Introduction
Next-generation sequencing (NGS) has introduced a
novel era, in which it has become possible to rapidly and
cost-efficiently obtain large amounts of sequence data for
patients. NGS analysis is therefore currently regarded as a
first-choice diagnostic tool, particularly in cases in which
phenotypical and clinical features are unclear or atypical, leaving
Sanger sequencing for the confirmation of detected variants
(1). The correct placement of a
preliminary clinical diagnosis and subsequent selection of
candidate genes for molecular analysis are challenging when
patients present with signs and symptoms that do not match any
known clinical condition. In these cases, the use of Sanger
sequencing to produce a molecular diagnosis is unreasonable due to
the high cost and effort required.
In these cases, whole exome sequencing (WES) can be
an appropriate tool to detect pathogenic mutations; however, this
requires an effective strategy for filtering results and in-depth
analysis to meaningfully interpret incidental findings (2,3).
Furthermore, in silico and functional studies may be
necessary to attribute the correct value to unexpected results.
The present study reports on a patient who was
selected, given the clinical phenotype complexity, for
proband-parents (trios) analysis using WES with the aim of
detecting all the possible genetic variants that may have generated
the patient's phenotype.
The present study confirmed that it is essential to
filter and confirm each step of the NGS based on the decision of a
team that has the necessary expertise to assess the impact of the
detected variants.
Materials and methods
Ethical approval
The technical and scientific review board of the
Institute for Maternal and Child Health, Istituto di Ricovero e
Cura a Carattere Scientifico (IRCCS) 'Burlo Garofolo' (Trieste,
Italy) (no. 185/08, 19/08/2008) approved the present study. For
enrolment of the child and the parents in the present study,
informed consent was obtained from the parents.
Patient
A six-year-old boy with non-consanguineous parents
was admitted to the Paediatric Department of the IRCCS 'Burlo
Garofolo' (Trieste, Italy) due to neurogenic diabetes insipidus,
mild hepatomegaly and recurrent episodes of fever with skin rash
and cervical adenitis.
DNA extraction
Genomic DNA (gDNA) was extracted from 1–2 ml
EDTA-anticoagulated blood obtained from the proband and his parents
using an EZ1 DNA Blood kit (Qiagen, Hilden, Germany) according to
manufacturer's instructions.
WES analysis
A total of 3 μg gDNA was used for library
preparation using TruSeq™ Exome Enrichment 62 Mb kit (Illumina
Inc., San Diego, CA, USA). A paired-end sequencing of each library
was performed on an Illumina HiSeq1000 (Illumina Inc.). Raw
sequencing data were collected as unmapped 100-bp reads in fastQ
format. CLC Genomics Workbench version 6.5 software (CLC Bio;
Qiagen) was employed to check the quality of the reads, to map
reads back to the human reference genome, version hg19, to
calculate the overall coverage, to perform local realignment and
base quality recalibration and, finally, to call single nucleotide
variants (SNVs) and small insertion/deletions (INDELs). All of
these data were collected into a standardized variant call format,
version 4.1 (4). SNVs/INDELs were
annotated using ANNOVAR software (http://www.openbioinformatics.org/annovar/; version
2013Aug23) referring to several public databases, including
National Center of Biotechnology Information dbSNP build138
(http://www.ncbi.nlm.nih.gov/SNP/), the
1000 Genomes Project (http://www.1000genomes.org/) and the National Heart,
Lung, and Blood Institute Exome Sequencing Project Exome Variant
Server (http://evs.gs.washington.edu/EVS/) (5).
Bioinformatic and statistical
analyses
SN Vs/INDELs were first analyzed with the aim of
evaluating all known mutations already associated with the clinical
manifestations of the patient. Subsequently, SNVs/INDELs were
filtered with the purpose of selecting variants which are poorly
represented within the general population and potentially
'pathogenic'; to this end, variants were selected according to the
following inclusion criteria: A) Minor allelic frequency <0.03
in residents with ancestry from northern and western Europe,
referring to the 1000 Genomes Project (http://www.1000genomes.rg/) and NHLBI Exome Sequencing
Project (ESP) Exome Variant Server (http://evs.gs.washington.du/EVS/); B) homozygous
variants carried by the proband; and C) variants predicted as
'pathogenic' based on Polyphen-2 (6), Mutation Taster and LRT scores, as
recorded in dbNSFP v2.0 (7,8).
