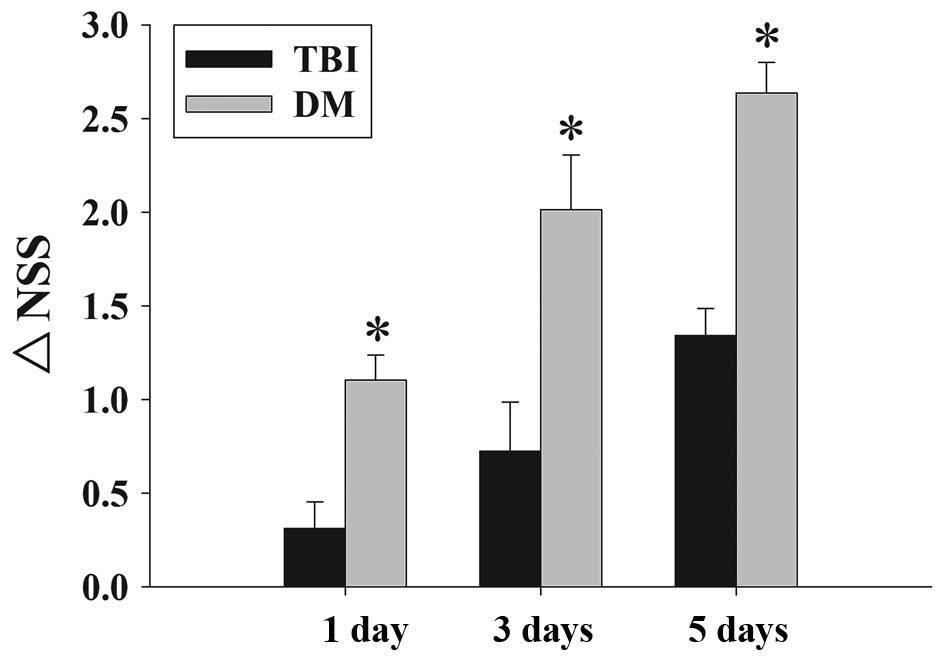
Fig. 2 depicts the
time-dependent changes in the functional recovery of the rats,
expressed as ΔNSS. The results clearly demonstrated that
post-injury administration of DM significantly improved motoric
function recovery at 1-5 days following TBI.
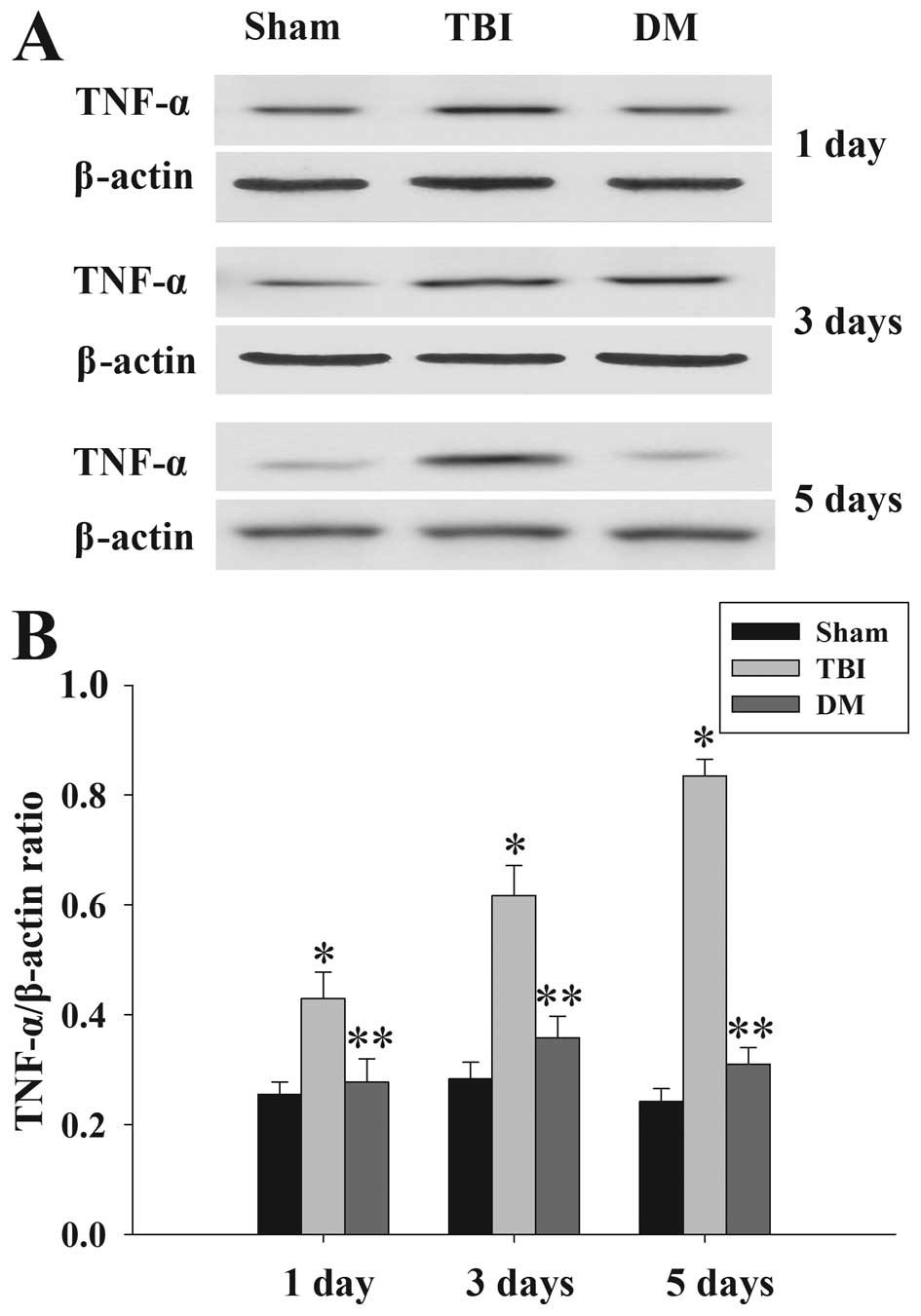
Treatment with DM attenuates TNF-α levels
in the cortex following TBI
The protein levels of TNF-α in the cortex at 1, 3
and 5 days were measured by western blot analysis (Fig. 3). TNF-α expression was
significantly increased at the various time-points in the TBI group
compared with that in the sham group. Of note, administration of DM
