Introduction
Oxygen supplementation is a common clinical
intervention for premature infants with respiratory failure,
however, prolonged exposure to hyperoxic conditions has long been
recognized as a potential risk factor for bronchopulmonary
dysplasia (BPD) (1). Exposing
premature newborn rats to high concentrations of ambient oxygen
induces the enlargement of distal air spaces with simplification of
lung structure, which recapitulates the histopathology of BPD
(2). Hyperoxia therapy produces
reactive oxygen species (ROS), which can induce cell death, disrupt
postnatal alveolarization, lead to abnormal lung repair and cause
lung tissue injury (3). Although
the specific mechanisms by which hyperoxia causes lung injury
remain to be fully elucidated, the generation of ROS and cellular
apoptosis appear to be important.
Increasing evidence has indicated that endoplasmic
reticulum (ER) stress is involved in apoptosis. The ER is an
important organelle in mammalian cells with numerous functions,
including the maintenance of intracellular calcium homeostasis,
protein secretion, lipid synthesis, and protein glycosylation and
folding (4). Interruption of ER
homeostasis leads to ER stress and triggers the unfolded protein
response (UPR), resulting in activation of three ER stress
receptors, inositol requiring protein-1, protein kinase RNA-like ER
kinase and activating transcription factor 6, which result in
apoptosis in persistently stressed cells (5,6).
The 78-kDa glucose-regulated/binding immunoglobulin
protein (GRP78) is a hallmark of the UPR and ER stress responses
(6). The UPR responds rapidly to
ER stress to enhance cell survival. However, if protein aggregation
is persistent and stress cannot be resolved, signaling switches
from the pro-survival to the pro-apoptotic ER stress response
(5,7,8).
Evidence has suggested that CCAAT/enhancer binding protein
homologous protein (CHOP) is recruited and is involved in ER
stress-induced apoptosis, following the phosphorylation of
eukaryotic initiation factor 2α and upregulation of activating
transcription factor 4 (6,9,10).
CHOP is a 29 kDa protein with 169 (human) or 168 (rodent)
amino-acid residues. CHOP is also termed growth arrest, DNA damage
inducible gene 153 and DNA damage-inducible transcript 3 (11). The induction of CHOP may trigger ER
stress-induced apoptosis, and the involvement of CHOP-mediated
apoptosis has been demonstrated in various diseases, including
diabetes, neurodegenerative diseases, brain ischemia and certain
cardiovascular diseases (12–15).
However, the importance of ER stress and the role of CHOP in the
pathogenesis of neonatal lung injury and hyperoxia-induced BPD
remain to be fully elucidated. The present study hypothesized that
hyperoxia-induced BPD results from persistent ER stress, leading to
increased expression of CHOP and cell death.
Materials and methods
Study animals and procedures
Adult Sprague-Dawley (SD) rats, including 21 females
(200–220 g) and seven males (220–250 g) were purchased from the
Experimental Animal Center of Jiang Su University (Zhenjiang,
China). The rats were maintained under controlled temperature
(23±1°C), humidity, and lighting (12 h light/12 h dark cycle), and
were fed a standard rodent chow diet. All procedures and protocols
were reviewed and approved by the Animal Care and Ethics Committee
of Jiang Su University.
Hyperoxia exposure
The rats were left overnight for mating in groups of
four consisting of three females and one male. The day on which the
vaginal plug was identified was considered to be the first day of
pregnancy. On the 21st day of pregnancy (term=22 days), the
pregnant rats were anesthetized with intraperitoneal injections of
10% chloral hydrate (350 mg/kg; Wuhan Boster Biological Technology,
Ltd., Wuhan, China), and then underwent delivery via
uterine-incision, resulting in pups deemed premature. Within 24 h
of birth, the newborn rats were placed in chambers in which the
oxygen concentration was maintained at FiO2=0.85
for 7, 14 and 21 days, or were maintained in standard room air in
which the oxygen concentratoin was maintained at
FiO2=0.21. The nursing dams were provided with
food and water ad libitum, were maintained on a 12:12-h
on-off light cycle and were fostered by rotating in and out of the
chamber every 24 h to avoid oxygen toxicity. The oxygen
concentration was monitored continuously using an oxygen analyzer
(model 572; Servomex Co., Inc., Norwood, MA, USA).
Lung histology and morphometric
analysis
Following continued exposure to oxygen (n=24) or
room air (n=24) for 7, 14 and 21 days, the premature rats were
sacrificed with intraperitoneal injections of pentobarbital (100
mg/kg; Tianjin Pharmaceutical Group Xinzheng Co., Ltd., Xinzheng,
China). A catheter was placed into the trachea and the lungs were
inflated and maintained at 30 cm H2O pressure with 4%
paraformaldehyde in phosphate-buffered saline (PBS) for 45 min. A
ligature was tightened around the trachea to maintain pressure
following removal of the tracheal cannula. The lungs were removed
and immersed in paraformaldehyde solution overnight, and the left
lower lobes were embedded in paraffin. Sections were then cut using
a microtome set at 5 µm and mounted on RNase free slides for
hematoxylin and eosin (HE) staining, immunohistochemistry and a
terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end
labeling (TUNEL) assay. At least three lung sections from each
animal were subjected to morphometric analysis. Alveolarization was
assessed by performing radial alveolar counts (RACs), according to
the method of Emery and Mithal (16). From the center of the respiratory
bronchiole, a perpendicular was drawn to the edge of the acinus,
defined by a connective tissue septum or the pleura, and the number
of septa intersected by this line were counted. A total of six
counts were performed for each animal.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The total RNA was extracted from the frozen lung
tissues using TRIzol reagent (Invitrogen Life Technologies,
Carlsbad, CA, USA) according to the manufacturer's instructions.
