Introduction
Tuberculosis (TB) is one of the most
life-threatening infectious diseases worldwide, second only to
human immunodeficiency virus. An estimated 2 billion individuals
are infected with the bacteria that cause TB, and each year 8
million individuals are diagnosed with the disease (1). Despite therapy, the mortality rate of
TB between ~2 and 3 billion annually (1). Adding to the considerable burden of
TB-associated morbidity and mortality rates are drug-resistant
strains of the disease. Multidrug-resistant (MDR) TB (MDR MTB) is a
strain of Mycobacterium tuberculosis, which expresses in
vitro resistance to at least rifampicin and isoniazid, which
are two of the most powerful antituberculosis agents available.
Resistance to these drugs results in longer, more complicated and
costly treatment for TB (1).
Developing countries account for 95% of all TB cases and 98% of all
TB-associated mortality worldwide (2). Of these, >12% are diagnosed with
MDR MTB (1).
The distinctive features of Mycobacterium
tuberculosis, including slow growth rate, dormancy, unique cell
wall composition and resistance towards phagocytosis by
macrophages, require detailed investigation at the molecular level
(3). A number of previous studies
have revealed significant differences in the transcription process
of mycobacteria, compared with Escherichia coli and other
bacteria (4,5).
MicroRNAs (miRNAs) are a subset of non-coding RNAs,
~22 nucleotide (nt) long, which post-transcriptionally regulate
gene expression by base-pairing with target mRNAs. miRNAs are
transcribed as pri-miRNAs in the nucleus and are then processed
into pre-miRNAs. Following translocation to the cytoplasm, a mature
22 nt duplex is formed. One miRNA strand is then incorporated into
the RNA-induced silencing complex, and interacts with its target
mRNA via base-pairing at binding sites, usually located within 3′
untranslated regions, whereas the other strand is usually degraded
(6). Depending on the level of
miRNA-mRNA complementarity, the target mRNA is either degraded or
its translation is repressed (7).
miRNAs constitute an evolutionarily conserved
system, which is associated with the regulation of biological
functions at the post-transcriptional level. The capability of
organisms to rapidly adapt their metabolism is essential for
survival, and miRNAs are used by cells to rapidly transfer and
internalize an external signal (8). Furthermore, in addition to affecting
translation, miRNAs can affect epigenetic processes. miRNAs have
been identified in almost all kingdoms of life, including archaea,
humans and plants (8). However,
the way in which miRNA regulates the expression of mRNA at the
translational level in TB remains to be elucidated. The present
study analyzed differences between the miRNA expression profiles of
MDR MTB and drug-sensitive TB, in order to identify novel mRNA
transcripts associated with drug resistance in TB.
Materials and methods
Mycobacterium tuberculosis strains
Mycobacterium tuberculosis strains were
isolated from lung tissue specimens of four patients, hospitalized
with TB, diagnosed at the Beijing Ditan Hospital (Beijing, China),
between April 2010 and October 2012. Lung tissue was obtained by
thoracoscopic lung biopsy. TB was diagnosed based on the Chinese
Pulmonary Tuberculosis Diagnostic Criteria (WS288-2008) and the
Chinese TB Volume of Clinical Diagnosis and Treatment Guidelines
(9).
The present study was approved by the Ethics
Committee of Beijing Ditan Hospital, Capital Medical University
(Beijing, China), according to the Declaration of Helsinki
(10). Written informed consent
was obtained from all of the patients.
Culture and identification of
Mycobacterium tuberculosis
Sputum/bronchoalveolar lavage fluid, pleural fluid
and tissue samples were collected from the patients with TB for
myco-bacterial assessment, by culturing the bacteria with either
Lowenstein-Jensen (L-J) culture medium (Roche Molecular Systems,
Inc., Branchburg, NJ, USA) or in a BACTEC 960 system (Roche
Molecular Systems, Inc.). Blood and sputum samples, were initially
cultured using the BACTEC 9120 Blood Culture system (Roche
Molecular Systems, Inc.), and the positive samples were then
cultured in L-J media at 37°C for 30 days. The Mycobacterium
strains were identified using multi-locus polymerase chain reaction
(PCR; Roche Molecular Systems, Inc.) (11,12).