produced a significant reduction in the TBI-induced upregulation of
TNF-α expression.
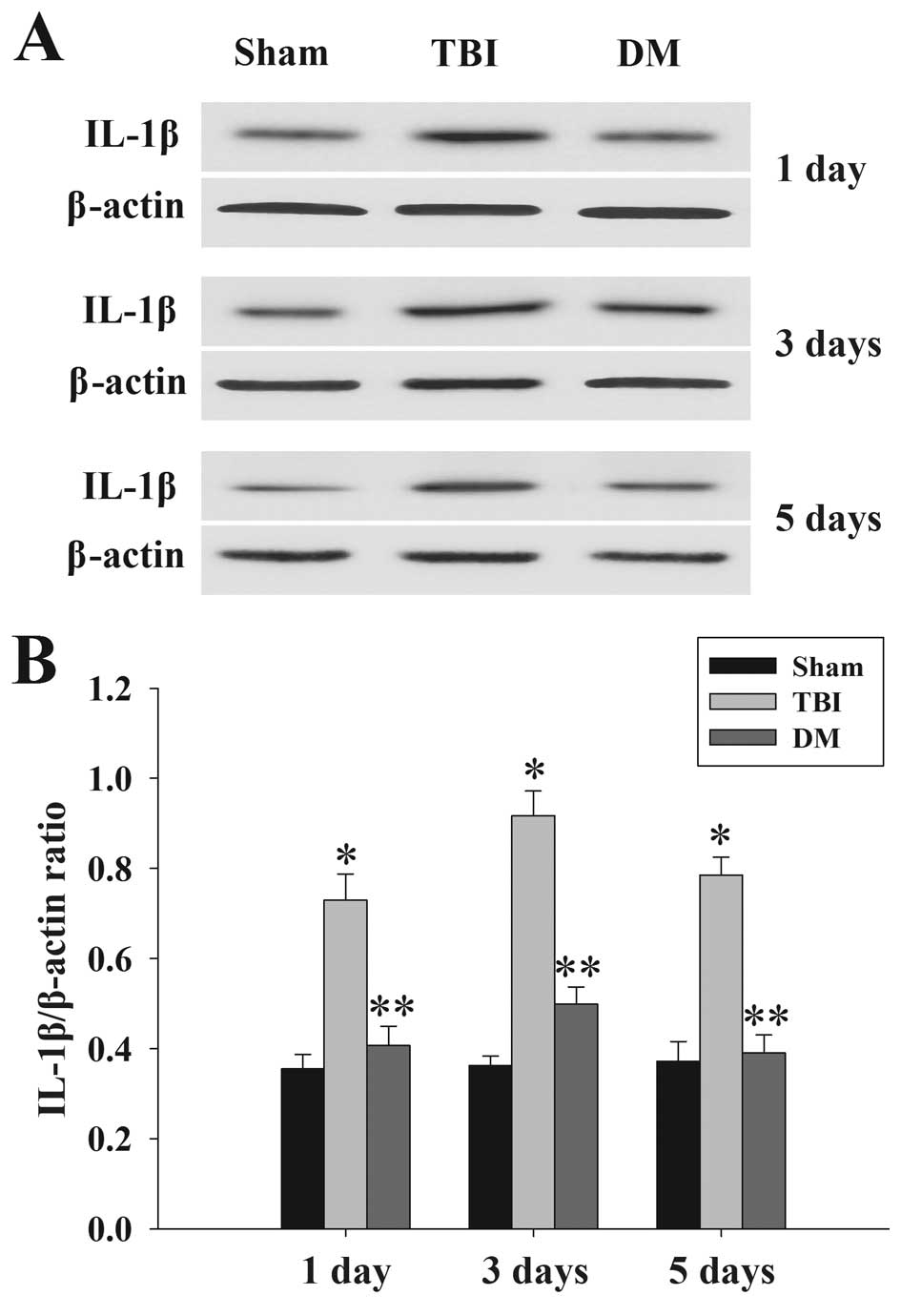
Treatment with DM attenuates IL-1β levels
in the cortex following TBI
The protein levels of IL-1β in the cortex at 1, 3
and 5 days were measured by western blot analysis. As shown in
Fig. 4, IL-1β expression in the
sham rat cortex at each time-point following injury was
consistently low, while being significantly increased in the TBI
group. Of note, the expression of IL-1β in the DM group was
significantly reduced compared with that in the TBI group at the
same time-points.