The concentration of total RNA was quantified using
spectrophotometry (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
RT was performed to synthesize the first-strand cDNA using a M-MLV
Reverse Transcriptase system (Promega Corporation, Madison, WI,
USA) and oligo (dT). The resulting cDNA was then subjected to
RT-qPCR for evaluation of the relative mRNA levels of GRP78, CHOP
and β-actin (internal control). The sequences for each primer
(Takara Biotechnology Co., Ltd., Dalian, China) and product are
shown in Table I. Gene-specific
amplification was performed using a 20 µl PCR mix containing
1 µl cDNA, 10 µl of 2X SYBR Green master mix
(Invitrogen Life Technologies) and 200 nmol sense and antisense
primers. The mix was preheated at 95°C (5 min) and then amplified
at 95°C (15 sec) and 60°C (20 sec) for 45 cycles. The relative
expression levels of GRP78 and CHOP mRNA were determined by
comparing with standard curves and normalization to β-actin. The
experiment was performed in triplicate.
 | Table ISequence for each primer and product
length. |
Table I
Sequence for each primer and product
length.
| Gene | Primer sequence
(5′-3′) | Length (bp) |
|---|
| GRP78 | F
CCGTAACAATCAAGGTCTACGA | 124 |
| R
AAGGTGACTTCAATCTGGGGTA | |
| CHOP | F
ACCACCACACCTGAAAGCA | 86 |
| R
AGCTGGACACTGTCTCAAAGG | |
| β-actin | F
GCAGAAGGAGATTACTGCCCT | 136 |
| R
GCTGATCCACATCTGCTGGAA | |
Western blot analysis
The homogenized lung tissue samples, were lysed in
ice-cold isolation solution containing 250 mmol/l sucrose, 10
mmol/l triethanolamine, 1 µg/ml leupeptin and 0.1 mg/ml
phenylmethylsulfonyl fluoride, and harvested following
centrifugation (12,000 × g) at 4°C for 30 min. The protein samples
(~40 µg) were then separated by electrophoresis on a 12%
sodium dodecyl sulfate polyacrylamide gel and transferred onto
polyvinylidene fluoride membranes (EMD Millipore, Bedford, MA,
USA). Following blocking of the non-specific binding sites for 60
min with 5% non-fat milk, the membranes were incubated with primary
antibodies overnight at 4°C. The following primary polyclonal
rabbit anti-rat antibodies were used: GRP78 (1:200; cat. no.
sc-13968; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA),
CHOP (1:500; cat. no. sc-793; Santa Cruz Biotechnology, Inc.) and
β-actin (1:1,000; cat. no. sc-1616-R; Santa Cruz Biotechnology,
Inc.). The membranes were then washed three times in Tris-buffered
saline with 0.1% Tween 20 (TBST) for 10 min and incubated with
biotinylated goat anti-rabbit immunoglobulin G (1:200; Wuhan Boster
Biological Technology, Ltd.) in PBS for 2 h at room temperature,
followed by washing in TBST three times, for 5 min each. The
membranes were then incubated for 1 min using an enhanced
chemiluminescence system (Applygen Technologies, Inc., Beijing,
China). An autoradiograph was obtained with an exposure time of 2–5
min. The relative band intensity was scanned and quantified using a
GDS-800 Complete Gel Documentation & Analysis system (Microtek
Lab, Inc., Carson, CA, USA).
Immunohistochemical staining
Immunohistochemical staining was performed on
tissues at 14 days after hyperoxia or air exposure. Following
deparaffinization, endogenous peroxidase activity was quenched with
3% hydrogen peroxide. Subsequently, the slides were boiled in
citrate buffer in a microwave for 15 min. Following blocking of
nonspecific binding using 5% bovine serum albumin (Wuhan Boster
Biological Technology, Ltd.), the slides were incubated with GRP78
antibody (1:150) or CHOP antibody (1:200) overnight at 4°C, and
then washed three times with PBS for 5 min. The sections were then
incubated for 1 h with a 500-fold dilution of secondary antibodies
conjugated with peroxidase, and incubated with 0.05%
3,3′-diaminobenzidine (DAB; Wuhan Boster Biological Technology,
Ltd.) solution for visualization. Finally, staining was performed
using hematoxylin for 3 sec and the slides were visualized under
microscopy (BX51; Olympus Corporation, Tokyo, Japan).
Detection of apoptosis
The present study investigated whether hyperoxic
exposure induces apoptosis in lung cells. Apoptosis was detected
using a TUNEL assay 14 days after hyperoxia or air exposure,
according to the manufacturer's instructions (Roche Applied
Science, San Francisco, CA, USA). Briefly, the sections were
deparaffinized, digested with proteinase K (1:200) at 37°C for 10
min, and soaked in PBS for 5 min. Each section was covered with a
terminal deoxynucleotidyl trans-ferase (TDT) enzyme solution,
containing 45 µl equilibration buffer, 1 µl
biotin-11-dUTP and 4 µl TDT enzyme, and incubated for 2 h at
37°C in a humidified chamber. The sections were immersed in stop
buffer to terminate the enzymatic reaction and gently rinsed with
PBS, followed by incubation with biotinylated anti-digoxin antibody
(1:200) at 37°C for 30 min, SABC (1:100) at 37°C for 30 min, DAB
coloration, and visualization using microscopy (BX51; Olympus
Corporation). Sections incubated with PBS, instead of TDT enzyme
solution served as the negative controls. The TUNEL-positive cells
were counted in six randomly selected visual fields at the same
magnification in the control and treated mice (magnification,
×400). The percentage of apoptotic cells was determined as the
percentage of the total cells positive for TUNEL.
