Introduction
Pulmonary arterial hypertension (PAH) is a
life-threatening disease, which contributes to the morbidity and
mortality rates of patients with various lung and heart diseases
(1). PAH has a multifactorial
pathology; and a variety of cell types, including endothelial
cells, smooth muscle cells, inflammatory cells and platelets, may
be implicated in the progression of PAH (2).
Vascular smooth muscle cells (VSMCs) are present in
the medial wall of blood vessels, which are normally quiescent and
express a differentiated phenotype to maintain vascular tone.
However, under pathological conditions, VSMCs switch to a
'synthetic' phenotype, secrete inflammatory cytokines and
contribute to vascular pathogenesis (3). VSMC proliferation of the pulmonary
artery has been considered as one of the major causes of pulmonary
arterial remodeling (4).
Progressive pulmonary arterial remodeling is a characteristic of
PAH, which is central to the persistent deterioration and the
irreversibility of the disease (5). At present, few therapeutic options
are effective for targeting pulmonary arterial structure remodeling
following development of PAH.
Nitric oxide (NO), one of the smallest known
bioactive products in mammalian cells, has an important role in
controlling vascular tone and structure, in which it mediates
relaxation of the vessels through the activation of cyclic
guanosine mono-phosphate-dependent pathways (6). NO has been reported as being central
in the pathogenesis of pulmonary hypertension (7,8). A
previous study demonstrated that the levels of NO in lung tissues
are lower in patients with PAH, compared wirth healthy controls
(9), and that NO inhalation may be
effective in pulmonary vasodilator therapy (10). NO synthase (NOS) transforms
l-arginine
(l-Arg) into NO
(11), and the production of NO
predominantly depends on the activity of NOS and protein
expression.
It has been reported that the oral administration of
l-Arg improves the
hemodynamics and exercise capacities of patients diagnosed with PAH
(12). l-Arg is a common catalyzing
substrate of NOS and arginase (Arg), and Arg is the enzyme in the
urea cycle, which converts l-Arg into urea and polyamines
(13). Thus, Arg and NOS have
reciprocal activities that may shift the metabolism of l-Arg towards polyamine
homeostasis or twoards NO production, respectively. Arg has two
isoforms, Arg I and Arg II. Arg I is predominantly expressed in the
SMCs, and it has been reported that Arg I contributes to human
aortic SMC proliferation (14).
The function of Arg in tissues has attracted increasing attention
(15).
At present, the function of Arg in the development
of PAH remains to be elucidated. The present study aimed to observe
the effects of Arg inhibition on PAH and investigate the associated
mechanisms. It was hypothesized that Arg inhibition may exert a
beneficial role in the prevention and treatment of PAH and, in
order to assess this hypothesis, a series of in vivo and
in vitro experiments were designed to investigate the
underlying roles and mechanisms. The results may support a novel
target for the treatment of PAH.
Materials and methods
Cell culture
Human pulmonary artery smooth muscle cells (HPASMCs)
were purchased from the American Type Culture Collection (Manassas,
VA, USA) and were cultured in Dulbecco's modified Eagle's medium
containing 10% fetal bovine serum at 37°C in a 5% CO2
and 95% air atmosphere. The cells up to the fourth passage were
used for the subsequent experiments. The HPASMCs were placed in
hypoxic conditions (1% O2; 5% CO2) in a cell
culture, which was either untreated or pretreated with
S-(2-boronoethyl)-l-cysteine (BEC) at 37°C for 48 h
(16). BEC (Abcam, Cambridge, UK),
an Arg inhibitor, was used as a positive control in the detection
of Arg interference.
Measurement of HPASMC proliferation
The proliferation of the HPASMCs was determined
using an MTT assay (Beyotime Institute of Biotechnology, Haimen,
China). Briefly, the HPASMCs were seeded into 96-well plates at a
density of 5,000 cells/well. Following exposure to hypoxic
conditions with or without treatment with BEC, the HPASMCs were
incubated with 10 µl MTT (5 mg/ml)/well for 4 h at 37°C. The
supernatant was carefully removed and 75 µl/well dimethyl
sulfoxide was added to dissolve the formazan crystals. The samples
were then analyzed at 570 nm using a Varioskan Flash Multifunction
plate reader (Thermo Fisher Scientific, Waltham, MA, USA).
For cell counting, the HPASMCs were seeded in a
6-well plate at the same density as for the MTT assay, and were
treated in the above-mentioned conditions. Subsequently, the cells
were washed with phosphate-buffered saline, harvested with trypsin
and were counted using a hemocytometer (BC-5300; Mindray Medical
International Limited, Shenzhen, China).
Rat model of chronic hypoxia
exposure
For the in vivo investigations, 8-week-old
male Sprague-Dawley rats were exposed to normoxic (21%
O2) or hypoxic (10% O2) conditions for 3
weeks. During the final 10 days of exposure to the conditions, each
animal was administered with either rosiglitazone (10 mg/kg/day;
R&D, Minneapolis, MN, USA) or an equal volume of vehicle
(methylcellulose; Fortune Biotech, Shanghai, China) daily by oral
gavage. It has been previously reported that this hypoxia regimen
stimulates increased right ventricular systolic pressures (RVSP),
right ventricular hypertrophy and pulmonary vascular remodeling,
and that these hypoxic derangements are attenuated by rosiglitazone
(17). All animals had access to
standard rat chow and water ad libitum and all procedures
were reviewed and approved by the Animal Care and Use Committee of
Shandong University (Jinan, China).
