Introduction
Chronic kidney disease (CKD) often begins with
urinary protein loss (proteinuria), an early sign of kidney injury
that constitutes a risk factor for further progressive destruction
of the kidney. Proteinuria stems from injury to podocytes. Loss of
podocytes or failure of podocyte function contributes to the
development of glomerulosclerosis, which is the final stage of
various renal diseases (1). A
study demonstrated that there may be a close association between
vascular endothelial growth factor (VEGF) and proteinuria (2). VEGF is an important regulator of
angiogenesis. In addition, podocytes are the major source of VEGF
production in the glomerulus (3,4). The
VEGF family incorporates five ligands that can bind differentially
to three receptor tyrosine kinases (VEGFR-1, -2 and -3). Recent
studies have demonstrated that plasma VEGF levels are increased in
CKD and podocyte-derived VEGF is upregulated in the early stages of
diabetic nephropathy (5–7). The therapeutic effects of anti-VEGF
strategies were partially shown to prevent albuminuria in diabetic
rodents and prevent the complications of CKD (8–11).
It was reported that primary or secondary elevations in VEGFR-1
(sFlt-1) may lead to decreased tissue levels of VEGF-A and the
organ defects observed in preeclampsia (12). By contrast, Eremina et al
(13) demonstrated that VEGF
production by podocytes is also required to maintain the integrity
of the glomerular basement membrane once fully formed, as
pharmacologic and genetic inhibition of VEGF in mature podocytes
resulted in glomerular endothelial cell damage and thrombotic
microangiopathy (13).
From existing literature, the role of VEGF
production by podocytes is well established; however, the
expression and/or function(s) of VEGF receptors within the podocyte
is less clear and is an area of discussion. It is also unclear
whether VEGF can directly affect podocytes or is central to the
pathogenesis of proteinuria. The aim of the present study was to
identify the role of VEGF and its receptors in podocytes.
Materials and methods
Antibodies and reagents
The following commercially available antibodies were
used: Anti-β-actin (cat. no. sc-8432), anti-synaptopodin (cat. no.
sc-50459), anti-VEGFR1 (cat. no. sc-271789),
anti-phosphorylated-VEGFR2 (cat. no. sc-16629), anti-VEGFR2 (cat.
no. sc-6251) and horseradish peroxidase-conjugated rabbit
anti-mouse immunoglobulin G secondary antibody (cat. no. sc-358918;
Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), horseradish
peroxidase-conjugated anti-rabbit secondary antibody (cat. no.
sc-2007; Santa Cruz Biotechnology, Inc.)
anti-phosphorylated-extracellular signal-regulated kinases (ERK;
Cell Signaling Technology Inc., Beverly, MA, USA). SU5416 an
inhibitor of VEGFR2 was obtained from Pfizer Inc. (New York, NY,
USA). A bicinchoninic acid (BCA) protein assay kit and
nitrocellulose membranes were obtained from Bio-Rad (Hercules, CA,
USA). ELISA for the Quantikine mouse VEGF-A immunoassay was
purchased from R&D Systems (Minneapolis, MN, USA). A RNAeasy
Mini kit was obtained from Qiagen (Hilden, Germany).
Cell cultures
A thermosensitive, SV40-transfected immortalized
mouse podocyte cell line was provided by Dr Peter Mundel (Mount
Sinai School of Medicine, New York, NY, USA). The conditionally
immortalized mouse podocytes carry a temperature-sensitive variant
of the SV-40 large T antigen (tsA58) that is stimulated by mouse
interferon (IFN)-γ and is stable at 33°C as described previously
(14). At 33°C, cells were left to
proliferate in RPMI-1640 medium (Sigma-Aldrich, St. Louis, MO, USA)
supplemented with 10–20 U/ml mouse recombinant IFN-γ (Peprotech,
Rocky Hill, NJ, USA) 100 U/ml penicillin/streptomycin
(Sigma-Aldrich) and 10% fetal calf serum (FCS; Fuzhou Maixin
Biotechnology Development Co., Ltd., Fuzhou, China). To induce
differentiation, cells were thermoshifted to 37°C for two weeks
without IFN-γ. These cells showed an epithelial morphology with a
polyhedral shape and were detected by synaptopodin, a
differentiated podocyte-specific marker, using immunofluorescence
staining (Fig. 1A) (15).
Experimental design
Stimulation experiments were performed with
Angiotensin II (Ang-II; Fuzhou Maixin Biotechnology Development
Co., Ltd). Ang-II concentrations (10−5, 10−6,
10−7, 10−8 and 10−9 M) resulting
in significant VEGF-A activation were used for time course
experiments (24, 36, 48 and 60 h). The synchronized differentiated
podocytes were either untreated or treated with Ang-II
(10−7 M) for 48 h to achieve a podocyte injury pattern.
To define the effect of the angiotensin type 1 receptor blocker
(ARB), irbesartan (10−4 M; Sigma-Aldrich), or a VEGF
receptor 2 inhibitor, SU5416, on Ang-II-stimulated podocytes
(16,17), these two compounds were added to
podocytes 1 h prior to treatment with Ang-II. All experimental
groups were collected for extraction of total RNA and protein after
exposure to experimental conditions for 48 h. Three independent
experiments were performed.
ELISA
VEGF-A concentrations were measured in the
supernatant of podocytes by ELISA using a Quantikine human VEGF-A
Immunoassay. The supernatant was left on cells for 48 h. All
experiments were performed in three independent experimental
setups.