Statistical analysis
All data are expressed as the mean ± standard
deviation and were analyzed using SPSS 13.0 (SPSS, Inc., Chicago,
IL, USA). Differences among groups were assessed using a one-way
analysis of variance and between pairs using Student's t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Lung histology and RAC
Compared with the control rats, the lung histology
of the rats exposed to hyperoxia was characterized by decreased
septation, distal air space enlargement and a reduction in
complexity. To quantitfy the apparent decreases in the number of
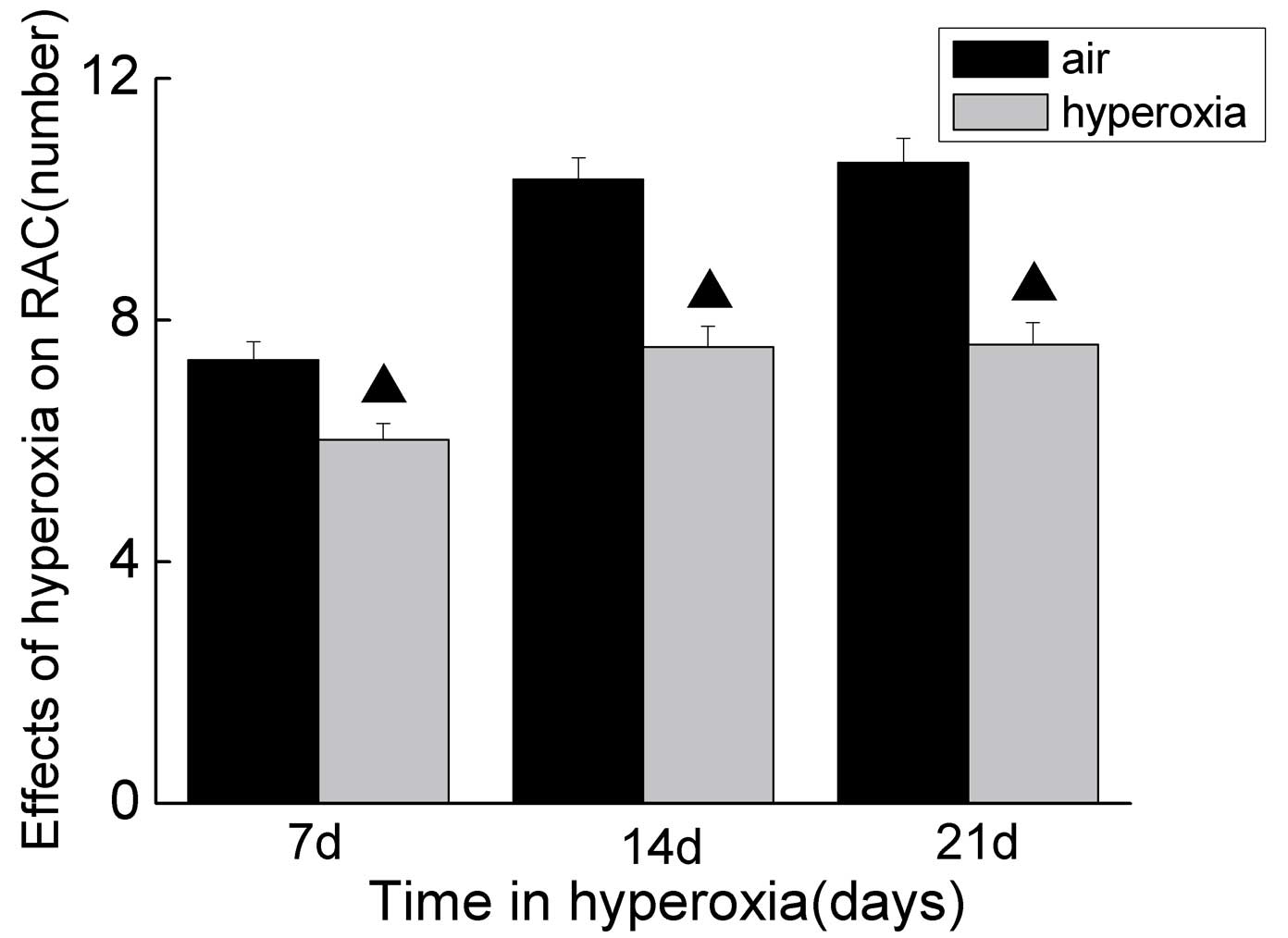
alveoli, RACs were performed. As shown in Fig. 1, the RACs were lower in the rats
exposed to hyperoxia, compared with the rats exposed to room air.
The neonatal rats exposed to hyperoxia exhibited a histological
pattern of alveolar simplification and a reduced RAC, which was
similar to the histology observed in human infants with BPD
(17). These results demonstrated
the animal model of BPD was successful.