Animals
A total of 45 male Sprague-Dawley rats were
purchased from the Animal Center of the Shandong University School
of Medicine and used for all experiments. All animals were kept
under a 12-h light/dark cycle at 25°C with five rats per cage. All
rats had free access to food and water and were randomly divided
into the following three groups, each containing 15 rats: Normal
group, control group and Arg group. The rats in the normal group
were exposed to normoxic conditions, while the control and Arg
groups were exposed to hypoxic or normoxic conditions, and the Arg
group was administrated with monocrotaline (Sigma-Aldrich, St.
Louis, MO, USA) and the Arg inhibitor, BEC. Subsequent to injection
for 24 days, the rats were anesthetized by intraperitoneal
injection of 2% pentobarbital sodium (0.3 ml/100 g; Sigma-Aldrich)
and the pulmonary arteries were isolated from the rats of the two
groups for the following experiments. All animal care and
experimental protocols complied with the animal management rules of
the Animal Care and Use Committee of Shandong University (Jinan,
China).
Measurement of RVSP
The presence of increased right ventricular pressure
confirms the successful establishment of PAH animal models. Prior
to sacrification of the rats (by intraperitoneal injection of
pentobarbital sodium (20 mg/100 g; Sigma-Aldrich), RVSP was
measured by right heart catheterization. The right jugular vein was
isolated, following which a small polyethylene catheter was passed
through a small transverse cut and advanced into the right
ventricle. RVSP was recorded using a miniature pressure transducer
digitized by a data acquisition system.
Measurement of Arg activity
The activities of Arg in the HPASMCs and the rats
were measured, as previously described (18). Briefly, the cells or tissues were
lysed for 30 min and Tris-HCl (25 mM) containing MnCl2
(5 mM; pH 7.4) was added. Arg was activated by heating for 10 min
at 56°C. The activated lysate was incubated with 0.5 M arginine (pH
9.7) at 37°C for 60 min, followed by termination of the reaction.
The concentration of urea was measured at 540 nm using a Varioskan
Flash Multifunction reader, with one unit of enzyme activity
defined as the quantity of enzyme that catalyzes the formation of 1
µmol urea/min.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The pulmonary arteries of the rats were isolated and
the HPASMCs were harvested, and the total RNA was extracted using
TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA),
according to the manufacturer's instructions. The concentrations of
the total RNA were tested using a spectrophotometer. 1 µg
mRNA was used for reverse transcription in a final volume of 20
µl. The reverse transcription from mRNA to cDNA was
performed using a iScript cDNA synthesis kit (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) containing a mixture of oligo(dT) and
random primers. Real-time PCR was performed with an iQ™ SYBR Green
Supermix kit (Bio-Rad Laboratories, Inc.) using 1 µl cDNA in
a 20-µl volume. The PCR was performed using an iCycler iQ
real-time PCR detection system (Bio-Rad Laboratories, Inc.) and the
program was performed for 40 cycles at 95°C for 30 sec, 55°C for 30
sec and 72°C for 30 sec. In the in vivo experiment, qPCR was
performed using the following primers (Biosune, Shanghai, China):
Cyclin D1, forward 5′-CAGACCAGCCTAACAGATTTC-3′ and reverse
5′-TGACCCACAGCAGAAGAAG-3′; cyclin-dependent kinase (CDK)4, forward
5′-GCTACCACTCGATATGAACCCGTGGCTGAA-3′ and reverse
5′-GGTGCTTTGTCCAGGTATGTCCGTAGGTCC-3′; p27, forward
5′-CTTGGAGAAGCACTGCCGAGAT-3′ and reverse
5′-CCCTGGACACTGCTCCGCTA-3′; and β-actin, forward
5′-ATCATGTTTGAGACCTTCAACA-3′ and reverse
5′-CATCTCTTGCTCGAAGTCCA-3′. In the in vitro experiment, the
sequences of primers were as follows: Cyclin D1, forward
5′-CTCCTCTCCGGAGCATTTTGATA-3′ and reverse
5′-TTAAAGACAGTTTTTGGGTAATCT-3′; CDK4, forward
5′-ATGGCTACCTCTCGATATGAGCCA-3′ and reverse
5′-TCACTCCGGATTACCTTCATCCTT-3′; and p27, forward
5′-CTTGGAGAAGCACTGCCGAGAT-3′ and reverse
5′-CCCTGGACACTGCTCCGCTA-3′. The relative expression levels of the
genes was obtained using the 2−ΔΔct calculation method
(19). Each sample was analyzed in
triplicate and the expression levels were normalized to that of
β-actin.
Western blot analysis
The total proteins were extracted from pulmonary
arteries of the rats and HPASMCs. HPASMCs were scraped off the
dish, and the cell suspension was transferred into a pre-cooled
tube. 30 mg pulmonary arteries of the rats were dissected and
placed in 300-µl cooled lysis buffer. Lysates were kept on
ice for immediate homogenization and maintained under constant
agitation for 30 min at 4°C. The mixtures were centrifuged for 20
min at 16,000 ×g at 4°C. The protein lysate was then transferred to
a fresh tube on ice and an equal volume of 2X loading buffer was
added. Each lysate was boiled in loading buffer at 99°C for 5 min
and subsequently stored at −20°C for western blotting. The protein
concentrations were assayed using a bicinchoninic acid method
(Beyotime Institute of Biotechnology, Haimen, China). The samples
were separated on a 10–12% SDS-polyacrylamide gel and
electrophoretically transferred onto a nitrocellulose membrane (EMD
Millipore, Billerica, MA, USA). Following blocking with 5% non-fat
milk for 2 h at room temperature, the membrane was washed in
Tris-buffered saline with Tween 20 (TBS-T; Beyotime Institute of
Biotechnology) three times for 10 min. Subsequently, the membrane
was incubated with primary antibodies, including rabbit polyclonal
cyclin D1 antibody (1:1,000; Cell Signaling Technology, Inc.,
Danvers, MA, USA), rabbit polyclonal CDK4 (1:500; Abcam, Cambridge,
MA, USA), rabbit monoclonal anti-p27 (1:500; Abcam), rabbit
monoclonal anti-Akt and monoclonal phosphorylated (p)-Akt (1:1,000;
Cell Signaling Technology, Inc.), rabbit monoclonal anti-ERK and
monoclonal p-ERK (1:1,000; Cell Signaling Technology, Inc.) and
rabbit monoclonal anti-β-actin (1:1,000; Cell Signaling Technology,
Inc.) antibodies at 4°C overnight. Following washing with TBS-T
three times, the membrane was incubated with a horseradish
peroxidase-conjugated secondary antibody. The bands were detected
using an enhanced chemiluminescent method (EMD Millipore) and
analyzed using Image-Pro Plus software, version 6.0 (Media
Cybernetics, Inc., Rockville, MD, USA).