Immunofluorescent staining
Differentiated mouse podocytes were seeded on
collagen I-coated glass coverslips (Shanghai Precision &
Scientific Instrument Co., Ltd., Shanghai, China) for two weeks at
37°C, washed three times in 1X phosphate-buffered saline (PBS) for
5 min, then fixed with 4% formaldehyde for 30 min at 37°C and
rinsed with PBS. Subsequently, 0.2% Triton X-100 (Sigma-Aldrich) in
PBS was added for 20 min to permeabilize the cells. Podocytes on
glass coverslips were washed with PBS again, blocked with 2% FCS
for 30 min at 37°C, then incubated with an anti-mouse synaptopodin
antibody in 1:200 dilution at 4°C overnight. Cells were then washed
with PBS and incubated with secondary antibody in the dark for 30
min at room temperature. Photomicrographs of each section of the
groups were observed at ×100 magnification with a fluorescence
microscope (BX61; Olympus Corporation, Tokyo, Japan).
Western blot analysis
Podocytes from five experimental groups (control,
Ang-II, SU5416, Ang-II + SU5416, and Ang-II + irbesartan) were
collected and total protein concentration was measured using a BCA
protein assay kit. Protein (30 μg) was electrophoresed on 5%
SDS-PAGE and transferred onto a nitrocellulose membrane. The
membrane was blocked in 5% non-fat milk in Tris-buffered
saline/0.15% Tween-20 (TBST) for 2 h at room temperature and
hybridized overnight at 4°C with one of the primary antibodies.
After three washes in TBST, membranes were incubated with the
secondary antibody for 1 h at room temperature. The specific
protein band on membranes was detected by computer-assisted video
densitometry and its density was measured by using the Image Reader
LAS-4000 (Fujifilm Corp., Tokyo, Japan).
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total mRNA was purified from differentiated mouse
podocytes using an RNAeasy Mini kit (Qiagen, Hilden, Germany)
according to the manufacturer's instructions. RNA concentration was
assessed by spectrophotometry at 260 and 280 nm. Total mRNA (1
μg) was used for reverse transcription to generate cDNA
following a reported protocol (18). The reverse transcription kits were
provised by Bioteke Corp. (Beijing, China). The resulting cDNA was
used as a template for PCR amplification. Next, cDNA was amplified
with the following PCR specific primers: Sense: 5′-GCC TGG TCT ACA
TAC AGA GTG AG-3′ and antisense: 5′-TCT AGT CCT CAG ACC CAG TCA
TA-3′ for mouse transforming growth factor (TGF)-β1; and sense:
5′-GTC CCT CAC CCT CCC AAA AG-3′ and antisense: 5′-GCT GCC TCA ACA
CCT CAA CCC-3′ for mouse β-actin. PCR conditions were as follows:
Denaturation at 92°C for 2 min, 35 cycles of denaturation at 94°C
for 30 sec, annealing at 55°C for 30 sec, and extension at 72°C for
30 sec, followed by final extension at 72°C for 10 min. The RT-qPCR
products were run on a 2% agarose gel with ethidium bromide
staining by electrophoresis. The predicted band sizes were 699 bp
for TGF-β1 and 266 bp for mouse β-actin. The abundance of TGF-β1
mRNA was normalized to β-actin.
Statistical analysis
All data are presented as the mean ± standard error
of the mean. Differences between groups were analyzed by a
two-sample t-test or one-way analysis of variance using GraphPad
Prism software version 3.03 (GraphPad Software, San Diego, CA,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
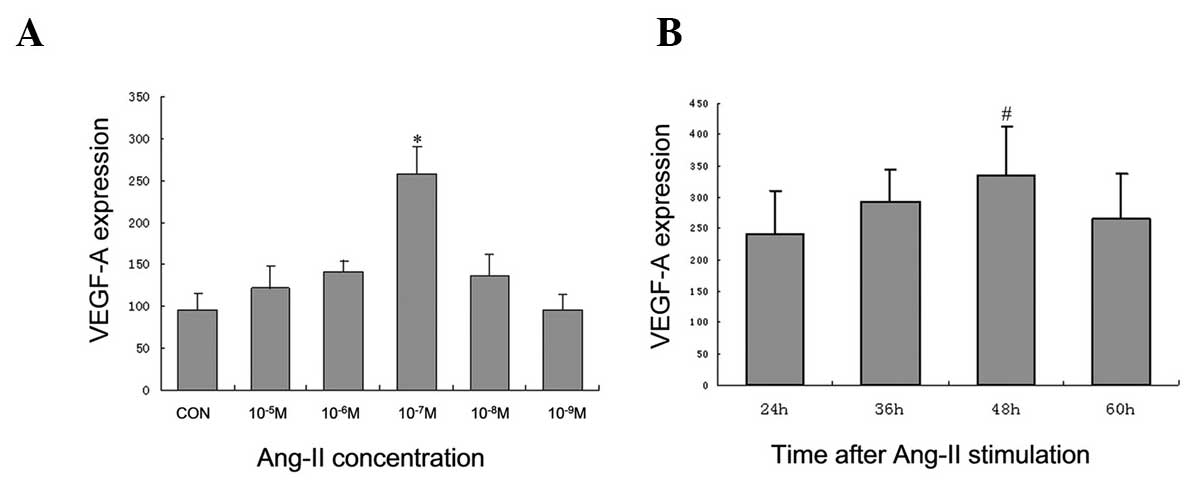
Ang-II induces the expression of VEGF-A
in podocytes
The integrity of VEGF-A was demonstrated by
stimulation of Ang-II in podocytes. Concentrations of
10−5, 10−6, 10−7, 10−8
and 10−9 M Ang-II were used for the dose response
experiment. Treatment with 10−7 M Ang-II led to a strong
activation of VEGF-A (increased 2.7-fold, P<0.001, in three
independent experiments) (Fig.