The total volume of the PCR reaction mixture was 25 µl (13
µl 2X PCR mix; 1 µl forward primer; 1 µl
reverse primer; 1 µl DNA template; 9 µl DNase free
water). The PCR amplification procedure was as follows:
Pre-degeneration at 94°C for 5 min; 35 cycles of degeneration at
94°C for 1 min, annealing at 60°C for 1 min and extension at 72°C
for 1 min; followed by a final extension step at 72°C for 10 min.
The Mastercycler Nexus (Eppendorf, Hamburg, Germany) was used to
conduct PCR. The sequences of the primers (synthesized by
Invitrogen Life Technologies, Carlsbad, CA, USA) were as follows
(lower case, plasmid; upper case, TB): Forward (F)1,
tgtaaaacgacggccagtCGGATMACCGCTTTCGCCG, reverse (R)1,
caggaaacagctatgaccGACATGTGTGAGCTGTTTGC; F2,
tgtaaaacgacggccagtGAAGGCGGTATTCAAGC, R2,
caggaaacagctatgaccGAGTCACCCTCCACAATGTA; F3,
tgtaaaacgacggccagtGAAACCATTTCAACGGGTTC, R3,
caggaaacagctatgaccCCATTGTAGCTGTACCAAGCACCC; F4,
tgtaaaacgacggccagtTGGCCATAACGACATTCTG, R4,
caggaaacagctatgaccGAGCACCAACGTGTTTAGC; F5,
tgtaaaacgacggccagtACGGCTACGCAAAAGAAATG; R5,
caggaaacagctatgaccTTGAGGCTGAGCCGATACTT; F6,
tgtaaaacgacggccagtAGCAACCGGTAAAATTGTCG, R6,
caggaaacagctatgaccCAGTGTAAGAACCGGCACAA; and F7,
tgtaaaacgacggccagtTGTACGAAATTGCCACCAAA, and R7,
caggaaacagctatgaccAATATTTTCGCCGCATCAAC. Drug sensitivity testing
(DST) of the Mycobacterium tuberculosis strains was
performed using the proportion method with four first-line
anti-tuberculosis drugs: Isoniazid, rifampicin, streptomycin and
ethambutol. Briefly, the tested bacteria liquid (20 mg/l) was
prepared, and then 0.01 ml bacteria liquid was inoculated in L-J
medium, with was 0.1 µg and 0.001 µg of each drug,
respectively. After inoculation in 37°C culture medium for 4 weeks,
the colony numbers were counted. If the percentage of colony
numbers compared to control is ≤1% the strain is considered
sensitive, and if it is >1% the strain is considered
resistant.
RNA extraction
RNA was extracted from the Mycobacterium
tuberculosis using the RNA isolation reagent TRIzol®
(Invitrogen Life Technologies). Initially, 30 mg cultured
Mycobacterium tuberculosis was added to a
methanol/chloroform (Sigma-Aldrich, St. Louis, MO, USA) suspension
(1:3). The suspension was then agitated with 5 ml
TRIzol®, centrifuged at 1,000 × g for 15 min at 4°C, and
the colorless upper phase was collected. An equivalent quantity of
isopropanol (0.5 ml) was mixed with this upper phase, centrifuged
and the collected sediment at the bottom of the tube was mixed with
ddH2O. To prevent DNA contamination, the total RNA was
treated with 20 µl RNase-free DNase II (Invitrogen Life
Technologies).
Next-generation sequencing (NGS)
Small RNA fractions, with a length ≤50 nt, were
subjected to hybridization and ligation using Adaptor mix (Agilent
Technologies, Santa Clara, CA, USA). Subsequently, the RNA samples
were reverse transcribed and sequenced using miRNA sequencing on an
Illumina HiSeq 2000 platform (Illumina, San Diego, CA, USA).