mRNA expression levels of GRP78 and
CHOP
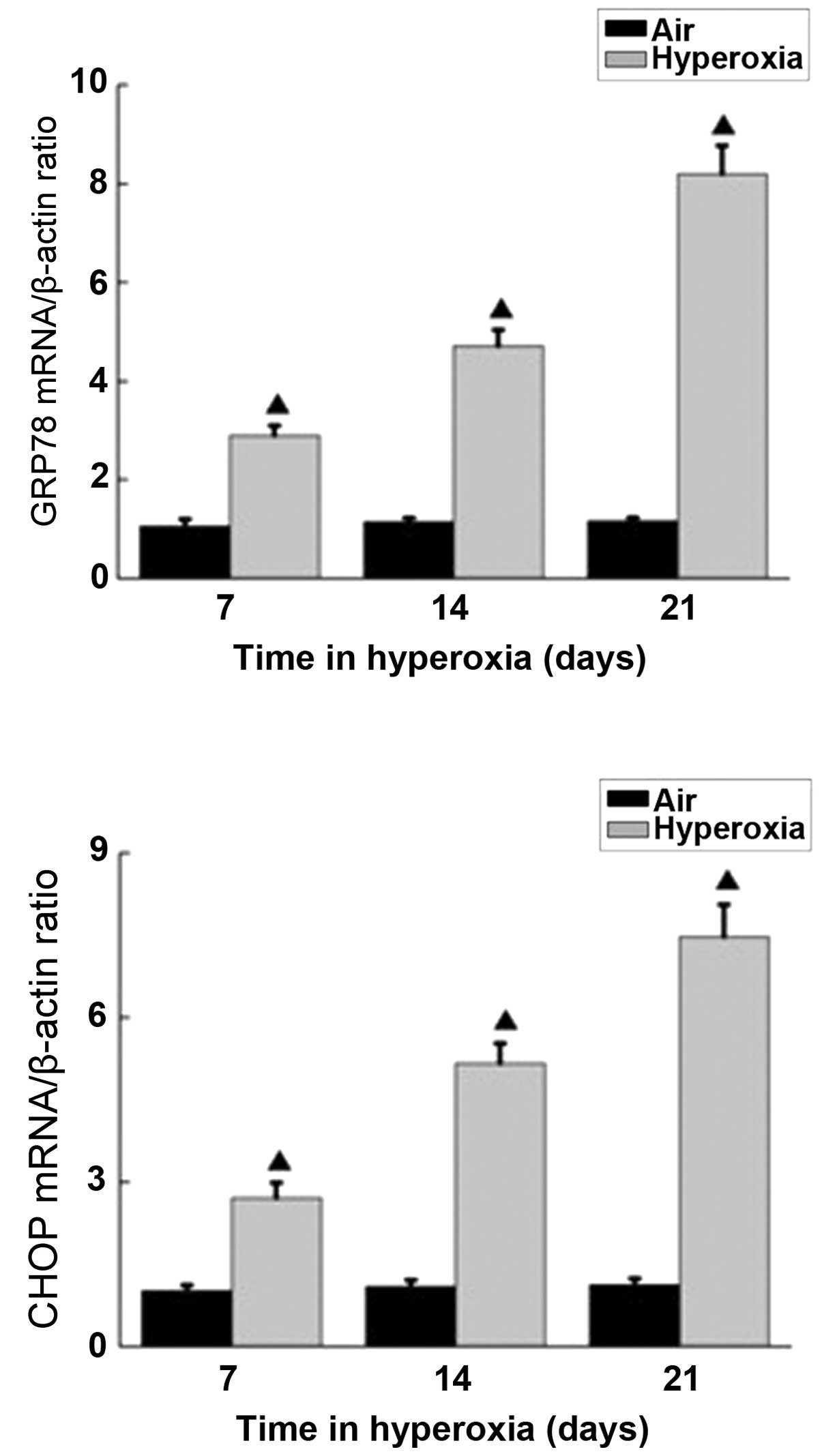
RT-qPCR was performed on the rat lungs exposed to
hyperoxia and room air to determine the mRNA levels of GRP78 and
CHOP. As shown in Fig. 2, the mRNA
expression levels of GRP78 and CHOP in the room air and
hyperoxia-exposed rat lungs were detectable, and no differences in
expression were observed in the lungs of the control rats over the
entire 21 day time-course. However, in the rats were exposed to 85%
hyperoxia, the mRNA expression levels of GRP78 and CHOP increased
gradually over 7, 14 and 21 days, and the expression levels were
significantly higher than those in the control group. In addition,
as the duration of hyperoxia exposure increased, the increased
differences between the two groups became more significant
(Fig. 2). These data demonstrated
that hyperoxia had similar effects on the expression levels of
GRP78 and CHOP, contributing to lung injury in BPD rats.
Expression of GRP78 and CHOP, determined
using western blot analysis
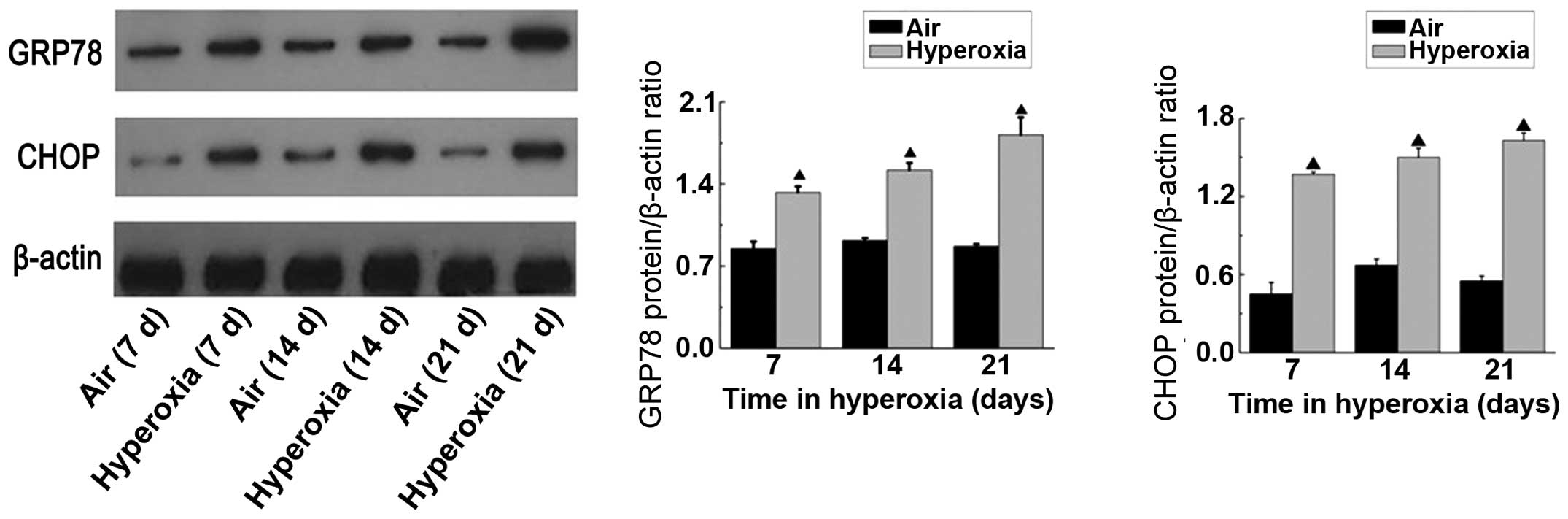
The protein levels of GRP78 and CHOP were determined
using western blot analysis. To determine whether hyperoxia induced
the expression of CHOP and GRP78, the newborn rats were exposed to
85% oxygen for 7, 14 or 21 days. The results of the western blot
analysis revealed a GRP78 band at 78 kDa and a CHOP band at 30 kDa.