1A). In addition, time course experiments were performed to
examine the time dependency of VEGF-A activation. Based on the
dose-response experiments, 10−6, 10−7, and
10−8 M Ang-II were used for the time course experiments.
VEGF-A peaked 48 h following stimulation with Ang-II (Fig. 1B). Thus, in the following
experiments, a treatment dose of 10−7 M Ang-II for 48 h
was used.
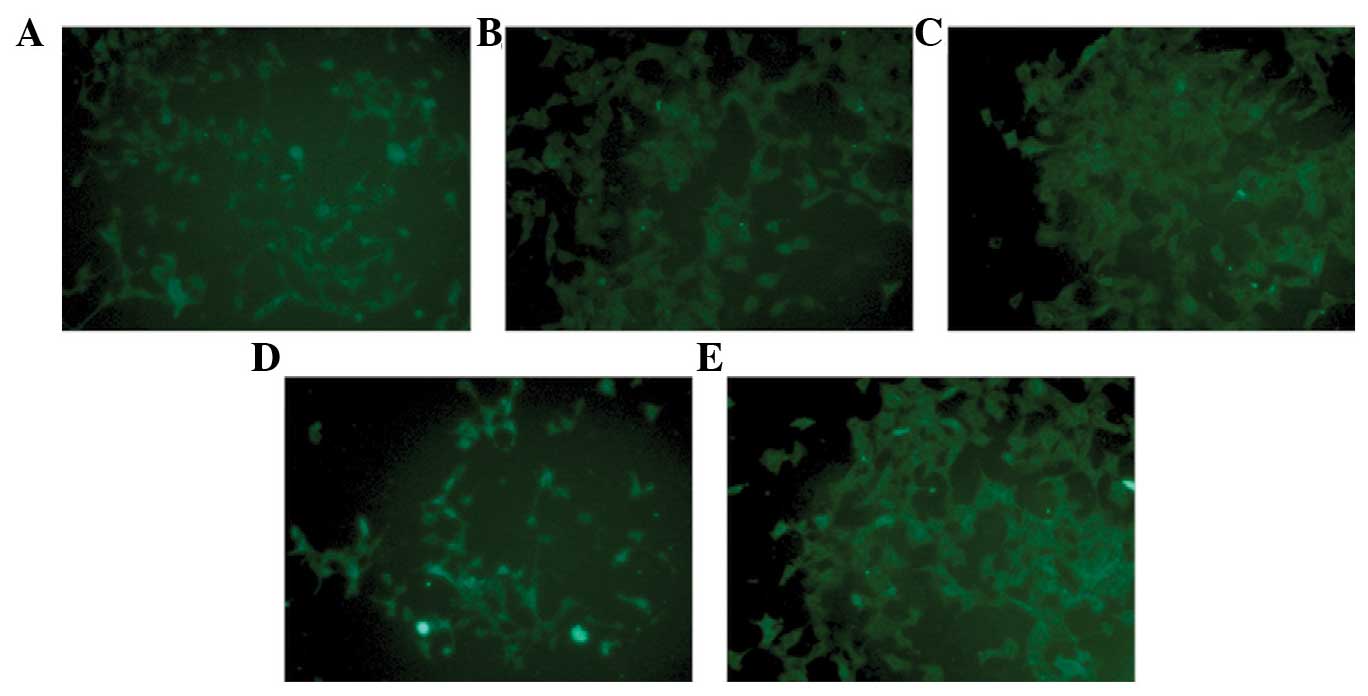
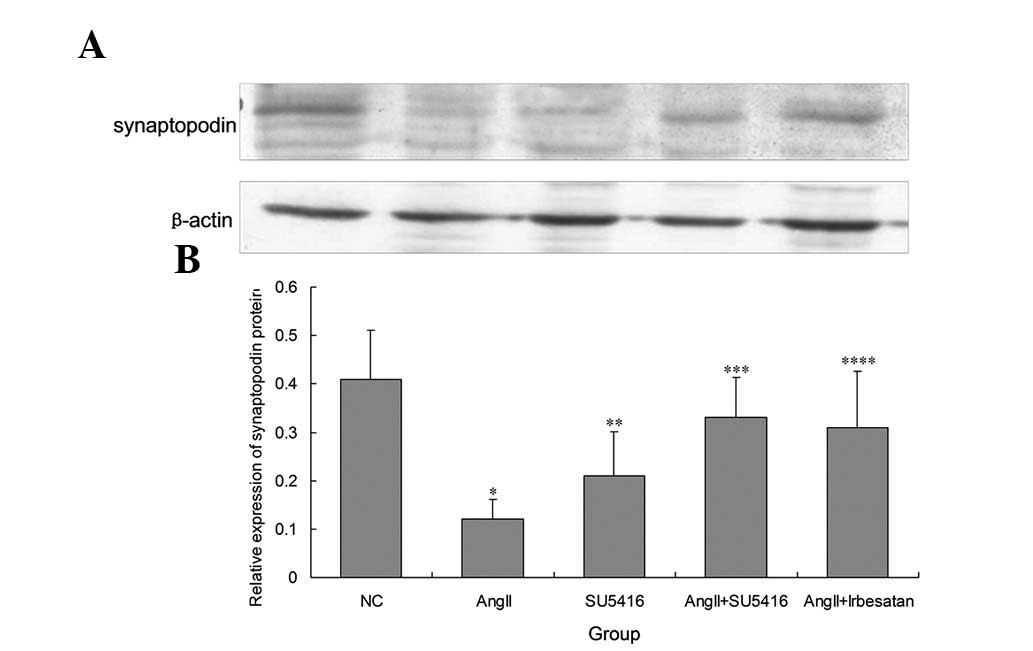
SU5416 is observed to ameliorate the
decrease in the protein level of synaptopodin in Ang-II-injured
podocytes
Following treatment with Ang-II (10−7 M)
for 48 h, the synchronized differentiated podocytes were injured.