Statistical analyses
Analysis of the data was performed using SPSS,
version 13.0 (SPSS, Inc., Chicago, IL, USA). Continuous variables
are expressed as the mean ± standard error of the mean. All
statistical comparisons were performed using Student's t-test or
one-way analysis of variance. P<0.05 was considered to indicate
a statistically significant difference.
Results
BEC reduces hypoxia-induced HPASMC
proliferation in vitro
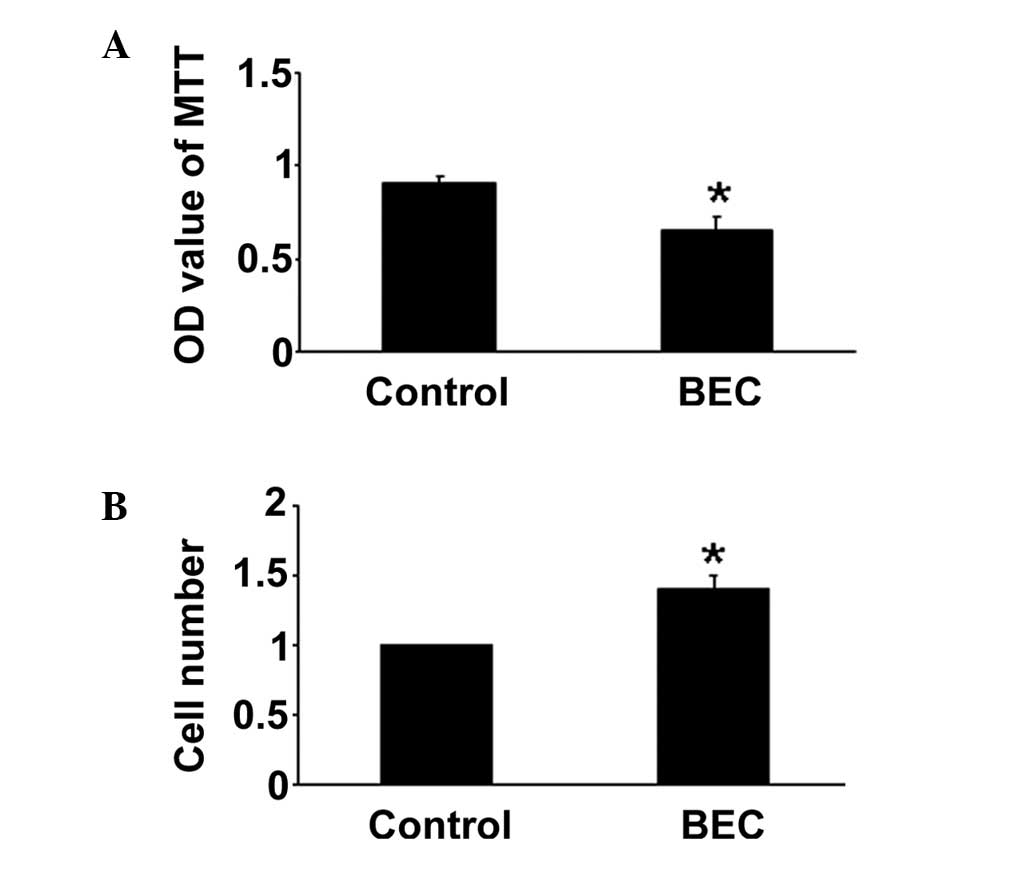
The effects of BEC on HPASMC proliferation were
evaluated under hypoxic conditions. The MTT assay demonstrated that
the inhibition of Arg by BEC inhibited HPASMC proliferation,
compared with the hypoxia group. The cell counting assay produced
similar results (P<0.05; Fig.
1). The regulation of Arg inhibition on HPASMC proliferation
may be one important mechanism of anti-PAH.