Polymerase chain reaction (PCR) and
Sanger analyses
PCR amplification was performed for the specific
genecoding sequences of the MVK and RAB40AL genes. 50
ng DNA was amplified using KAPA 2G Fast Hot Start Readymix (initial
concentration, 2X; final concentration, 1X; respective primer
initial concentration, 10 μM; final concentration, 0.2
μM; final volume, 25 μl) (Resnova, Roma, Italy) and a
2720 Thermal Cycler (Applied Biosystems, Thermo Fisher Scientific,
Waltham, MA, USA). Amplification products were purified with
ExoSAP-IT enzyme (Affymetrix, Inc., Santa Clara, CA, USA) at 37°C
for 20 sec, subsequently inactivated at 85°C for 10 sec, and
directly sequenced by the Sanger method using ABI Prism 3.1 Big Dye
terminator chemistry (Invitrogen Life Technologies, Carlsbad, CA,
USA) and BigDye XTerminator Purification kit (Applied Biosystems).
Runs were performed on the ABI Prism 3130XL automated DNA sequencer
(Applied Biosystems). Sequences were analyzed with Seqman II
Software (DNASTAR I Lasergene 7.0; DNASTAR, Madison, WI, USA). PCR
amplification and Sanger sequencing primers to confirm the
MVK (NM_000431) mutations (c.1129G>A; p.V377I) and
(c.404C>T; p.S135L) were: MVK5 forward,
5′-GTTGAGAAAACTGGACCAGATGC-3′ and MVK5 reverse,
5′-CTCAGCTTCCTCATGTTAAAATG-3′; and MVK11 forward,
5′-GGCTTTTGCCTTGAATATGATGA-3′ and MVK11 reverse,
5′-GGGCCTCTCCAGCAGTGTC-3′. PCR amplification and Sanger sequencing
primers to confirm the RAB40AL (NM_001031834) known
dinucleotide missense changes c.A176G and c.C177A, leading top.D59G
were: RAB40AL forward, 5′-CCTCTGCGCACAACCTTGC-3′ and
RAB40AL reverse, 5′-CTGGAGCGATTCCAGCTTG-3′. Primers were
obtained from Integrated DNA Technologies (Coralville, IA, USA).
PCR amplification cycle: Initial denaturation (95°C for 1 sec), 30
cycles of denaturation (95°C for 10 sec), annealing (60°C for 10
sec) and elongation (72°C for 5 sec), followed by a final
elongation (72°C for 30 sec). Sequence reaction cycle: Initial
denaturation (96°C for 3 sec), 26 cycles of denaturation (96°C for
30 sec), annealing (53°C for 15 sec) and elongation (60°C for 4
sec).
RAB40AL inactivation analysis
300 ng genomic DNA of the patient was digested with
40 U HpaII methylation-sensitive restriction enzyme and 20 U RsaI
methylation-insensitive restriction enzyme (New England Biolabs,
Ipswich, MA, USA) in a total volume of 20 μl for 24 h at
37°C. After digestion, the mixtures were incubated at 95°C for 10
min. 50 ng digested and undigested DNA were amplified with
RAB40AL forward and reverse primers as previously described
to verify the inactivation of the gene.
X-chromosome inactivation (XCI) analysis
via androgen receptor
300 ng genomic DNA of the patient's mother was
digested as described above and incubated at 95°C for 10 min. 50 ng
of digested and undigested DNA was amplified with primers
5′-GCTGTGAAGGTTGCTGTTCCTCAT-3′ and
5′–56-FAM/TCCAGAATCTGTTCCAGAGCGTGC-3′ according to the protocol of
a previous study (9) for
amplification of a region of the human androgen receptor (HAR)
gene, localized on Xq12 and characterized by a polymorphic number
of CAG repeats. Since this fragment has two HpaII restriction
enzyme sites, only methylated DNA was amplified.