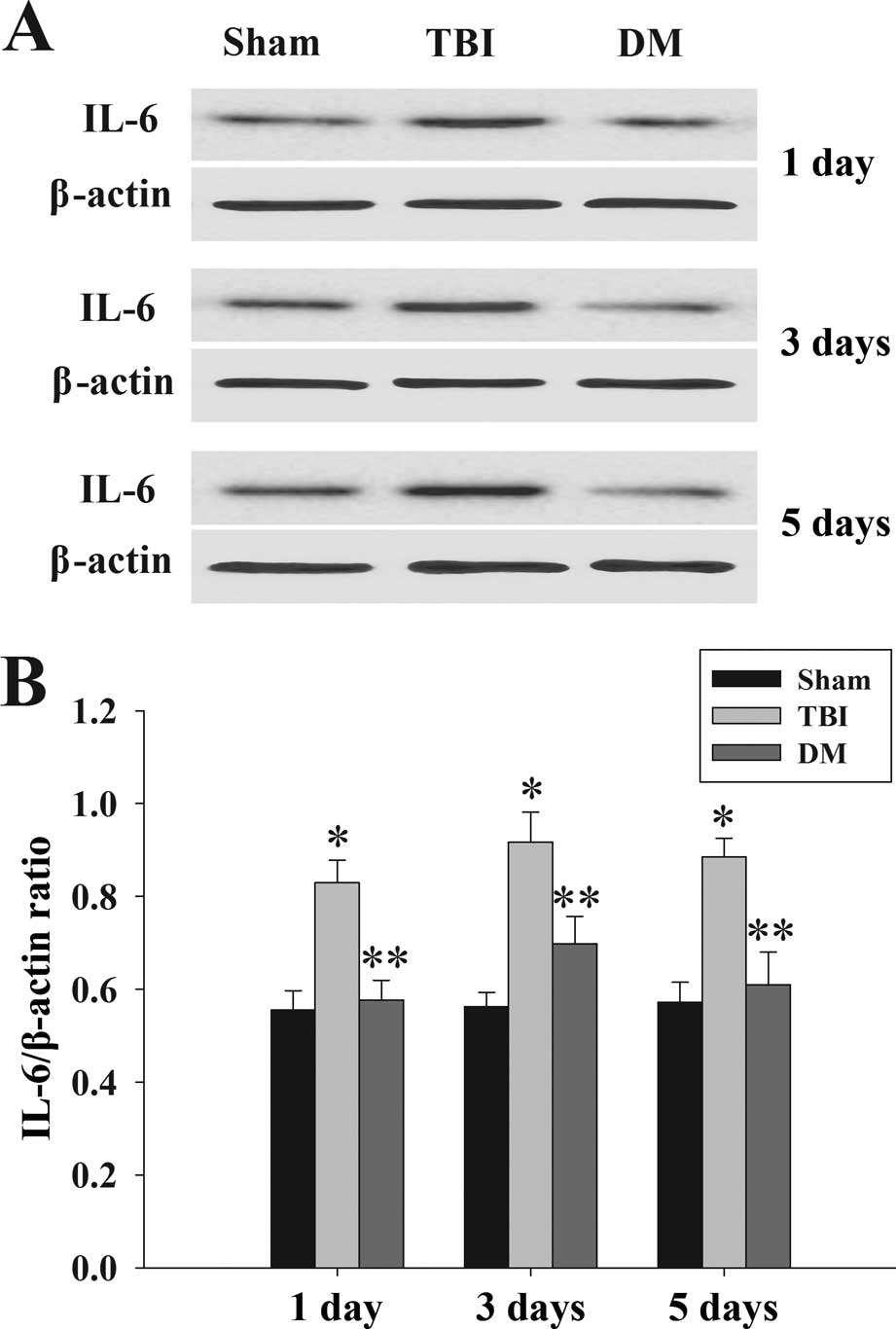
Treatment with DM attenuates IL-6 levels
in the cortex following TBI
The protein levels of IL-6 in the cortex at 1, 3 and
5 days were determined by western blot analysis. As shown in
Fig. 5, IL-6 expression was
significantly increased at various time-points in the TBI group
compared with that in the sham group. By contrast, treatment with
DM produced a significant reduction of IL-6 expression compared
with that in the TBI group.