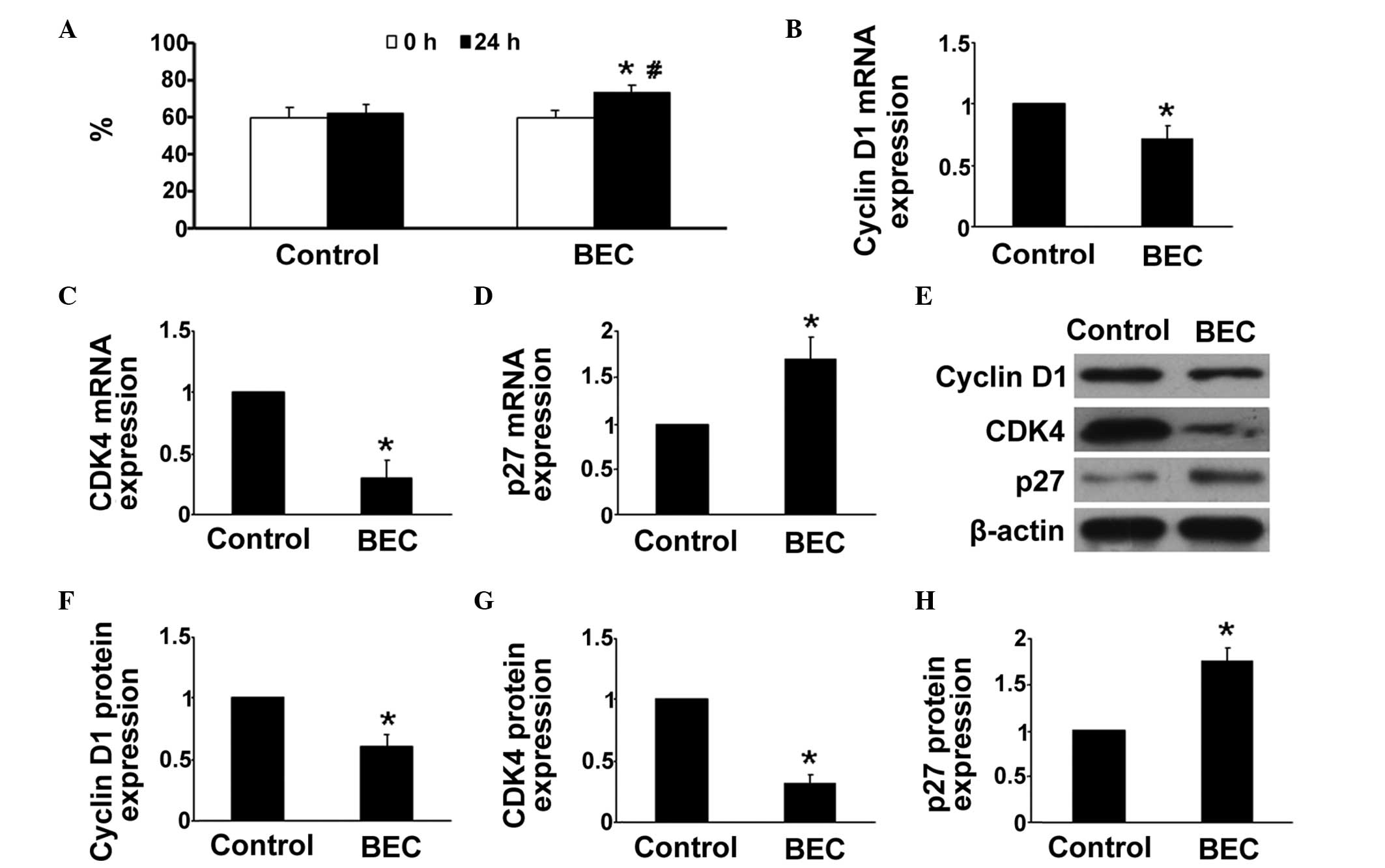
Arg inhibition arrests HPASMCs in the
G1/G0-phase under hypoxic conditions
The proliferation of cells is dependent on the cell
cycle transition between the G1/G0 and the
G2/S phases. In the present study, whether the Arg
inhibitor affected the cell cycle distribution of HPASMCs was
investigated. As shown in Fig. 2A,
compared with the control group, BEC treatment arrested a higher
percentage of HPASMCs in the G1/G0 phase
(P<0.05).
Arg inhibition reduces the expression
levels of cyclin D1 and CDK4, and increases the expression of
p27
The mechanism underlying the effect of hypoxia was
then investigated. Previous studies have demonstrated that cyclin
D1, CDK4 and p27 are key in regulation of cell proliferation and
the cell cycle, therefore, the effects of hypoxia and BEC on their
levels of expression were evaluated in the HPASMCs. RT-qPCR
analysis revealed that the gene expression levels of cyclin D1 and
CDK4 were significantly reduced in the Arg inhibitor group,
compared with those in the control group, whereas treatment with
the Arg inhibitor treatment significantly enhanced hypoxia-induced