The protein level of synaptopodin in injured podocytes was
significantly reduced (0.41±0.101 vs. 0.12±0.041, P=0.003)
(Figs. 2 and 3). It was demonstrated that SU5416
restored the protein level of synaptopodin for blockade of VEGFR2,
which was demonstrated by immunofluorescence (Fig. 2) and western blot analysis
(0.12±0.041 vs. 0.33±0.084, P=0.002; Fig. 3). Compared with the control, the
expression of synaptopodin was decreased in the normal podocytes
treated with SU5416 alone (0.41±0.101 vs. 0.21±0.092, P=0.008;
Fig. 2).
In order to evaluate whether ARBs have beneficial
effects on injured podocytes, 10−4 M irbesartan was
added to Ang-II treated podocytes for 48 h. It was observed that
irbesartan also partially restored the protein level of
synaptopodin (0.12±0.041 vs. 0.31±0.117, P=0.013; Figs. 2 and 3).
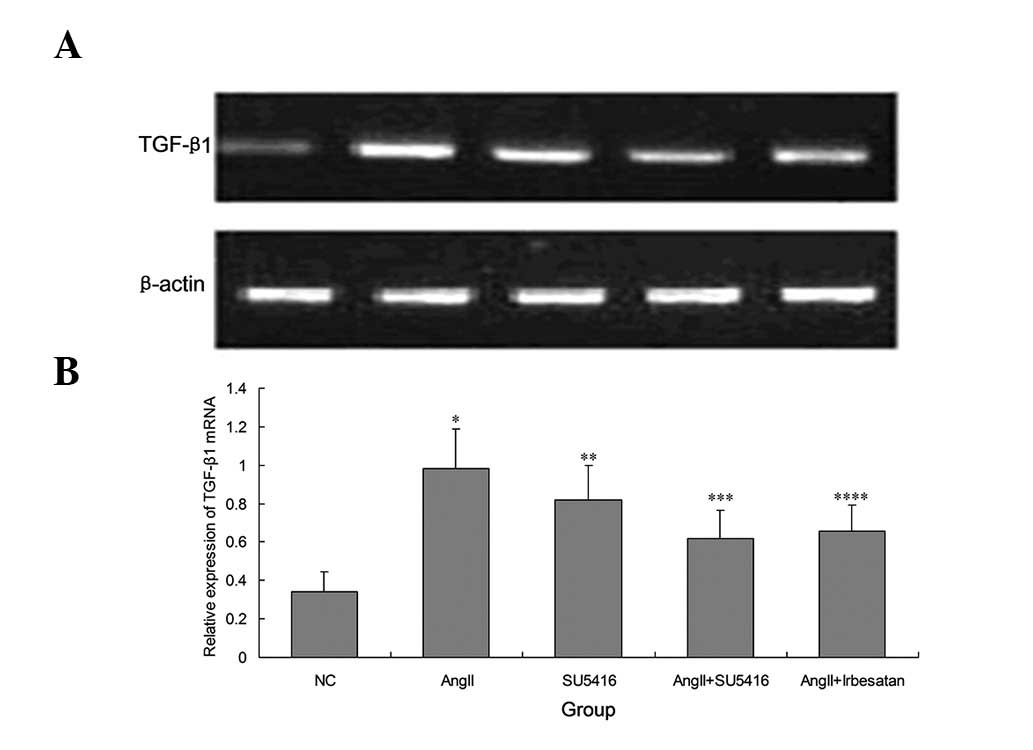
SU5416 decreases TGF-ß1 mRNA expression
in Ang-II-injured podocytes
It was then examined whether blocking the
VEGF/VEGFR2 pathway effects TGF-β1, a cytokine involved in
extracelluar fibrosis. Thus, SU5416 (1 μmol/l) was added to
Ang-II injured podocytes for 48 h in order to analyze TGF-β1 mRNA
expression. RT-qPCR revealed that the high expression of TGF-β1
mRNA in Ang-II-injured podocytes was decreased by SU5416 through
the blockade of VEGFR2 (0.98±0.211 vs. 0.62±0.146, P=0.011;
Fig. 4). Furthermore, it was
observed that 10−4 M irbesartan reversed the expression
of TGF-β1 mRNA in injured podocytes (0.98±0.211 vs. 0.65±0.134,
P=0.009). In the normal state, treatment with SU5416 alone was
observed to increase the mRNA expression of TGF-β1 (0.34±0.104 vs.
0.82±0.178, P=0.012; Fig. 4).