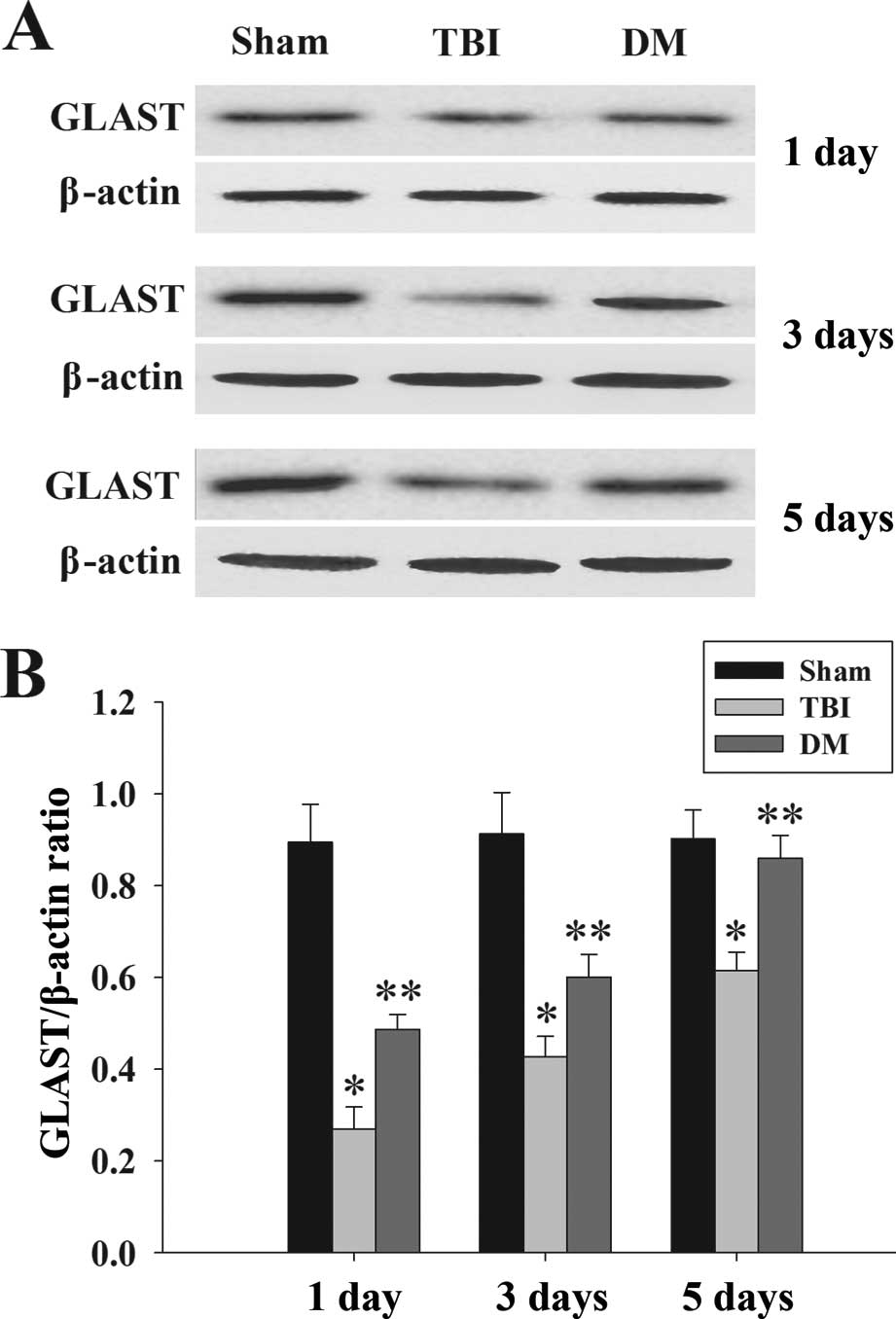
Treatment with DM increases GLAST protein
expression in the cortex following TBI
GLAST protein expression in the cortex was
determined by western blot analysis at 1, 3 and 5 days. As
demonstrated in Fig. 6, there was
a significant downregulation of GLAST expression in the TBI group
compared with that in the sham group. Of note, administration of DM
caused a marked elevation of GLAST at 1, 3 and 5 days compared to
that in the TBI group.