gene expression of p27 (P<0.05; Fig. 2B–D). Similar results were obtained
in the western blot analyses (P<0.05; Fig. 2E–H).
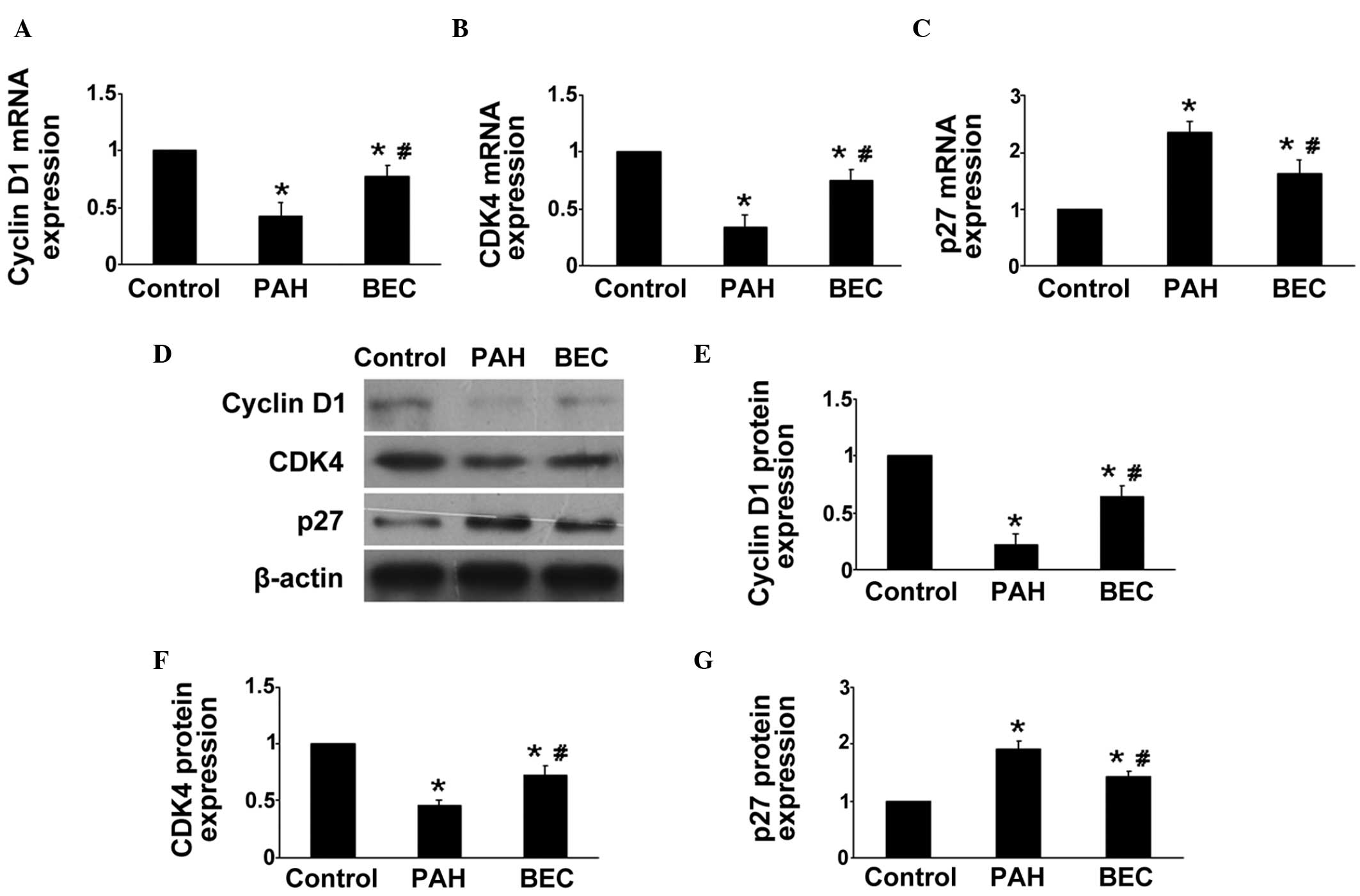
The levels of cyclin D1, CDK4 and p27 were measured
in vivo in the pulmonary arteries of the rats. As
hypothesized, compared with the control group, the gene and protein
expression levels of cyclin D1 and CDK4 were markedly reduced, and
the expression of p27 was markedly increased in the BEC-treated
rats (P<0.0; Fig. 3). These
results indicated that the regulation of Arg inhibition on the cell
cycle may be another mechanism of its anti-PAH effects.
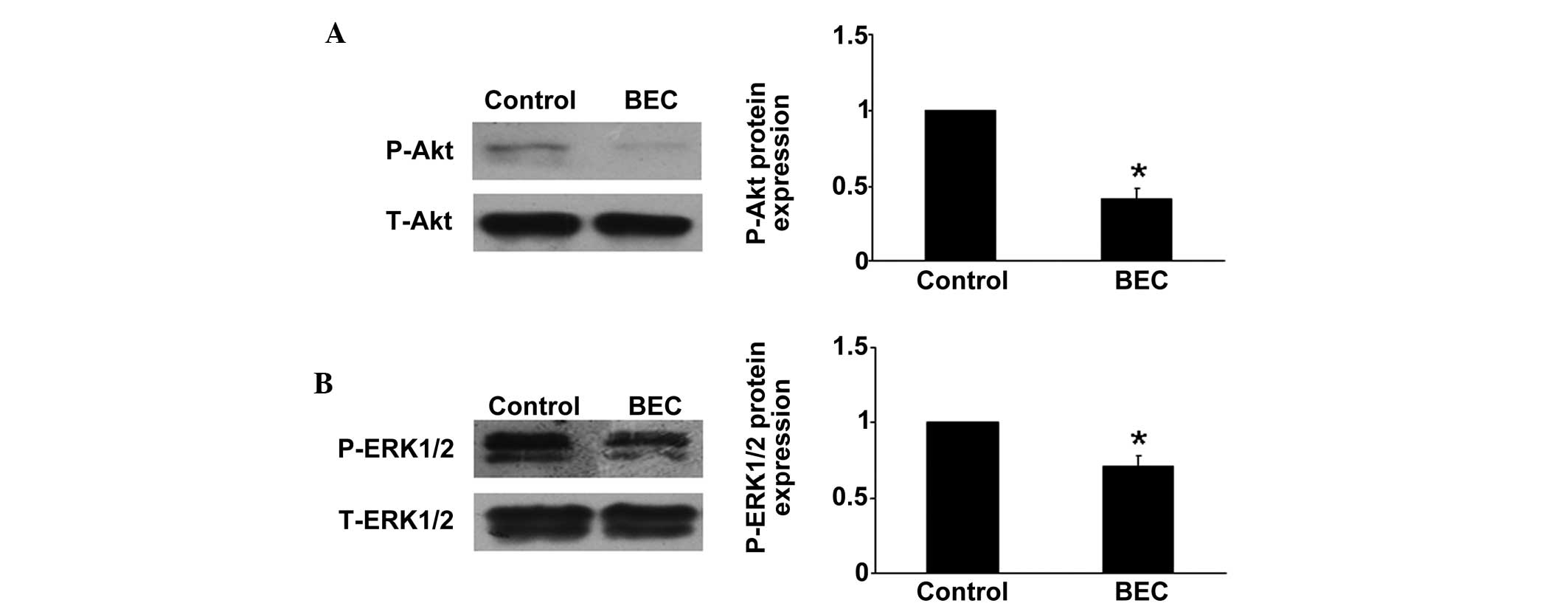
Arg inhibition decreases the
phosphorylation of Akt and ERK1/2
It has been reported that the Akt and ERK pathways
are involved in the proliferation pfHPASMCs and progression of PAH
(20). Thus, in the present study,
the protein expression levels of Akt and ERK in vitro were
assessed using western blot analysis. Compared with the hypoxia
group, Arg inhibition downregulated the phosphorylation of Akt and
ERK, which may be another mechanism of the protective effects of
Arg inhibition (P<0.05; Fig.