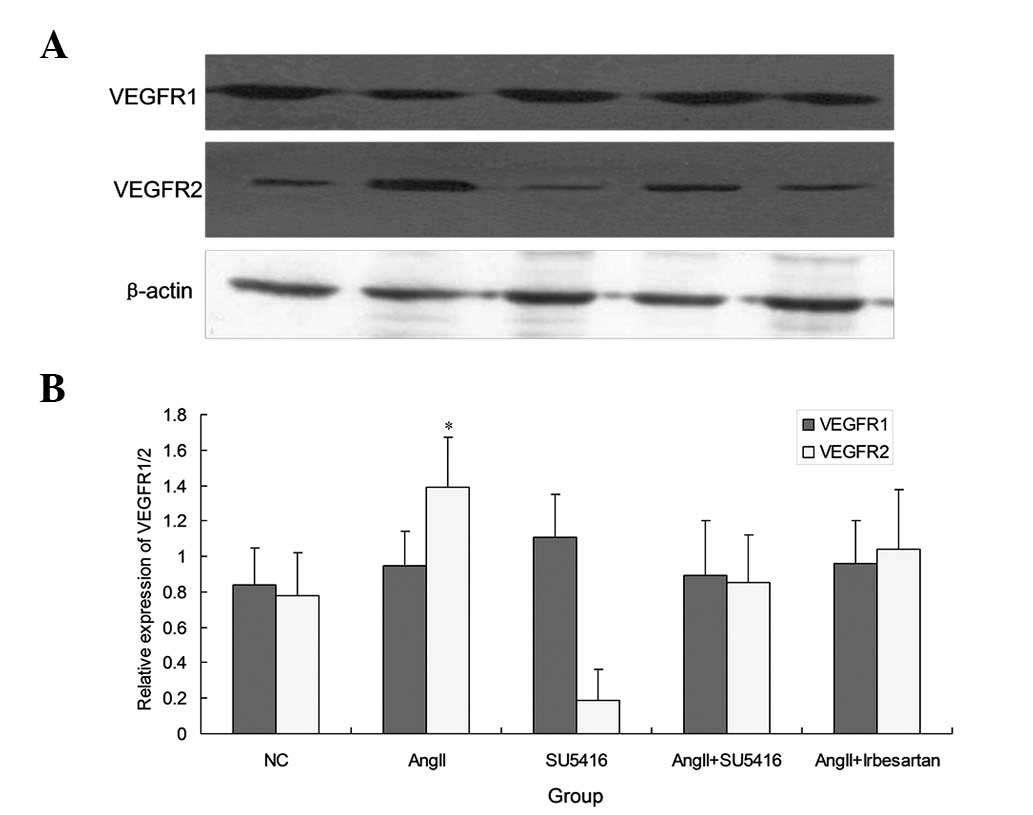
Ang-II exhibits no effect on VEGFR1 and
enhances the expression of VEGFR2
In addition to the expression of synaptodin and
TGF-β1, the expression of two major VEGF-A receptors VEGFR1 (flt-1)
and VEGFR2 (flk-1) were investigated following treatment with
Ang-II and SU5416. The VEGFR2 (flk-1) level was enhanced markedly
by Ang-II stimulation compared with control as observed using
western blot analysis (1.39±0.327 vs. 0.78±0.246, P=0.024). By
contrast, VEGFR1 (flt-1) levels were not affected by treatment with
Ang-II and SU5416 (P>0.05; Fig.
5)
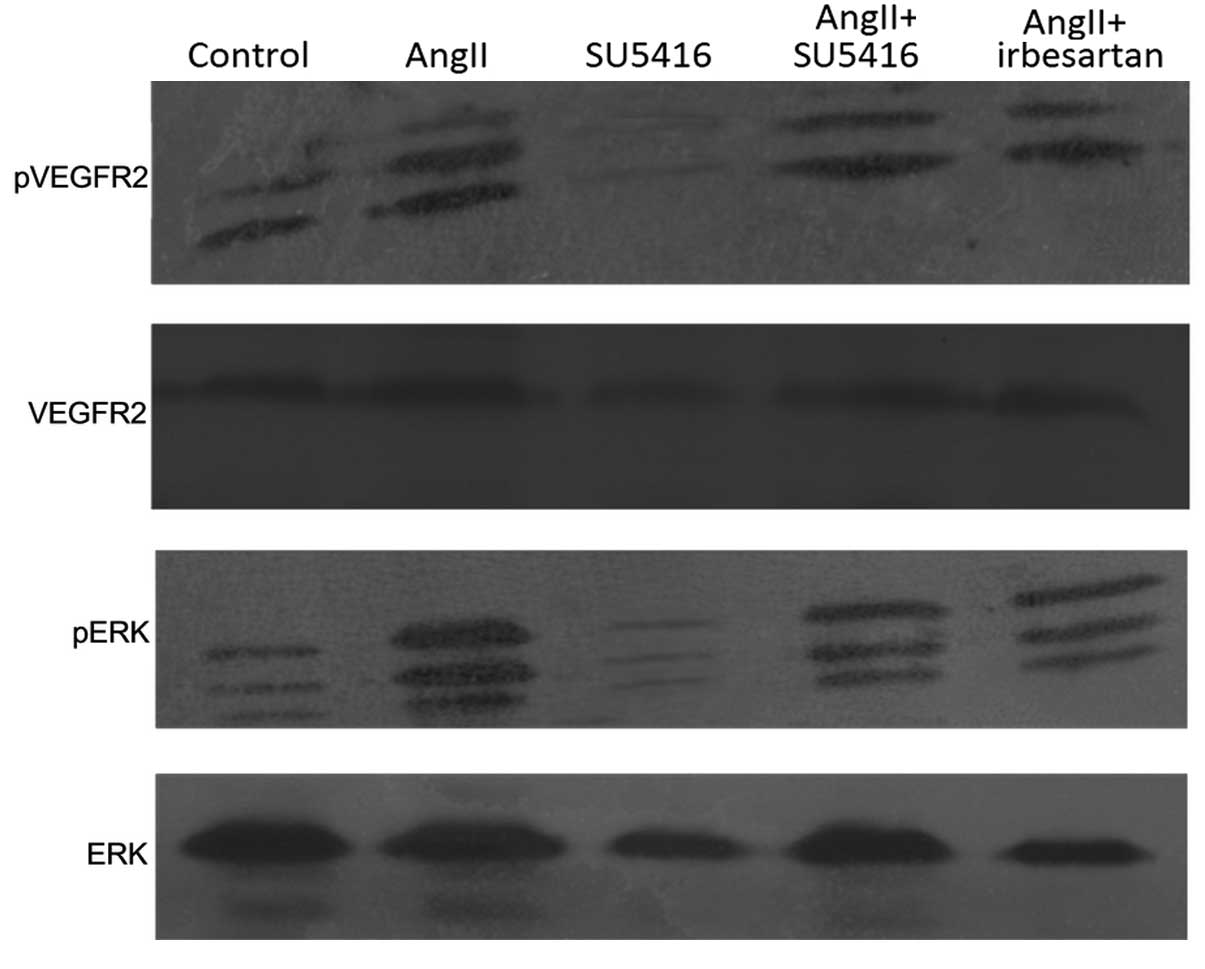
SU5416 downregulates the phosphorylation
of VEGFR2 and ERK
Based on the results that demonstrated that the
expression of VEGFR2 was enhanced by Ang-II, the level of
phosphorylated VEGFR2 and its downstream factor, ERK, were
examined. It was observed that the expression of phosphorylated
VEGFR2 was significantly increased by Ang-II, and was decreased by
SU5416. Compared with control, phosphorylation of ERK was also
upregulated by treatment with Ang-II. Blockade of VEGFR2 decreased
the level of phosphorylated-ERK (Fig.
6).
Irbesartan showed the same effect on the
phosphorylation of VEGFR2 and ERK as SU5416 (Fig. 6).
Discussion
Recently, studies have shown that modulating
angiogenesis-related factors has potential therapeutic effects in
CKD (19–21). In the present study, the increased
expression of VEGF-A was observed in cultured podocytes stimulated
with Ang-II. Ang-II was observed to increase the expression of
VEGFR2 (flk-1) and its level of phosphorylation; however, it had a
lower effect on VEGFR1 (flt-1). Blockade of VEGFR2 by SU5416
downregulated the phosphorylation of VEGFR2 and ERK.
Simultaneously, it recovered the expression of synaptopodin and
decreased the level of TGFβ1 in Ang-II-induced podocytes. It was
also demonstrated that the normal podocytes were injured by
treatment with SU5416 alone. Irbesartan showed the same effect on
the Ang-II stimulated podocytes as SU5416.
VEGF-A is required for the development of the
glomerular filtration barrier (22). Although a previous study (22) established a link between increased
VEGF-A, and glomerular injury and proteinuria, it remains unknown