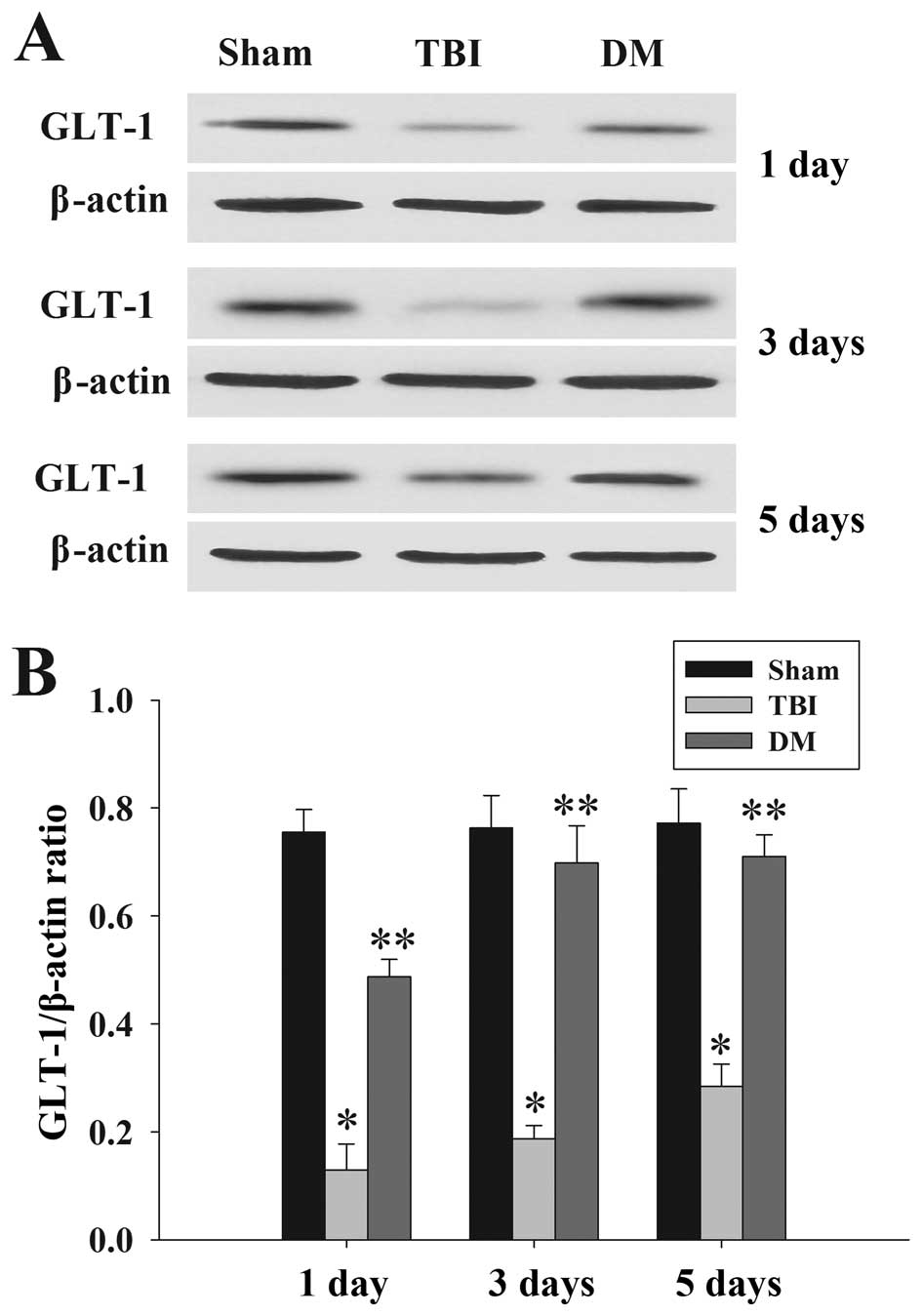
DM treatment increases GLT-1 expression
in the cortex following TBI
GLT-1 protein expression in the cortex was assessed
by western blot analysis at 1, 3 and 5 days. As demonstrated in
Fig. 7, there was a significant
downregulation of GLT-1 expression in the TBI group compared to
that in the sham group at 1, 3 and 5 days. DM produced a marked
elevation of GLT-1 expression compared with that in the TBI group
at the same time-points.