4).
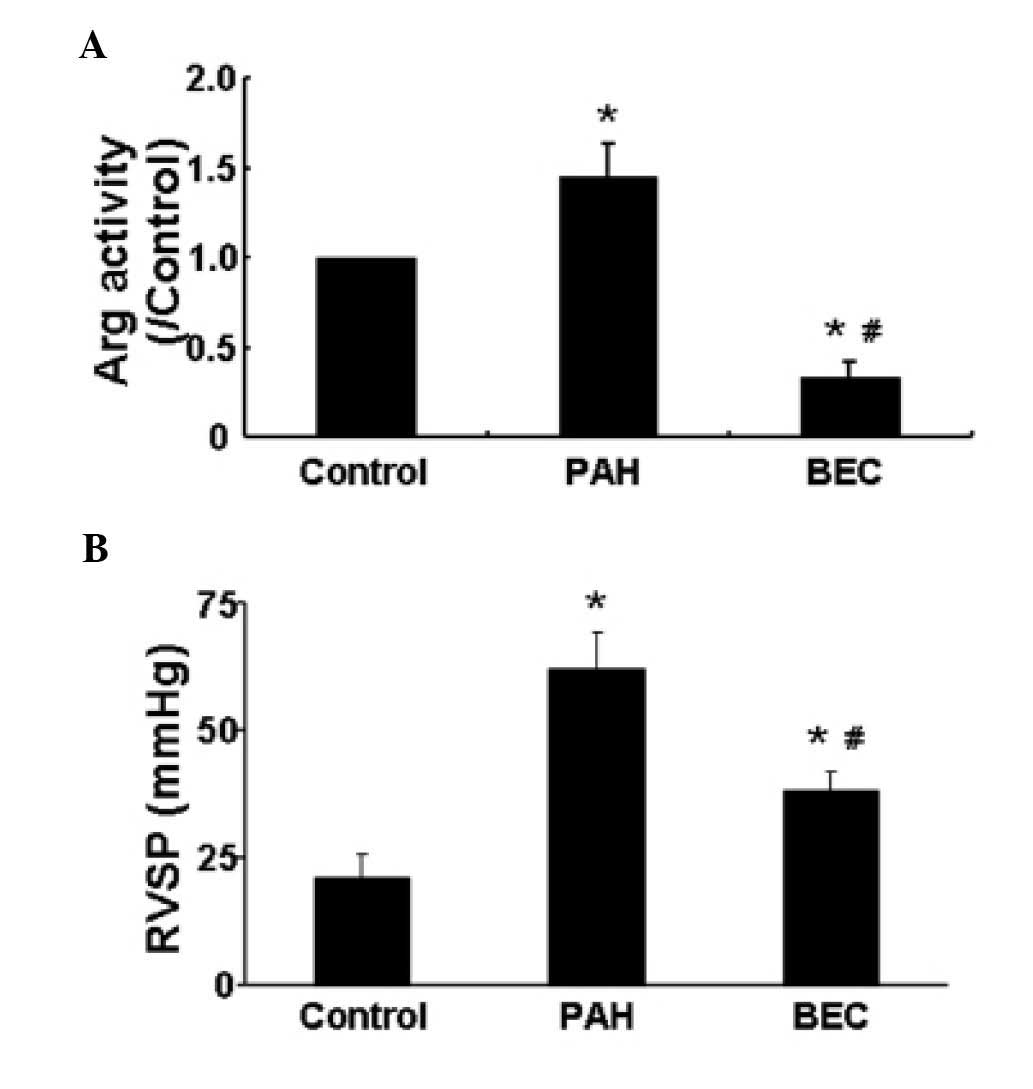
Arg inhibition reduces the PAH-induced
increase of RVSP
The hemodynamic parameters of rats were measured
prior to sacrifice. No significant differences were observed in the
mean blood pressure or heart rate among the groups (data not
shown). Compared with the control group, the rats in the PAH group
exhibited a higher RVSP. However, following the inhibition of Arg
with BEC, the RVSP was significantly reduced (P<0.05; Fig. 5). These results suggested that Arg
inhibition reduced the PAH-induced increase of RVSP.
Hypoxia increases the activity of Arg in
vivo
The activity of Arg in vivo was also
assessed, as previously described (18). The results demonstrated that,
compared with the control group, hypoxia significantly increased
the activity of Arg, and this was reduced following Arg inhibition
by BEC (Fig. 5).
Discussion
PAH is a life-threatening disease, the etiology of
which remains to be elucidated. Although a number of studies have
focused on the development and treatment of PAH, few effective
therapies have been developed. The most important finding of the
present study was that Arg inhibition prevented the progression of
PAH in the rat model. The major mechanisms may involve significant
inhibition of HPASMC proliferation, regulation of the cell cycle
and reduced expression levels of Akt and ERK by BEC. To the best of
our knowledge, this is a novel observation regarding the Arg
inhibitor BEC.