XCI analysis via the polymorphic marker
DXS1214
300 ng genomic DNA of the patient's mother was
digested as described above and incubated at 95°C for 10 min. 50 ng
digested and undigested DNA was a mplified with specific
DXS1214-labeled primers.
The labeled PCR products were denatured and run on
the automated sequencer (ABI 3730XL Platform; Applied Biosystems)
using the POP7 polymer and the ROX size standard as size markers
(Invitrogen Life Technologies, Inc.). GeneMapper software (v. 4.0;
Applied Biosystems) was used for fragment analysis.
Biochemical analysis
Urinary mevalonic acid was determined by means of
gas chromatography/mass spectrometry. Spectra were obtained using a
Hewlett Packard gas chromatograph 6890 series equipped with a
Hewlett Packard 5973 quadrupole operating in electron-impact mode
at 70 eV (Hewlett Packard, Palo Alto, CA, USA) (10).
Results and Discussion
WES revealed two missense mutations of the
MVK gene (heterozygous): NM_000431, c.1129G>A; p.V377I
(rs28934897); and NM-000431, c.404C>T; p.S135L (rs104895297). In
addition, the known dinucleotide missense mutations of the
RAB40AL (NM_001031834) gene were detected: c. A176G
(rs145606134) and c. C177A (rs138133927), leading to p.D59G. Sanger
sequencing analysis confirmed all mutations. No mutations were
detected in genes associated with diabetes insipidus.
The MVK gene encodes mevalonate kinase, the
third enzyme on the pathway leading to the synthesis of cholesterol
from acetyl-CoA (11). Mutations
of the MVK gene cause a recessive inherited disease called
mevalonate kinase deficiency (MKD), which manifests as a clinical
and biochemical continuum with mevalonic aciduria at the most
severe end [MA; online mendelian of inheritance in man (OMIM)
#610377] and hyper immunoglobulin D and periodic fever syndrome at
the mildest end (HIDS; OMIM #260920) (12).
The D59G variant of RAB40AL has already been
reported as causative of the Martin-Probst syndrome (MPS; OMIM
#300519), a recessive X-linked disease characterized by congenital
sensorineural hearing loss, mental retardation, short stature,
congenital umbilical hernia, facial dysmorphism, abnormal teeth,
widely spaced nipples and abnormal dermatoglyphics (13,14).
In the present study, a multidisciplinary team discussed the
relevance of the genetic results in order to compare the patient's
clinical features with those pertaining to MKD and MPS, based on
clinical, genetic and functional evaluations.
The diagnosis of MKD was supported not only by the
fact that the two mutations detected have a well known role in the
disease, but also by a biochemical analysis that demonstrated the
accumulation of mevalonic acid [5,496 μg/ml; the normal
range of mevalonic acid levels is 34–323 μg/ml (internal
laboratory reference)] in urine during febrile crisis, as a proof
of MK enzyme deficiency (10).
In the case of the present study, the diagnosis of
MKD adequately explained the proband's condition, apart from the
presence of neurogenic diabetes insipidus (Table I).
 | Table IMain symptoms for MP syndrome and MK
deficiency compared with the symptoms of patient of the present
study. |
Table I
Main symptoms for MP syndrome and MK
deficiency compared with the symptoms of patient of the present
study.