Consistent with the RT-qPCR results, increases in the protein
expression levels of GRP78 and CHOP were observed in the lungs from
the rats exposed to hyperoxia, compared with the age-matched
controls exposed to normal air (Fig.
3). Over the time-course of hyperoxic exposure, the protein
levels of GRP78 and CHOP gradually increased. These data
demonstrated that GRP78 and CHOP were activated in the
hyperoxia-exposed newborn rat lungs, further supporting the
hypothesis that hyperoxia leads to persistent ER stress, resulting
in increased expression of CHOP and subsequent cell death.
Immunohistochemical analysis of GRP78 and
CHOP distribution
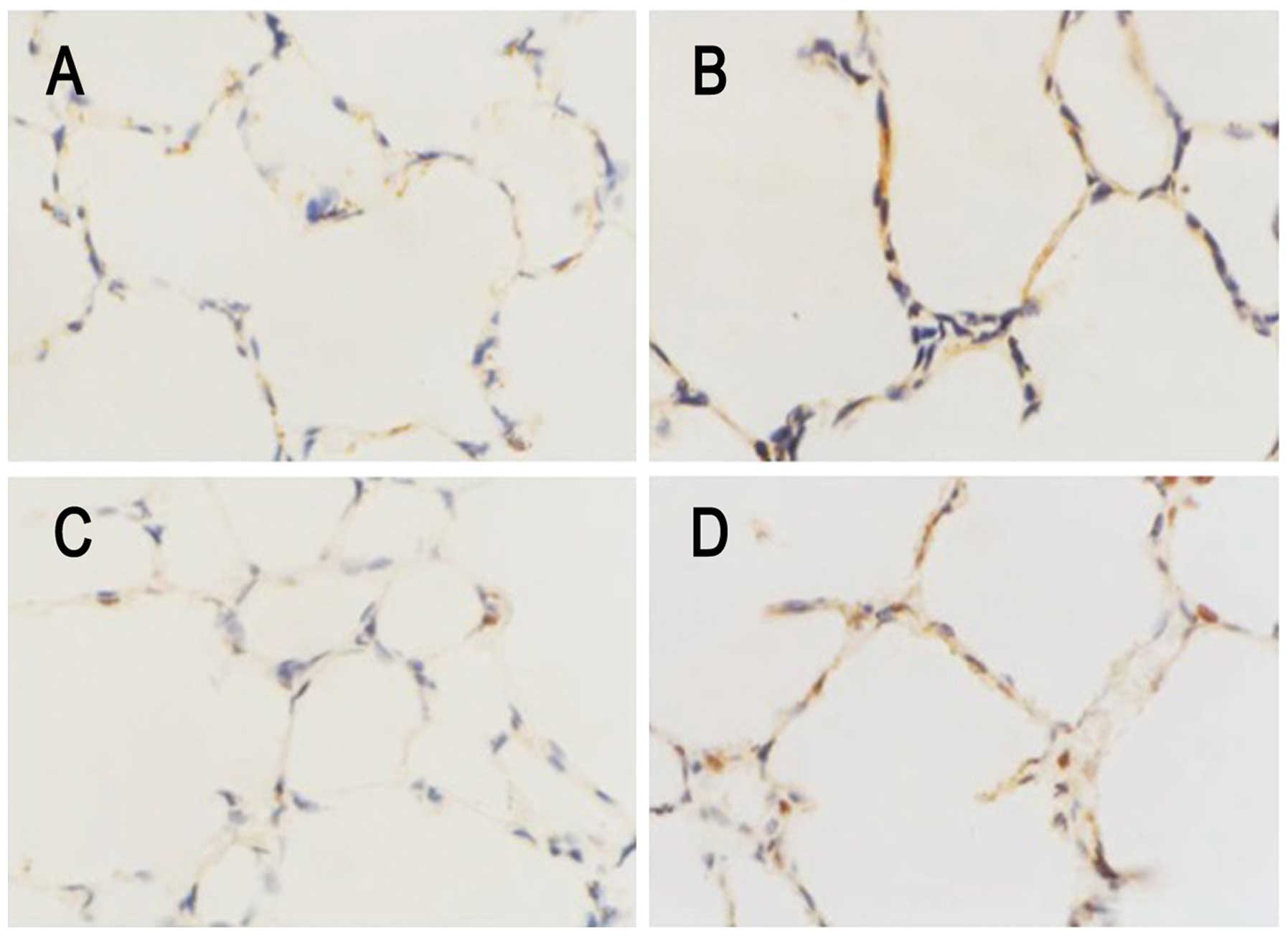
As shown in Fig. 4,
the immunohistochemical investigation revealed the arrest of
alveolar septation, char-acterized as enlarged alveoli and a lack
of homogenous small and middle-sized alveoli, in the lung tissues
from rats exposed to hyperoxia, compared with those from rats
exposed to normal air. GRP78 and CHOP were abundantly expressed in
the hyperoxia-exposed rat tissues, whereas the control rats
exhibited modest or weak immunoreactivity for these molecules.