Increased pulmonary artery constriction and
remodeling is a key characteristic of PAH. NO, synthesized by NOS,
is considered to be critical in maintaining pulmonary arterial
pressure and vascular resistance, and it has been reported that NO
is involved in the pathogenesis of pulmonary hypertension (21). A reduction in NO promotes the
development of pulmonary hypertension (22). It was reported previously that, in
an animal model of NOS-deficient mice, increased mean pulmonary
arterial pressure was observed, and pulmonary arterial pressure was
partially restored following the transfer of NOS to the mice
(23). NO inhalation has been
considered as an effective method in PAH therapy (10). l-Arg is a common catalyzing
substrate of NOS and Arg, and the metabolic pathway of l-Arg is an important mechanism
of NO synthesis, where the presence of l-Arg augments NO synthesis and
endothelium-dependent vasodilation. However, Arg has been reported
to compete with NOS for the common catalyzing substrate, thus
shifting the metabolism of arginine to urea (19). Therefore, inhibition of Arg may
inhibit the conversion of l-Arg to urea and increase NO
synthesis.
The principal phenotype of SMCs is contraction,
which preserves vasodilation and blood flow regulation in
physiological conditions. However, SMCs exhibit a 'synthetic'
phenotype in pathological conditions, and increase the capacity of
proliferation and generation of matrix components of the blood
vessel wall, contributing to vascular remodeling (24). Aberrant HPASMC proliferation leads
to pulmonary arterial remodeling and contributes to the progression
of PAH, whereas effective inhibition the aberrant HPASMCs can delay
and even halt the deteriorative progression of PAH (4). In the present study, the role of Arg
inhibition in the proliferation of hypoxia-induced HPASMCs was
investigated, which revealed that Arg inhibition effectively
inhibited the proliferation of HPASMCs. Therefore, the anti-PAH
properties of Arg inhibition in the rats may have been attributed
to its role in HPASMCs proliferation.
Under hypoxic conditions, more HPASMCs enter cell
mitosis, and acceleration of the cell cycle is an initial factor in
cell proliferation. Hypoxia has been reported to result in low cell
numbers in the G0/G1 phase and an increase in
HPASMCs entering G2/S phase (25). In the present study, the effects of
Arg inhibition on the cell cycle of HPASMCs were assessed, and it
was demonstrated that Arg inhibition reversed the effect of
hypoxia.
A previous study reported that the balance between
cell quiescence and proliferation is regulated by cyclin-dependent
kinases (CDKs) and CDK inhibitors (26). Cyclin D1 and CDKs, predominantly
CDK4, are key genes controlling the cell cycle, are associated with
cell proliferation and facilitate the transition of cells between
the G1 phase and the S phase (27). The overexpression of CDK4 promotes
cell proliferation, whereas inhibition of the expression of CDK4
can lead to arrest at the G1 phase and the suppression
of cell proliferation (28). In
the present study, Arg inhibition significantly reduced the
expression levels of cyclin D1 and CDK4 in vivo and in
vitro. p27, as one of the key CDK inhibitors, effectively
inhibits cyclin D1-CDK4 protein kinase activity and negatively
regulates G1 progression in cells, and verexpression of
p27 results in G1 arrest and reduces the proliferation
of HPASMCs (29). The results of
the present study demonstrated that Arg inhibition increased the
mRNA and protein expression levels of p27 in vivo and in
vitro. Thus, it was suggested that Arg inhibition promoted
G1 phase arrest, which may be the direct mechanism of
Arg inhibition against HPASMCs proliferation and PAH.
The activation of Akt and ERK by diverse
extracellular signals triggers cellular cascade responses,
including cell growth, proliferation, survival and motility,
prompting investigation of their expression in the present study.
The levels of p-Akt and p-ERK were higher in the hypoxic HPASMCs,
compared with the control cells, and Arg inhibition inhibited the
activation of the Akt and ERK pathways.
The findings of the present study provide support
for inhibition of Arg as a useful therapeutic intervention for the
treatment of pulmonary hypertensive disorders.
References
|
1
|
Frumkin LR: The pharmacological treatment
of pulmonary arterial hypertension. Pharmacol Rev. 64:583–620.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Humbert M, Morrell NW, Archer SL, Stenmark
KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O and
Voelkel NF: Cellular and molecular pathobiology of pulmonary
arterial hypertension. J Am Coll Cardiol. 43(Suppl 12): 13S–24S.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Orlandi A, Bochaton-Piallat ML, Gabbiani G
and Spagnoli LG: Aging, smooth muscle cells and vascular
pathobiology: Implications for atherosclerosis. Atherosclerosis.
188:221–230. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Luo Y, Xu DQ, Dong HY, Zhang B, Liu Y, Niu
W, Dong MQ and Li ZC: Tanshinone iia inhibits hypoxia-induced
pulmonary artery smooth muscle cell proliferation via
Akt/Skp2/p27-associated pathway. PLoS One. 8:e567742013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stenmark KR, Fagan KA and Frid MG:
Hypoxia-induced pulmonary vascular remodeling: Cellular and
molecular mechanisms. Circ Res. 99:675–691. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vasa M, Fichtlscherer S, Adler K, Aicher
A, Martin H, Zeiher AM and Dimmeler S: Increase in circulating
endothelial progenitor cells by statin therapy in patients with
stable coronary artery disease. Circulation. 103:2885–2890. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sudar E, Dobutovic B, Soskic S, Mandusic
V, Zakula Z, Misirkic M, Vucicevic L, Janjetovic K, Trajkovic V,
Mikhailidis DP, et al: Regulation of inducible nitric oxide
synthase activity/expression in rat hearts from ghrelin-treated
rats. J Physiol Biochem. 67:195–204. 2011. View Article : Google Scholar
|
|
8
|
Isenovic ER, Meng Y, Divald A, Milivojevic
N and Sowers JR: Role of phosphatidylinositol 3-kinase/akt pathway
in angiotensin ii and insulin-like growth factor-1 modulation of
nitric oxide synthase in vascular smooth muscle cells. Endocrine.