| Feature | MP syndrome | MK deficiency | Patient |
|---|
| Growth | Short stature | From failure to
thrivea to normal; hyper
immunoglobulin D; periodic fever syndrome | Normal |
| Head | Microcephaly | Microcephalya | Normal |
| Ears | Hearing loss | Normal | Normal |
| Eye | Normal | Central
cataractsa | Normal |
| Dysmorphisms | Low-set
ears
Telecanthus
Hypertelorism
Epicanthal folds
Broad nasal root
Broad mouth
Full lower lip
Abnormal teeth | Triangular
facea, Hypoplastic alae
nasia | Normal |
| Genitourinary | Bifid
scrotum
Small phallus
Cryptorchidism
Hypoplastic kidneys
Renal insufficiency | Normal | Normal |
| Skin | Telangiectasias | Skin rashes | Skin rashes |
| Central nervous
system | Mental retardation,
mild to severe | Psychomotor
retardationa, Cerebella
ataxiaa | Neurogenic diabetes
insipidus |
| Osteoarticular
system | Normal |
Arthralgia/arthritis | Normal |
| Endocrine
features | Hypothyroidism | Normal | Normal |
| Hematology | Pancytopenia | Anemia | Mild anemia |
| Lymphoid organs | Leukopenia | Lymphadenopathy,
hepato-splenomegaly | Lymphadenopathy,
hepato-splenomegaly |
| Molecular Basis | RAB40AL | MVK | MVK
V377I/S135L
RAB40AL D59G |
In fact, most of the clinical features of the
patient are characteristics of MKD, although a large number of
MKD-associated symptoms were not present in the patient. In
particular, diabetes insipidus, which was one of the main
complaints of the patient, is not characteristic of MKD. By
contrast, no sign or symptom of MPS was present in the patient.
Since MPS is described as an X-recessive disease with complete
penetrance, it was unexpected that the patient had not developed
any of the severe impairments, which are typical of the disease. It
was thus hypothesized that either the defect in MK has a protective
effect against the development of MPS or that the variant detected
in RAB40AL is not the actual cause of MPS. Of note, the
variant identified in RAB40AL was predicted to affect
protein farnesylation; however, MKD is also associated with an
abnormal pattern of protein prenylation due to a shortage of
mevalonate-derived isoprenoids (15,16).
However, considering that the two defects can reduce protein
farnesylation, the combined effect of the two defects would have
been expected to be even more severe. On the contrary, the National
Heart, Lung and Blood Institute-exome sequencing project (ESP)6500
database (http://evs.gs.washington.edu/EVS/) lists two healthy
males carrying the D59G variant of RAB40AL, suggesting that