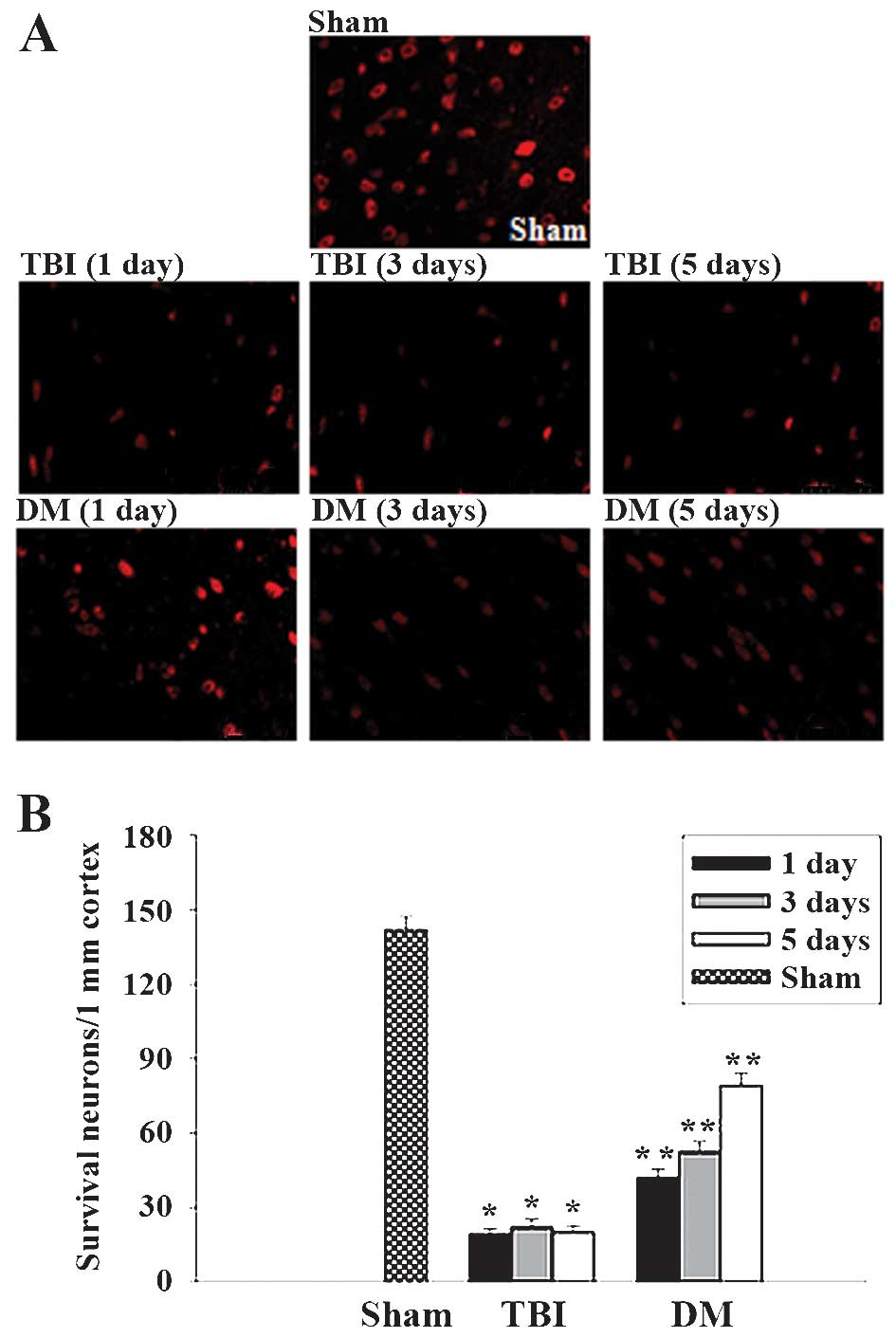
Treatment with DM increases neuronal
survival in the cortex following TBI
The cortex regions of brains were collected at 1, 3
and 5 days after TBI and subjected to immunostaining with the
neuronal marker NeuN. Neuronal survival was quantified by counting
the number of NeuN-positive cells per 1 mm length in the cortexes
of all rats. Representative images of NeuN-stained sections are
presented in Fig. 8A, and
quantified results from all rats are presented in Fig. 8B. As demonstrated in Fig. 8, at 1, 3 and 5 days following TBI,
administration of DM significantly increased neuronal survival in
the cortex compared to that in the TBI group.
Discussion
TBI is the main cause of death in children and young
adults worldwide (1). Studies
using animal models of TBI are important in the process of
understanding and evaluating the complex physiological, behavioral
and histopathological changes associated with TBI (21). However, head injury is an
unpredictable and spontaneous event, and no single animal model is
entirely successful in reproducing the complete pathological
changes observed following TBI in humans. In the present study, a
CCI model of TBI was used. This model is an invasive impact method
that was adapted from similar methods employed in experimental
spinal cord injury studies. A number of advantages of this model
include the ability to control deformation parameters, including
velocity, time and depth of impact. In addition, this model can
also mimic the whole spectrum of focal-type damage and diffuse
axonal injury. Therefore, any mice not showing moderate to severe
neurological deficits consistent with the surgery were excluded
from further study.
In the present study, the effectiveness of the
common anti-tussive agent DM as a therapeutic option for the
treatment of TBI was tested. The results showed that a single
injection of DM immediately following TBI significantly reduced
brain edema and neurological deficits as well as increased neuronal
survival. These effects correlated with a decrease of TNF-α, IL-1β
and IL-6 protein expression and an increase of GLAST and GLT-1 in
the cortex of the brain. Previous studies have demonstrated that DM
provides neuroprotection in a variety of experimental injury
models, including epilepsy, cerebral ischemia, neurodegenerative
disorders and acute brain injury (17–19).