19:287–292. 2002. View Article : Google Scholar
|
|
9
|
Kaneko FT, Arroliga AC, Dweik RA, Comhair
SA, Laskowski D, Oppedisano R, Thomassen MJ and Erzurum SC:
Biochemical reaction products of nitric oxide as quantitative
markers of primary pulmonary hypertension. Am J Respir Crit Care
Med. 158:917–923. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pepke-Zaba J, Higenbottam TW, Dinh-Xuan
AT, Stone D and Wallwork J: Inhaled nitric oxide as a cause of
selective pulmonary vasodilatation in pulmonary hypertension.
Lancet. 338:1173–1174. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ribeiro MO, Antunes E, de Nucci G,
Lovisolo SM and Zatz R: Chronic inhibition of nitric oxide
synthesis. A new model of arterial hypertension. Hypertension.
20:298–303. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nagaya N, Uematsu M, Oya H, Sato N,
Sakamaki F, Kyotani S, Ueno K, Nakanishi N, Yamagishi M and
Miyatake K: Short-term oral administration of l-arginine improves
hemodynamics and exercise capacity in patients with precapillary
pulmonary hypertension. Am J Respir Crit Care Med. 163:887–891.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mori M and Gotoh T: Regulation of nitric
oxide production by arginine metabolic enzymes. Biochem Biophys Res
Commun. 275:715–719. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wei LH, Wu G, Morris SM Jr and Ignarro LJ:
Elevated arginase I expression in rat aortic smooth muscle cells
increases cell proliferation. Proc Natl Acad Sci USA. 98:9260–9264.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ckless K, Lampert A, Reiss J, Kasahara D,
Poynter ME, Irvin CG, Lundblad LK, Norton R, van der Vliet A and
Janssen-Heininger YM: Inhibition of arginase activity enhances
inflammation in mice with allergic airway disease, in association
with increases in protein s-nitrosylation and tyrosine nitration. J
Immunol. 181:4255–4264. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu X, Murphy TC, Nanes MS and Hart CM:
Ppar{gamma} regulates hypoxia-induced nox4 expression in human
pulmonary artery smooth muscle cells through nf-{kappa B. Am J
Physiol Lung Cell Mol Physiol. 299:L559–L566. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Crossno JT Jr, Garat CV, Reusch JE, Morris
KG, Dempsey EC, McMurtry IF, Stenmark KR and Klemm DJ:
Rosiglitazone attenuates hypoxia-induced pulmonary arterial
remodeling. Am J Physiol Lung Cell Mol Physiol. 292:L885–L897.
2007. View Article : Google Scholar
|
|
18
|
Corraliza IM, Campo ML, Soler G and
Modolell M: Determination of arginase activity in macrophages: A
micromethod. J Immunol Methods. 174:231–235. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang XP, Chen YG, Qin WD, Zhang W, Wei SJ,
Wang J, Liu FQ, Gong L, An FS and Zhang Y: Arginase i attenuates
inflammatory cytokine secretion induced by lipopolysaccharide in
vascular smooth muscle cells. Arterioscler Thromb Vasc Biol.
31:1853–1860. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kiss T and Kovacs K, Komocsi A, Tornyos A,
Zalan P, Sumegi B, Gallyas F Jr and Kovacs K: Novel mechanisms of
sildenafil in pulmonary hypertension involving
cytokines/chemokines, MAP kinases and Akt. PLoS One. 9:e1048902014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu W, Kaneko FT, Zheng S, Comhair SA,
Janocha AJ, Goggans T, Thunnissen FB, Farver C, Hazen SL and
Jennings C: Increased arginase ii and decreased no synthesis in
endothelial cells of patients with pulmonary arterial hypertension.
FASEB J. 18:1746–1748. 2004.PubMed/NCBI
|
|
22
|
Fagan KA, Morrissey B, Fouty BW, Sato K,
Harral JW, Morris KG Jr, Hoedt-Miller M, Vidmar S, McMurtry IF and
Rodman DM: Upregulation of nitric oxide synthase in mice with
severe hypoxia-induced pulmonary hypertension. Respir Res.
2:306–313. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Champion HC, Bivalacqua TJ, Greenberg SS,
Giles TD, Hyman AL and Kadowitz PJ: Adenoviral gene transfer of
endothelial nitric-oxide synthase (enos) partially restores normal
pulmonary arterial pressure in enos-deficient mice. Proc Natl Acad
Sci USA. 99:13248–13253. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Owens GK: Regulation of differentiation of
vascular smooth muscle cells. Physiol Rev. 75:487–517.
1995.PubMed/NCBI
|
|
25
|
Kadowaki M, Mizuno S, Demura Y, Ameshima
S, Miyamori I and Ishizaki T: Effect of hypoxia and Beraprost
sodium on human pulmonary arterial smooth muscle cell
proliferation: the role of p27kip1. Respir Res. 8:772007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu L, Quinn DA, Garg HG and Hales CA: Gene
expression of cyclin-dependent kinase inhibitors and effect of
heparin on their expression in mice with hypoxia-induced pulmonary
hypertension. Biochem Biophys Res Commun. 345:1565–1572. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dong Y, Sui L, Sugimoto K, Tai Y and
Tokuda M: Cyclin D1-cdk4 complex, a possible critical factor for
cell proliferation and prognosis in laryngeal squamous cell
carcinomas. Int J Cancer. 95:209–215. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sakamoto K, Ohki K, Saito M, Nakahara T
and Ishii K: Small molecule cyclin-dependent kinase inhibitors
protect against neuronal cell death in the ischemic-reperfused rat
retina. J Ocul Pharmacol Ther. 27:419–425. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Toyoshima H and Hunter T: P27, a novel
inhibitor of g1 cyclin-cdk protein kinase activity, is related to
p21. Cell. 78:67–74. 1994. View Article : Google Scholar : PubMed/NCBI
|