it represents an irrelevant albeit rare polymorphism. In addition,
recent studies described the same RAB40AL variant in a
patient with a distinctly different phenotype from that reported in
MPS patients (17,18).
To ascertain the possibility that RAB40AL is
not the causative gene of MPS, the present study exploited the
opportunity of studying the mother of the patient, who is a
heterozygous carrier of the same gene variant. Bedoyan et al
(13) demonstrated that, while
males affected by MPS develop progressive loss of white blood
cells, female carriers have a normal leukocyte count and display a
skewed XCI in peripheral blood cells. The present study then
attempted to analyze the inactivation profile of RAB40AL by
amplifying the gene after digestion with a methylation-sensitive
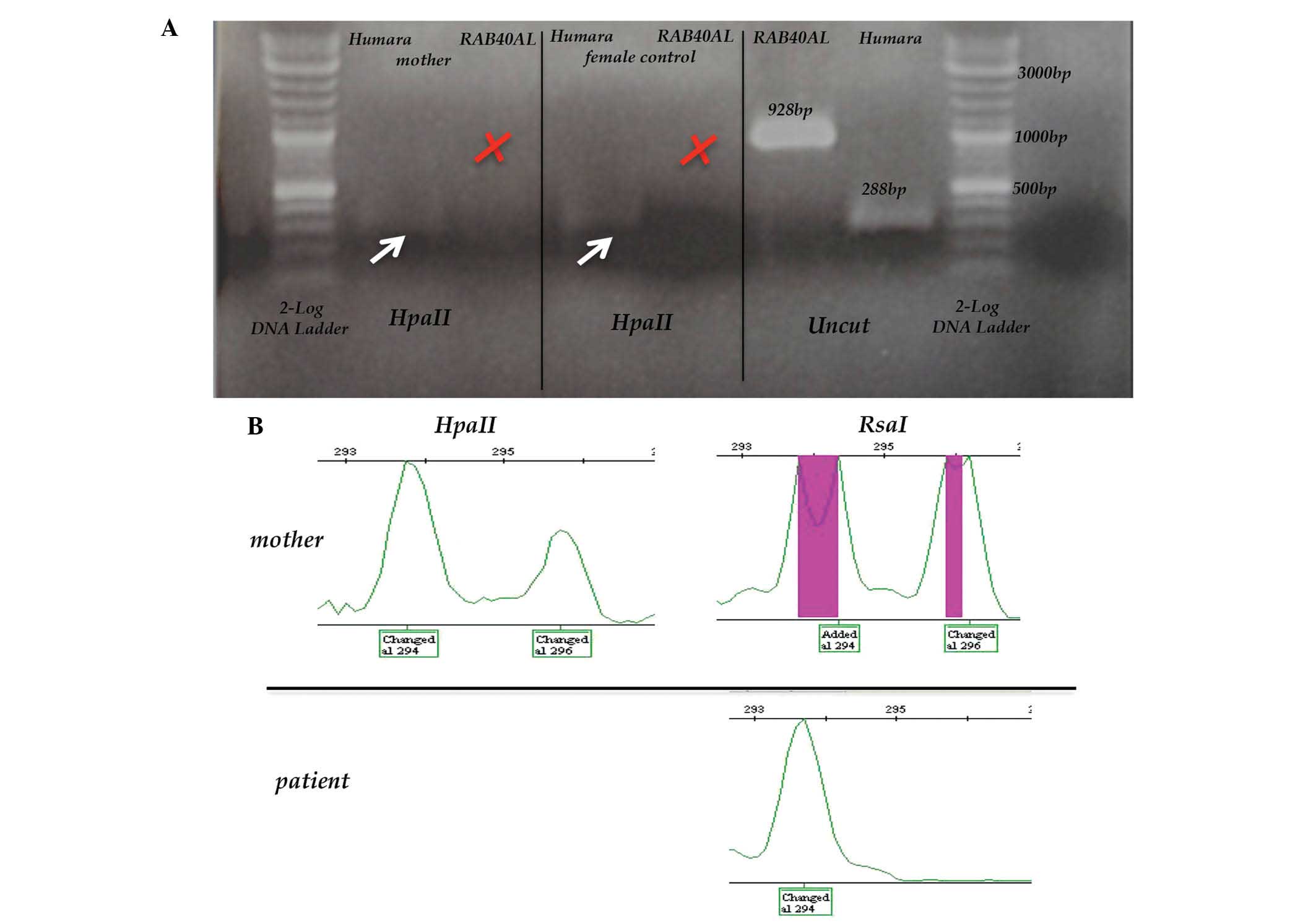
enzyme (19,20). However, it was not possible to
amplify RAB40AL after HpaII digestion, suggesting
that this gene escapes inactivation (Fig. 1A). In order to verify the
hypothesis of a skewed XCI, the present study then used polymorphic
X-chromosome markers known to undergo methylation and inactivation.
As the most widely used androgen receptor humara locus was
uninformative in the patient's family, the polymorphic marker
DXS1214 was then analyzed. After digestion with HpaII, which
is selective for active unmethylated DNA, the two DXS1214
polymorphic alleles remained present to similar extents, confirming
a balanced XCI (Fig. 1B). These
results suggested that the pattern of skewed XCI in the family
examined by Bedoyan et al (13) is not due to the D59G variant in
RAB40AL, but to another unidentified X-chromosome gene
mutation. In conclusion, these results suggested that mutations of
RAB40AL are not the cause of MPS.
The case of the present study confirmed the
potential of the NGS technique to identify pathogenic and causative
mutations, but also highlighted the crucial role of an expert team
performing the critical revision of all detected WES variants.
Acknowledgments
The authors would like to thank the patient and his
family for participating in this study. This work was supported by
an Intramural grant of the Institute for Maternal and Child Health
(IRCCS 'Burlo Garofolo', Trieste, Italy). The present study was
also supported by the 'Associazione Azzurra Malattie Rare' and
'Beneficentia Stiftung in Vaduz, Liechtenstein'.
References
|
1
|
van Dijk EL, Auger H, Jaszczyszyn Y and
Thermes C: Ten years of next-generation sequencing technology.