Using the rat model of TBI, the present study confirmed and
extended these previous observations and demonstrated, for the
first time, that post-injury administration of DM provides a
neuroprotective function via attenuation of pro-inflammatory
cytokines and upregulation of glutamate transporter proteins
following experimental TBI in rats.
Inflammatory response induced following TBI is a
major contributing factor to secondary injury and has been shown to
be an important therapeutic target for reducing the extent of
tissue damage after injury (3).
TNF-α and IL-1β are potent enhancers of inflammatory reactions via
activating blood elements, capillary endothelial cells and glia, as
well as through enhancing the expression of downstream inflammatory
factors (22). Moreover, these two
cytokines also trigger the upregulation of IL-6, which has been
suggested to act in concert with TNF-α and IL-1β to mediate
multiple biological effects (23).
The present study showed, for the first time, that DM suppressed
the induction of pro-inflammatory cytokines in the injured brain
following TBI. This finding was in agreement with previous studies,
which indicated that DM inhibits several inflammatory processes
using other animal models (24,25).
Although the present study did not establish a direct causal
association between cytokine reduction and functional deficits,
several lines of evidence implied that TNF-α, IL-1β and IL-6 are
detrimental in the acute post-injury period and that inhibiting
cytokine activation or blocking cytokine receptors may have a
neuroprotective effect (3,10).
Excitotoxicity is widely recognized as a crucial
process in nerve cell death after acute brain injury (2). Previous studies demonstrated that the
neuroprotective properties of DM appear to be functionally
associated with its inhibitory effects on glutamate-induced
neurotoxicity via its N-methyl-d-aspartate receptor antagonist
(26), sigma-1 receptor agonist
functions (27) and voltage-gated
calcium channel antagonist (28).
Furthermore, it is worth mentioning that a recent study
demonstrated that GLAST and GLT-1 appeared to be inhibited by MeHg
exposure, and these alterations were significantly prevented by
pre-treatment with DM (29).
However, no previous study has assessed GLAST and GLT-1 expression
after DM treatment in a TBI model.
The present study found that GLAST and GLT-1
downregulation was induced after TBI and that this phenomenon was
attenuated by administration of DM. These findings emphasized that
DM exerts its neuroprotective effects, at least in part via its
anti-excitotoxicity effects following experimental TBI in rats.
In conclusion, the present study demonstrated that
administration of DM reduced brain edema and neurological deficits
as well as increased neuronal survival in a rat model of TBI.
Furthermore, DM decreased TNF-α, IL-1β and IL-6 protein expression
and upregulated GLAST and GLT-1 protein expression in the cortex of
the brain. These findings emphasized that DM exerts its
neuroprotective function via anti-inflammatory and
anti-excitotoxicity effects following experimental TBI in rats. The
present study has shed light on the potential use of DM as a
neuroprotective agent in the treatment of cerebral injuries.
Abbreviations:
|
DM
|
dextromethorphan
|
|
TBI
|
traumatic brain injury
|
|
CCI
|
controlled cortical impact
|
|
NSS
|
neurologic severity score
|
|
NeuN
|
neuron-specific nuclear protein
|
|
TNF-α
|
tumor necrosis factor α
|
|
IL-6
|
interleukin-6
|
|
IL-1β
|
interleukin-1β
|
|
GLAST
|
glutamate/aspartate transporter
|
|
GLT-1
|
glutamate transporter-1
|
References
|
1
|
Luo CL, Li BX, Chen XP, et al: Autophagy
is involved in traumatic brain injury-induced cell death and
contributes to functional outcome deficits in mice. Neuroscience.
184:54–63. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cui C, Cui Y, Gao J, et al:
Neuroprotective effect of ceftriaxone in a rat model of traumatic
brain injury. Neurol Sci. 35:695–700. 2014. View Article : Google Scholar
|
|
3
|
Morganti-Kossmann MC, Rancan M, Stahel PF
and Kossmann T: Inflammatory response in acute traumatic brain
injury: a double-edged sword. Curr Opin Crit Care. 8:101–105. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Feuerstein GZ, Liu T and Barone FC:
Cytokines, inflammation and brain injury: role of tumor necrosis
factor-alpha. Cerebrovasc Brain Metab Rev. 6:341–360.