whether increased VEGF-A is causative or simply a consequence of
these pathologic processes. There remains controversy regarding the
beneficial and deleterious effects of VEGF-A on the kidney as
certain studies suggested that VEGF may protect against renal
injury (7,8). In the present study, it was observed
that blockade of VEGF-A/VEGFR2 may injure podocytes, suggesting
that VEGF-A/VEGFR2 is essential for the normal growth of podocytes.
When the expression of VEGF-A/VEGFR2 was lower than the normal
level, the podocytes were damaged. This is consistent with the
findings of Eremina et al (13).
Simultaneously increased VEGF-A expression was
observed in Ang-II-stimulated podocytes. Blockade of VEGFR2 was
shown to reduce the phosphorylation of VEGFR2 and ERK, and
ammeliorate the change in expression of synaptopodin and TGF-ßI.
Thus, VEGF A autocrined by podocytes treated with Ang II may have
an important effect on the extracellular matrix (ECM) and cell
morphology through VEGFR2. Simultaneously, it may be postulated
that the proportion balance of phosphorylated VEGFR2 has been
broken when the podocytes is injured by Ang-II. Hohenstein et
al (23) found that the
majority of cells at sites of prominent injury, such as crescents,
demonstrated high expression levels of VEGFR1 in a large number of
renal biopsies from patients with glomerulonephritis (GN). However,
numerous glomeruli had less intense VEGFR1 expression, despite
obvious morphological changes such as increased cellularity or
matrix. By contrast podocytic VEGFR2 expression was more prominent
in biopsies with GN. The results of the present study showed that
VEGFR1 had not been influenced by Ang-II and suggested that
blockade of VEGFR2 had a beneficial effect on Ang-II-stimulated
podocytes. It could be deduced that the roles of VEGR1/2 were
different in podocytes. While VEGFR1 may be important in cell
proliferation and survival, VEGFR2 appeared to be involved in the
podocytes architecture and morphology. When phosphorylation of
VEGFR2 was impaired, cytokines involved in morphology and matrix,
such as synaptopodin and TGF-β1 exhibited altered levels of
expression. These data reveal that VEGF-A activation of VEGFR2, not
VEGFR1, directly resulted in the damage of podocyte ECM and
morphology by Ang-II.