Trends Genet. 30:418–426. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pelak K, Shianna KV, Ge D, Maia JM, Zhu M,
Smith JP, Cirulli ET, Fellay J, Dickson SP, Gumbs CE, et al: The
characterization of twenty sequenced human genomes. PLoS Genet.
6:e10011112010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Robinson PN, Krawitz P and Mundlos S:
Strategies for exome and genome sequence data analysis in disease
gene discovery projects. Clin Genet. 80:127–132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Danecek P, Auton A, Abecasis G, Albers CA,
Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST,
et al 1000 Genomes Project Analysis Group: The variant call format
and VCF tools. Bioinformatics. 27:2156–2158. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schwarz JM, Rödelsperger C, Schuelke M and
Seelow D: MutationTaster evaluates disease-causing potential of
sequence alterations. Nat Methods. 7:575–576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu X, Jian X and Boerwinkle E: dbNSFP
v2.0: A database of human non-synonymous SNVs and their functional
predictions and annotations. Hum Mutat. 34:E2393–E2402. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tilley WD, Marcelli M, Wilson JD and
McPhaul MJ: Characterization and expression of a cDNA encoding the
human androgen receptor. Proc Natl Acad Sci USA. 86:327–331. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shoemaker JD and Elliott WH: Automated
screening of urine samples for carbohydrates, organic and amino
acids after treatment with urease. J Chromatogr A. 562:125–138.
1991. View Article : Google Scholar
|
|
11
|
Goldstein JL and Brown MS: Regulation of
the mevalonate pathway. Nature. 343:425–430. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Haas D and Hoffmann GF: Mevalonate kinase
deficiencies: From mevalonic aciduria to hyperimmunoglobulinemia D
syndrome. Orphanet J Rare Dis. 1:132006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bedoyan JK, Schaibley VM, Peng W, Bai Y,
Mondal K, Shetty AC, Durham M, Micucci JA, Dhiraaj A, Skidmore JM,
et al: Disruption of RAB40AL function leads to Martin-Probst
syndrome, a rare X-linked multisystem neurodevelopmental human
disorder. J Med Genet. 49:332–340. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee J, Wong S and Boles RG: Mutation in
the X-linked RAB40AL gene (Martin-Probst syndrome) with mental
retardation, sensorineural hearing loss, and anomalies of the
craniofacies and genitourinary tract: A second case report. Eur J
Pediatr. 173:967–969. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mandey SH, Schneiders MS, Koster J and
Waterham HR: Mutational spectrum and genotype-phenotype
correlations in mevalonate kinase deficiency. Hum Mutat.
27:796–802. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Marcuzzi A, Pontillo A, De Leo L,
Tommasini A, Decorti G, Not T and Ventura A: Natural isoprenoids
are able to reduce inflammation in a mouse model of mevalonate
kinase deficiency. Pediatr Res. 64:177–182. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ołdak M, Ścieżyńska A, Młynarski W,
Borowiec M, Ruszkowska E, Szulborski K, Pollak A, Kosińska J,
Mueller-Malesińska M, Stawiński P, et al: Evidence against RAB40AL
being the locus for Martin-Probst X-linked deafness-intellectual
disability syndrome. Hum Mutat. 35:1171–1174. 2014. View Article : Google Scholar
|
|
18
|
Kalscheuer V, Iqbal Z, Hu H, et al:
RAB40AL loss-of-function mutation does not cause X-linked
intellectual disability. J Med Genet: Reply to reference.
1:2013.
|
|
19
|
Allen RC, Zoghbi HY, Moseley AB,
Rosenblatt HM and Belmont JW: Methylation of HpaII and HhaI sites
near the polymorphic CAG repeat in the human androgen-receptor gene
correlates with X chromosome inactivation. Am J Hum Genet.
51:1229–1239. 1992.PubMed/NCBI
|
|
20
|
Wolf SF, Jolly DJ, Lunnen KD, Friedmann T
and Migeon BR: Methylation of the hypoxanthine
phosphoribosyltrans-ferase locus on the human X chromosome:
Implications for X-chromosome inactivation. Proc Natl Acad Sci USA.
81:2806–2810. 1984. View Article : Google Scholar
|