GRP78 and CHOP were preferentially localized in the alveolar and
bronchiolar epithelial cells. These results suggested that
hyperoxia increased GRP78 and CHOP content, predominantly in the
enlarged and simplified alveolar epithelium.
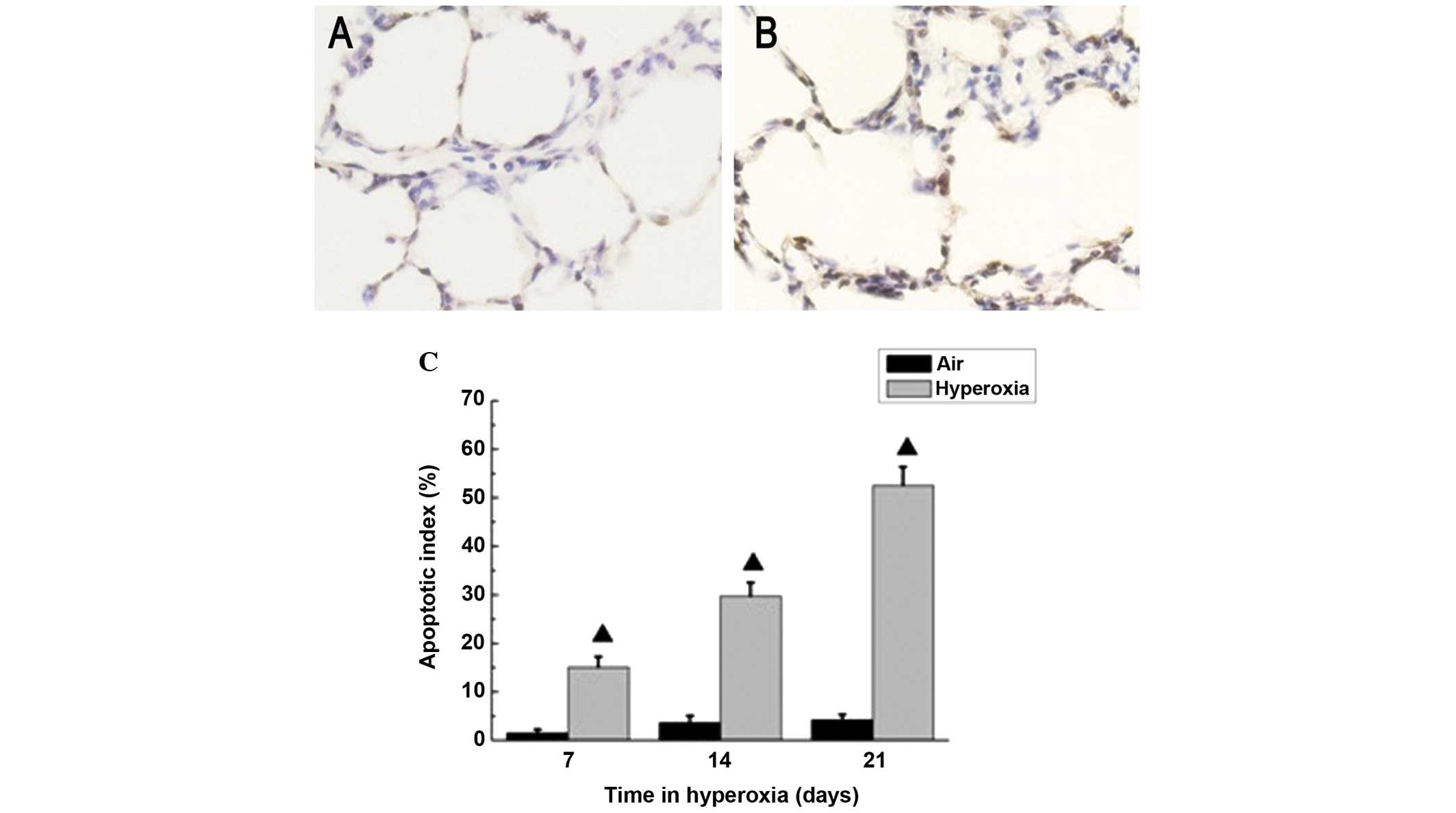
Localization of apoptosis using a TUNEL
assay
The present study determined whether hyperoxia
exposure induces CHOP-mediated apoptosis in the lung. Apoptosis was
analyzed using a TUNEL assay following exposure to hyperoxia or
room air for 14 days. As shown in Fig.
5, apoptosis was induced in the lungs of the rats exposed to
hyperoxic conditions. A few TUNEL-positive cells were observed in
the control group, however, numerous positive cells were observed
in the hyperoxia group. The increase in the number of apoptotic
cells paralleled with the increase in GRP78 and CHOP-positive
cells, which further indicated that activation of the ER stress
pathway, involving CHOP, led to cellular apoptosis in the rat lungs
following hyperoxic exposure.
Discussion
Infants born prematurely are at substantial risk for
the development of BPD (18). The
pathogenesis of BPD remains to be fully elucidated, and multiple
contributing injurious factors are considered to be involved.
Previous studies have demonstrated that rat alveolar development
comprises four stages; pseudoglandular, canalicular, saccular and
alveolar; suggesting that the process of alveolar development has a
time sequence in the rat is similar to that in humans. The alveolar
stage (5–21 days gestation) is a key period of lung development,
which is particularly affected by mechanical factors. Oxygen
toxicity caused by oxygen therapy/hyperoxia is considered to be one
of the major contributing factors (1,19).