To identify the mechanisms by which VEGF-A activated
of VEGFR2 in stimulated Ang-II injured podocytes, VEGF-A-induced
signaling pathways were analyzed by blockade of VEGF-A/VEGFR2 using
SU5416. It was shown that VEGF-A/VEGFR2 activation in
Ang-II-injured podocytes stimulated phosphorylation of ERK. To the
best of our knowledge, phosphorylation of ERK is the upstream
regulator of TGFβ1. Thus, this suggested that the phosphorylation
of ERK was required for the VEGF-A/VEGFR2-induced damage of
podocytes treated with Ang-II.
Synaptopodin, an actin-associated protein, is
expressed only in completely differentiated podocytes and in the
telencephalic synapses (24).
Podocytes are important in the maintenance of renal glomerular
function and in the pathogenesis of glomerulosclerosis (25). It is postulated that synaptopodin
modulates the actin-based contractile microfilament apparatus of
the podocyte foot processes and the integrity of matured podocytes.
Synaptopodin may be involved in the development of proteinuric
renal diseases and maintenance of the glomerular filtration barrier
(26,27). The increasing level of synaptopodin
in podocytes has been revealed with proteinuria (28). The present study examined that
blockade of VEGF-A/VEGFR2 by the VEGFR2 inhibitor ameliorated the
decrease in the protein level of synaptopodin in Ang-II-injured
podocytes. This result is in line with anti-VEGF-related
proteinuria in non-diabetic CKD (29).
Although it is now evident that damage is
transmitted from podocyte to podocyte, and from podocyte to other
glomerular cells, it remains largely unknown how these
transmissions occur, and what factors are involved (30). Possible mechanisms include
increased toxic substance(s) secreted in an autocrine or paracrine
manner, such as basic fibroblast growth factor, TGF-β, Ang-II and
macrophage migration inhibitory factor, and decreased levels of
supportive substances for survival of the podocytes. In the present
study it was observed that inhibition of VEGF-A/VEGFR2 increased
the expression of TGF-β1 mRNA in Ang-II-injured podocytes,
indicating that blockade of the VEGF pathway may delay the
progression of glomerulosclerosis. The observation that blockade of
VEGF-A/VEGFR2 was associated with decreased expression of TGF-β1
mRNA suggested that podocyte damage may induce further podocyte
damage in a positive feedback mechanism, which drives local spread
of glomerulosclerosis.
Renin-angiotensin system (RAS) inhibitors have been
shown to reduce glomerular permeability and proteinuria. Local
elevations in Ang-II signaling mediated by angiotensin type I
receptors led to elevations in VEGF expression in endothelial cells
in rats (31). Several studies
have demonstrated a close correlation between RAS and the level of
VEGF in vitro, showing that Ang-II can induce VEGF
expression (32–34). In the present study, the
therapeutic effects of VEGFR2 inhibition on TGF-β1 and synaptopodin
were observed compared with ARB in cultured podocytes. It was shown
that pretreatment with irbesartan lowered the expression of TGF-β1,
which is a major mediator of the hypertrophic and prosclerotic
changes in the kidney. Furthermore, the results suggested another
anti-albuminuric molecular mechanism of irbesartan, which restored
the levels of synaptopodin in Ang-II-injured podocytes.
In conclusion, the pathway of VEGF-A/VEGFR2 is
essential for podocytes in the physiological state. The expression
of VEGF-A and VEGFR2 was increased in Ang-II-injured podocytes. The
VEGFR2 inhibitor appeared to restore the level of synaptopodin and
decrease the expression of TGF-β1. It suggested that blockade of
VEGF-A/VEGFR2 exhibits beneficial effects on the extracellular
matrix and cell morphology in Ang-II-stimulated podocytes.
References
|
1
|
Ma J, Rossini M, Yang HC, Zuo Y, Fogo AB
and Ichikawa I: Effects of podocyte injury on glomerular
development. Pediatr Res. 62:417–421. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kim NH, Oh JH, Seo JA, et al: Vascular
endothelial growth factor (VEGF) and soluble VEGF receptor FLT-1 in
diabetic nephropathy. Kidney Int. 67:167–177. 2005. View Article : Google Scholar
|
|
3
|
Bates DO and Curry FE: Vascular
endothelial growth factor increases hydraulic conductivity of
isolated perfused microvessels. Am J Physiol. 271:H2520–H2528.
1996.PubMed/NCBI
|
|
4
|
Siemann DW, Brazelle WD and Jürgensmeier
JM: The vascular endothelial growth factor receptor-2 tyrosine
kinase inhibitor cediranib (Recentin; AZD2171) inhibits endothelial
cell function and growth of human renal tumor xenografts. Int J
Radiat Oncol Biol Phys. 73:897–903. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maeshima Y and Makino H: Angiogenesis and
chronic kidney disease. Fibrogenesis Tissue Repair. 3:13–30. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Foster RR, Hole R, Anderson K, et al:
Functional evidence that vascular endothelial growth factor may act
as an autocrine factor on human podocytes. Am J Physiol Renal
Physiol. 284:F1263–F1273. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baharivand N, Zarghami N, Panahi F, et al:
Relationship between vitreous and serum vascular endothelial growth
factor levels, control of diabetes and microalbuminuria in
proliferative diabetic retinopathy. Clin Ophthalmol. 6:185–191.