1994.PubMed/NCBI
|
|
5
|
Aibiki M, Maekawa S, Ogura S, Kinoshita Y,
Kawai N and Yokono S: Effect of moderate hypothermia on systemic
and internal jugular plasma IL-6 levels after traumatic brain
injury in humans. J Neurotrauma. 16:225–232. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Taupin V, Toulmond S, Serrano A, Benavides
J and Zavala F: Increase in IL-6, IL-1 and TNF levels in rat brain
following traumatic lesion: Influence of pre-and post-traumatic
treatment with Ro5 4864, a peripheral-type (p site) benzodiazepine
ligand. J Neuroimmunol. 42:177–185. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Goodman JC, Robertson CS, Grossman RG and
Narayan RK: Elevation of tumor necrosis factor in head injury. J
Neuroimmunol. 30:213–217. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Csuka E, Morganti-Kossmann MC, Lenzlinger
PM, Joller H, Trentz O and Kossmann T: IL-10 levels in
cerebrospinal fluid and serum of patients with severe traumatic
brain injury: relationship to IL-6, TNF-α, TGF-β1 and blood-brain
barrier function. J Neuroimmunol. 101:211–221. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hayakata T, Shiozaki T, Tasaki O, et al:
Changes in CSF S100B and cytokine concentrations in early-phase
severe traumatic brain injury. Shock. 22:102–107. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lu KT, Wang YW, Yang JT, Yang YL and Chen
HI: Effect of interleukin-1 on traumatic brain injury-induced
damage to hippocampal neurons. J Neurotrauma. 22:885–895. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Globus MY, Alonso O, Dietrich WD, Busto R
and Ginsberg MD: Glutamate release and free radical production
following brain injury: effects of posttraumatic hypothermia. J
Neurochem. 65:1704–1711. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yi JH and Hazell AS: Excitotoxic
mechanisms and the role of astrocytic glutamate transporters in
traumatic brain injury. Neurochem Int. 48:394–403. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kanai Y and Hediger MA: Primary structure
and functional characterization of a high-affinity glutamate
transporter. Nature. 360:467–471. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pawlak J, Brito V, Küppers E and Beyer C:
Regulation of glutamate transporter GLAST and GLT-1 expression in
astrocytes by estrogen. Brain Res Mol Brain Res. 138:1–7. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tanaka K, Watase K, Manabe T, et al:
Epilepsy and exacerbation of brain injury in mice lacking the
glutamate transporter GLT-1. Science. 276:1699–1702. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tortella FC, Britton P, Williams A, Lu XC
and Newman AH: Neuroprotection (focal ischemia) and neurotoxicity
(electroen-cephalographic) studies in rats with AHN649, a 3-amino
analog of dextromethorphan and low-affinity N-methyl-D-aspartate
antagonist. J Pharmacol Exp Ther. 291:399–408. 1999.PubMed/NCBI
|
|
17
|
Werling LL, Lauterbach EC and Calef U:
Dextromethorphan as a potential neuroprotective agent with unique
mechanisms of action. Neurologist. 13:272–293. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang W, Wang T, Qin L, et al:
Neuroprotective effect of dextromethorphan in the MPTP Parkinson's
disease model: role of NADPH oxidase. FASEB J. 18:589–591.
2004.PubMed/NCBI
|
|
19
|
Duhaime AC, Gennarelli LM and Boardman C:
Neuroprotection by dextromethorphan in acute experimental subdural
hematoma in the rat. J Neurotrauma. 13:79–84. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Beni-Adani L, Gozes I, Cohen Y, et al: A
peptide derived from activity-dependent neuroprotective protein
(ADNP) ameliorates injury response in closed head injury in mice. J
Pharmacol Exp Ther. 296:57–63. 2001.
|
|
21
|
Povlishock JT, Hayes RL, Michel ME and
Mcintosh TK: Workshop on animal models of traumatic brain injury. J
Neurotrauma. 11:723–732. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang CX and Shuaib A: Involvement of
inflammatory cytokines in central nervous system injury. Prog
Neurobiol. 67:161–172. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Allan SM and Rothwell NJ: Cytokines and
acute neurodegeneration. Nat Rev Neurosci. 2:734–744. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu Y, Qin L, Li G, et al:
Dextromethorphan protects dopa-minergic neurons against
inflammation-mediated degeneration through inhibition of microglial
activation. J Pharmacol Exp Ther. 305:212–218. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gao HM, Liu B, Zhang W and Hong JS: Novel
anti-inflammatory therapy for Parkinson's disease. Trends Pharmacol
Sci. 24:395–401. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tortella FC, Pellicano M and Bowery NG:
Dextromethorphan and neuromodulation: old drug coughs up new
activities. Trends Pharmacol Sci. 10:501–507. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Klette KL, Lin Y, Clapp LE, DeCoster MA,
Moreton J and Tortella FC: Neuroprotective sigma ligands attenuate
NMDA and trans-ACPD-induced calcium signaling in rat primary
neurons. Brain Res. 756:231–240. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Carpenter CL, Marks SS, Watson DL and
Greenberg DA: Dextromethorphan and dextrorphan as calcium channel
antagonists. Brain Res. 439:372–375. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Feng S, Xu Z, Liu W, Li Y, Deng Y and Xu
B: Preventive effects of dextromethorphan on methylmercury-induced
glutamate dyshomeostasis and oxidative damage in rat cerebral
cortex. Biol Trace Elem Res. 159:332–345. 2014. View Article : Google Scholar : PubMed/NCBI
|