In the present study, it was demonstrated that prolonged exposure
to hyperoxia disrupted postnatal alveolarization and led to the
development of BPD in rats.
Hyperoxic therapy produces ROS, which can induce
apoptosis in the lungs of neonatal rats (3). In the present study, the results of
the TUNEL staining in the lung cells suggested that a higher
proportion of epithelial and endothelial cells were apoptotic in
the BPD model rat lungs, compared with the control group lungs.
These results demonstrated that lung cells may undergo increased
apoptosis, and apoptotic cell death may be important in
hyperoxia-induced lung injury and, therefore, in the development of
BPD.
Emerging data have revealed that oxidative
stress-induced ER stress may be crucial in the regulation of
apoptotic cell death (20,21). ER stress occurs when proteins
become misfolded, as can occur with increased oxidative stress or
when the capacity of the ER is exceeded by new protein synthesis
(20,22,23).
Lung epithelial cells secrete a significant quantity of protein,
including surfactants, and these cells may be prone to ER stress.
In previous investigations, ER stress was reported to be involved
in the pathogenesis of several diseases, including in the lungs of
lipopolysaccharide (LPS)-treated mice, chronic obstructive
pulmonary disease and idiopathic pulmonary fibrosis (24–26).
It was also found that CHOP, an apoptosis-associated molecule, is
critical in LPS-induced lung damage (24,27).
The present study hypothesized that ER stress is also associated
with hyperoxia-induced BPD. To investigate this hypothesis, the
expression levels of GRP78 and CHOP were examined in lung cells of
a hyperoxia-induced preterm newborn rat BPD model. The results of
the present study revealed that, following exposure of the rats to
85% hyperoxia, the expression of GRP78 in the lung tissues
increased gradually at 7, 14 and 21 days, and the expression levels
were significantly higher than those in the control air group.
GRP78 is a central regulator of ER homeostasis, and its
upregulation is widely used as a sentinel marker for ER stress
under pathological conditions (28,29).
Therefore, increased expression of GRP78, which indicated ER
stress, was induced in hyperoxia-induced BPD.
GRP78 facilitates protein folding, preventing
intermediates from aggregating and promoting the proteasomal
degradation of misfolded proteins (6,28).
However, if ER homeostasis cannot be restored, a prolonged ER
stress response may induce apoptosis. CHOP is critical in ER
stress-induced apoptosis (5,6). The
induction of CHOP has previously been reported in the lungs of mice
exposed to hyperoxia, with immunohistochemical and in situ
hybridization investigations localizing its expression
predominantly to the bronchiolar epithelium and, to a lesser
extent, throughout the lung parenchyma (30). The present study found that
persistent hyperoxic exposure upregulated the expression of CHOP
and also demonstrated that the enhanced expression levels of GRP78
and CHOP were associated with an increase in the number of
apoptotic cells. These results suggested a crucial role for CHOP in
the apoptosis in hyperoxia-induced BPD.
The present study demonstrated that hyperoxia
increased the expression of CHOP in the lung and that CHOP was
involved in the ER stress-mediated apoptosis pathway, accelerating
the process of hyperoxia-induced lung injury. In contrast to these
findings, Lozon et al (31)
observed that the increase in the expression of CHOP was
independent of ER stress and conferred protection, rather than
increased susceptibility, to hyperoxia-induced lung injury.
Differences between the two studies included the use of preterm
rats, rather than adult mice, in the present study, and the animal
model of hyperoxic lung injury in the present study was established
following a longer duration of exposure to oxygen. Therefore, the
present study hypothesized that the ER stress pathway, involving
CHOP, and ER stress-mediated apoptosis were involved in the
pathogenesis of BPD. Several studies have demonstrated that
decreasing the levels of the anti-apoptotic facto, B-cell lymphoma
2, and increasing the levels of ROS are important in the activation
of CHOP (21,32). Therefore, further investigation of
the precise mechanism of ER stress-mediated apoptosis downstream of
CHOP induction is required.
In conclusion, the present study demonstrated that
exposure to 85% hyperoxia induced the expression of CHOP in preterm
rats by a mechanism dependent on the ER stress pathway. Thus, the
activation of CHOP may be involved in the pathogenesis of BPD under
conditions of hyperoxic stress or inflammatory responses. Whether
inhibition of the protein expression of CHOP is protective for
hyperoxia-induced lung injury remains to be elucidated.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81370746), the
Natural Science Foundation of Jiangsu Province, China (grant no.
BK2011485) and the Social Development Foundation of Zhenjiang,
China (grant no. SH2011022).