2012.PubMed/NCBI
|
|
8
|
Ho C, Hsu YC, Tseng CC, et al: Simvastatin
alleviates diabetes-induced VEGF-mediated nephropathy via the
modulation of Ras signaling pathway. Ren Fail. 30:557–565. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Doi K, Leelahavanichkul A, Hu X, et al:
Pre-existing renal disease promotes sepsis-induced acute kidney
injury and worsens outcome. Kidney Int. 74:1017–1025. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Celletti FL, Waugh JM, Amabile PG, et al:
Vascular endothelial growth factor enhances atherosclerotic plaque
progression. Nat Med. 7:425–429. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ferrara N: Role of vascular endothelial
growth factor in physiologic and pathologic angiogenesis:
therapeutic implications. Semin Oncol. 29(Suppl 16): 10–14. 2002.
View Article : Google Scholar
|
|
12
|
Sugimoto H, Hamano Y, Charytan D, et al:
Neutralization of circulating vascular endothelial growth factor
(VEGF) by anti-VEGF antibodies and soluble VEGF receptor 1 (sFlt-1)
induces proteinuria. J Biol Chem. 278:12605–12608. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Eremina V, Jefferson JA, Kowalewska J, et
al: VEGF inhibition and renal thrombotic microangiopathy. N Engl J
Med. 358:1129–1136. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shankland SJ, Pippin JW, Reiser J and
Mundel P: Podocytes in culture: past, present, and future. Kidney
Int. 72:26–36. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Krtil J, Pláteník J, Kazderová M, et al:
Culture methods of glomerular podocytes. Kidney Blood Press Res.
30:162–174. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liang XB, Ma LJ, Naito T, et al:
Angiotensin type 1 receptor blocker restores podocyte potential to
promote glomerular endothelial cell growth. J Am Soc Nephrol.
17:1886–1895. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mezrich JD, Nguyen LP, Kennedy G, et al:
SU5416, a VEGF receptor inhibitor and ligand of the AHR, represents
a new alternative for immunomodulation. PLoS ONE. 7:e445472012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Westenskow PD, Kurihara T, Aguilar E, et
al: Ras pathway inhibtion prevents neovascularization by repressing
endothelial cell sprouting. J Clin Invest. 123:4900–4908. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Veron D, Bertuccio CA, Marlier A, et al:
Podocyte vascular endothelial growth factor (Vegf164)
overexpression causes severe nodular glomerulosclerosis in a mouse
model of type 1 diabetes. Diabetologia. 54:1227–1241. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nasu T, Maeshima Y, Kinomura M, et al:
Vasohibin-1, a negative feedback regulator of angiogenesis,
ameliorates renal alterations in a mouse model of diabetic
nephropathy. Diabetes. 58:2365–2375. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim W, Moon SO, Lee SY, et al:
COMP-angiopoietin-1 ameliorates renal fibrosis in a unilateral
ureteral obstruction model. J Am Soc Nephrol. 17:2474–2483. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Eremina V and Quaggin SE: The role of
VEGF-A in glomerular development and function. Curr Opin Nephrol
Hypertens. 13:9–15. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hohenstein B, Colin M, Foellmer C, et al:
Autocrine VEGF-R loop on podocytes during glomerulonephritis in
humans. Nephrol Dial Transplant. 25:3170–3180. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yanagida-Asanuma E1, Asanuma K, Kim K, et
al: Synaptopodin protects against proteinuria by disrupting
Cdc42:IRSp53: Mena signaling complexes in kidney podocytes. Am J
Pathol. 171:415–427. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
de Zoysa JR and Topham PS: Podocyte
biology in human disease. Nephrology (Carlton). 10:362–367. 2005.
View Article : Google Scholar
|
|
26
|
Asanuma K, Kim K, Oh J, et al:
Synaptopodin regulates the actin-bundling activity of alpha-actinin
in an isoform-specific manner. J Clin Invest. 115:1188–1198. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Faul C, Asanuma K, Yanagida-Asanuma E, Kim
K and Mundel P: Actin up: regulation of podocyte structure and
function by components of the actin cytoskeleton. Trends Cell Biol.
17:428–437. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Faul C, Donnelly M, Merscher-Gomez S, et
al: The actin cytoskeleton of kidney podocytes is a direct target
of the anti-proteinuric effect of cyclosporine A. Nat Med.
14:931–938. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Izzedine H, Massard C, Spano JP, et al:
VEGF signaling inhibition-induced proteinuria: Mechanisms,
significance and management. Eur J Cancer. 46:439–448. 2010.
View Article : Google Scholar
|
|
30
|
Ichikawa I, Ma J, Motojima M and Matsusaka
T: Podocyte damage damages podocytes: autonomous vicious cycle that
drives local spread of glomerular sclerosis. Curr Opin Nephrol
Hypertens. 14:205–210. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhao Q, Egashira K, Inoue S, et al:
Vascular endothelial growth factor is necessary in the development
of arteriosclerosis by recruiting/activating monocytes in a rat
model of long-term inhibition of nitric oxide synthesis.
Circulation. 105:1110–1115. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pan P1, Fu H, Zhang L, et al: Angiotensin
II upregulates the expression of placental growth factor in human
vascular endothelial cells and smooth muscle cells. BMC Cell Biol.
11:362010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pupilli C, Lasagni L, Romagnani P, et al:
Angiotensin II stimulates the synthesis and secretion of vascular
permeability factor/vascular endothelial growth factor in human
mesangial cells. J Am Soc Nephrol. 10:245–255. 1999.PubMed/NCBI
|
|
34
|
Kang YS, Park YG, Kim BK, et al:
Angiotensin II stimulates the synthesis of vascular endothelial
growth factor through the p38 mitogen activated protein kinase
pathway in cultured mouse podocytes. J Mol Endocrinol. 36:377–388.
2006. View Article : Google Scholar : PubMed/NCBI
|