References
|
1
|
Jobe AH and Bancalari E: Bronchopulmonary
dysplasia. Am J Respir Crit Care Med. 163:1723–1729. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen Y, Martinez MA and Frank L: Prenatal
dexamethasone administration to premature rats exposed to prolonged
hyperoxia: A new rat model of pulmonary fibrosis (bronchopulmonary
dysplasia). J Pediatr. 130:409–416. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang X, Shan P, Sasidhar M, Chupp GL,
Flavell RA, Choi AM and Lee PJ: Reactive oxygen species and
extracellular signal-regulated kinase 1/2 mitogen-activated protein
kinase mediate hyperoxia-induced cell death in lung epithelium. Am
J Respir Cell Mol Biol. 28:305–315. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Malhotra JD and Kaufman RJ: The
endoplasmic reticulum and the unfolded protein response. Semin Cell
Dev Biol. 18:716–731. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lai E, Teodoro T and Volchuk A:
Endoplasmic reticulum stress: Signaling the unfolded protein
response. Physiology (Bethesda). 22:193–201. 2007. View Article : Google Scholar
|
|
8
|
Zhang K and Kaufman RJ: Signaling the
unfolded protein response from the endoplasmic reticulum. J Biol
Chem. 279:25935–25938. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marciniak SJ, Yun CY, Oyadomari S, Novoa
I, Zhang Y, Jungreis R, Nagata K, Harding HP and Ron D: CHOP
induces death by promoting protein synthesis and oxidation in the
stressed endoplasmic reticulum. Genes Dev. 18:3066–3077. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zinszner H, Kuroda M, Wang X, Batchvarova
N, Lightfoot RT, Remotti H, Stevens JL and Ron D: CHOP is
implicated in programmed cell death in response to impaired
function of the endoplasmic reticulum. Genes Dev. 12:982–995. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar
|
|
12
|
Song B, Scheuner D, Ron D, Pennathur S and
Kaufman RJ: Chop deletion reduces oxidative stress, improves beta
cell function, and promotes cell survival in multiple mouse models
of diabetes. J Clin Invest. 118:3378–3389. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tessitore A, del P Martin M, Sano R, Ma Y,
Mann L, Ingrassia A, Laywell ED, Steindler DA, Hendershot LM and
d'Azzo A: GM1-ganglioside-mediated activation of the unfolded
protein response causes neuronal death in a neurodegenerative
gangliosidosis. Mol Cell. 15:753–766. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yuan Y, Guo Q, Ye Z, Pingping X, Wang N
and Song Z: Ischemic postconditioning protects brain from
ischemia/reperfusion injury by attenuating endoplasmic reticulum
stress-induced apoptosis through PI3K-Akt pathway. Brain Res.
1367:85–93. 2011. View Article : Google Scholar
|
|
15
|
Miyazaki Y, Kaikita K, Endo M, Horio E,
Miura M, Tsujita K, Hokimoto S, Yamamuro M, Iwawaki T, Gotoh T, et
al: C/EBP homologous protein deficiency attenuates myocardial
reperfusion injury by inhibiting myocardial apoptosis and
inflammation. Arterioscler Thromb Vasc Biol. 31:1124–1132. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Emery JL and Mithal A: The number of
alveoli in the terminal respiratory unit of man during late
intrauterine life and childhood. Arch Dis Child. 35:544–547. 1960.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Coalson JJ: Pathology of new
bronchopulmonary dysplasia. Semin Neonatal. 8:73–81. 2003.
View Article : Google Scholar
|
|
18
|
Merritt TA, Deming DD and Boynton BR: The
'new' broncho-pulmonary dysplasia: Challenges and commentary. Semin
Fetal Neonatal Med. 14:345–357. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Saugstad OD: Oxygen and oxidative stress
in bronchopulmonary dysplasia. J Perinat Med. 38:571–577.
2010.PubMed/NCBI
|
|
20
|
Malhotra JD and Kaufman RJ: Endoplasmic
reticulum stress and oxidative stress: A vicious cycle or a
double-edged sword? Antioxid Redox Signal. 9:2277–2293. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shore GC, Papa FR and Oakes SA: Signaling
cell death from the endoplasmic reticulum stress response. Curr
Opin Cell Biol. 23:143–149. 2011. View Article : Google Scholar :
|
|
23
|
Jäger R, Bertrand MJ, Gorman AM,
Vandenabeele P and Samali A: The unfolded protein response at the
crossroads of cellular life and death during endoplasmic reticulum
stress. Biol Cell. 104:259–270. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Endo M, Oyadomari S, Suga M, Mori M and
Gotoh T: The ER stress pathway involving CHOP is activated in the
lungs of LPS-treated mice. J Biochem. 138:501–507. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Greene CM and McElvaney NG: Protein
misfolding and obstructive lung disease. Proc Am Thorac Soc.
7:346–355. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Korfei M, Ruppert C, Mahavadi P, Henneke
I, Markart P, Koch M, Lang G, Fink L, Bohle RM, Seeger W, et al:
Epithelial endoplasmic reticulum stress and apoptosis in sporadic
idiopathic pulmonary fibrosis. Am J Respir Crit Care Med.
178:838–846. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakayama Y, Endo M, Tsukano H, Mori M,
Oike Y and Gotoh T: Molecular mechanisms of the LPS-induced
non-apoptotic ER stress-CHOP pathway. J Biochem. 147:471–483. 2010.
View Article : Google Scholar
|
|
28
|
Rao RV, Peel A, Logvinova A, del Rio G,
Hermel E, Yokota T, Goldsmith PC, Ellerby LM, Ellerby HM and
Bredesen DE: Coupling endoplasmic reticulum stress to the cell
death program: Role of the ER chaperone GRP78. FEBS Lett.
514:122–128. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee AS: The ER chaperone and signaling
regulator GRP78/BiP as a monitor of endoplasmic reticulum stress.
Methods. 35:373–381. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
O'Reilly MA, Staversky RJ, Watkins RH,
Maniscalco WM and Keng PC: p53-independent induction of GADD45 and
GADD153 in mouse lungs exposed to hyperoxia. Am J Physiol Lung Cell
Mol Physiol. 278:L552–L559. 2000.PubMed/NCBI
|
|
31
|
Lozon TI, Eastman AJ, Matute-Bello G, Chen
P, Hallstrand TS and Altemeier WA: PKR-dependent CHOP induction
limits hyperoxia-induced lung injury. Am J Physiol Lung Cell Mol
Physiol. 300:L422–L429. 2011. View Article : Google Scholar :
|
|
32
|
Sung B, Ravindran J, Prasad S, Pandey MK
and Aggarwal BB: Gossypol induces death receptor-5 through
activation of the ROS-ERK-CHOP pathway and sensitizes colon cancer
cells to TRAIL. J Biol Chem. 285:35418–35427. 2010. View Article : Google Scholar : PubMed/NCBI
|