The total RNA was isolated from each sample using
TRIzol®, and the degradation and contamination of RNA
was assessed using agarose gel (Abcam, Cambridge, MA, USA)
electrophoresis. The RNA was purified using a KingFisher™ Pure RNA
Tissue kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA), and
the ratio of the absorbance at 260 and 280 nm (A260/280) was
determined to confirm purity. The RNA integrity was assessed using
an RNA Nano 6000 Assay kit on a Bioanalyzer 2100 system (Agilent
Technologies). A total of 3 g RNA per sample was used as input
material for the RNA sample preparations, and all samples had RNA
integrity number values >8. The samples of three individuals
were then pooled within each group in equal quantities to generate
two mixed samples. The pooled samples were then used to prepare six
separate Illumina sequencing libraries, containing three technical
replicates for each condition. cDNA libraries were generated using
an Illumina TruSeq™ RNA Sample Preparation kit (Illumina),
according to the manufacturer's instructions, and index codes were
added to attribute sequences to each sample. Briefly, the mRNA was
purified from the total RNA using poly-T oligo-attached magnetic
beads (Thermo Fisher Scientific, Inc., Rockford, IL, USA).
Fragmentation was performed using divalent cations under elevated
temperature (55°C) in Illumina proprietary fragmentation buffer
(Illumina). First strand cDNA was synthesized using random
oligonucleotides and SuperScript II (Abcam). Second strand cDNA
synthesis was subsequently performed using DNA Polymerase I and
RNase H (Abcam). The remaining overhangs were converted into blunt
ends via exonuclease/polymerase activities and the enzymes were
removed. Following adenylation of the 3′ ends of the DNA fragments,
the Illumina PE adapter oligo-nucleotides were ligated to prepare
for hybridization. In order to select cDNA fragments of 200 bp in
length, the library fragments were purified using an AMPure XP
system (Beckman Coulter Genomics, Danvers, MA, USA). DNA fragments
with ligated adaptor molecules on each end were selectively
enriched using Illumina PCR Primer Cocktail (Illumina) in a 10
cycle PCR reaction. Primersequences were as follows: Forward:
5′-AATGATACGGCGACCACCGAGA-3′ and reverse:
5′-CAAGCAGAAGACGGCATACGAGT-3′). Products were purified using AMPure
XP system (Beckman Coulter, Beverly, MA, USA) and quantified using
the Agilent high sensitivity DNA assay on the Agilent Bioanalyzer
2100 system (Agilent Technologies). Prior to sequencing, all the
individual libraries were normalized and pooled together in a
single lane on an Illumina HiSeq 2000 platform, and 0–100 bp
paired-end reads were generated.
Data extraction and analysis
To detect standard Mycobacterium tuberculosis
strains in PubMed, particularly CDC1551 (http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=83331),
a library of reference sequences was prepared by identifying the
sequences of mature miRNAs, together with five flanking
nucleotides, within the hairpins deposited in miRBase version 19
(http://www.mirbase.org/) (13). The Illumina HiSeq platform requires
a minimum read length of 50 nt, therefore, all small RNAs were
extended using specific adapters (forward,
5′-AATGATACGGCGACCACCGAGA-3′; reverse,
5′-CAAGCAGAAGACGGCATACGAGT-3′), which were annealed to their 3′
ends during library preparation. Removal of the adapters was
performed in silico on the raw, 50 nt Illumina sequence
reads, using Cutadapt software (14). The resulting 15–30 nt long
sequences were subjected to further analysis as potential miRNAs.
The sequences were mapped on the prepared reference library using
Bowtie version 0.12.7 (15), with
the requirement of perfect matching. The numbers of mapped reads
were subsequently calculated for each miRNA and provided as a
number of each of the unique reads mapped to each reference
sequence, and as a number of all the reads mapped to each reference
sequence. Data obtained for each sample was normalized using reads
per million (RPM) normalization, according to the following
formula: RPM = (Nref/Nall) × 106.
Nref indicates the number of reads mapped to the miRNA
reference, and Nall indicates the total number of reads
mapped in the sample (16).
Statistical analysis
Selection of miRNAs and isomiRs deregulated between
the analyzed groups was performed using a Welch t-test, paired for
comparison between sensitive MTB and MDR MTB samples. False
discovery rate was used to assess multiple testing errors.
Statistical analyses were conducted using SPSS version 13.0 (SPSS
Inc., Chicago, IL, USA) Hierarchical clustering of samples, based
on the expression profiles of the selected miRNAs, was performed
using Ward's agglomeration method, operated on Euclidean distance
measures. Identification of target genes for each miRNA seed
sequence, with significantly deregulated expression between the
isoniazid-sensitive MTB and MDR MTB samples was performed using
Target Rank version 3.2 software (16,17).
Results
Capacity and quality of sequencing
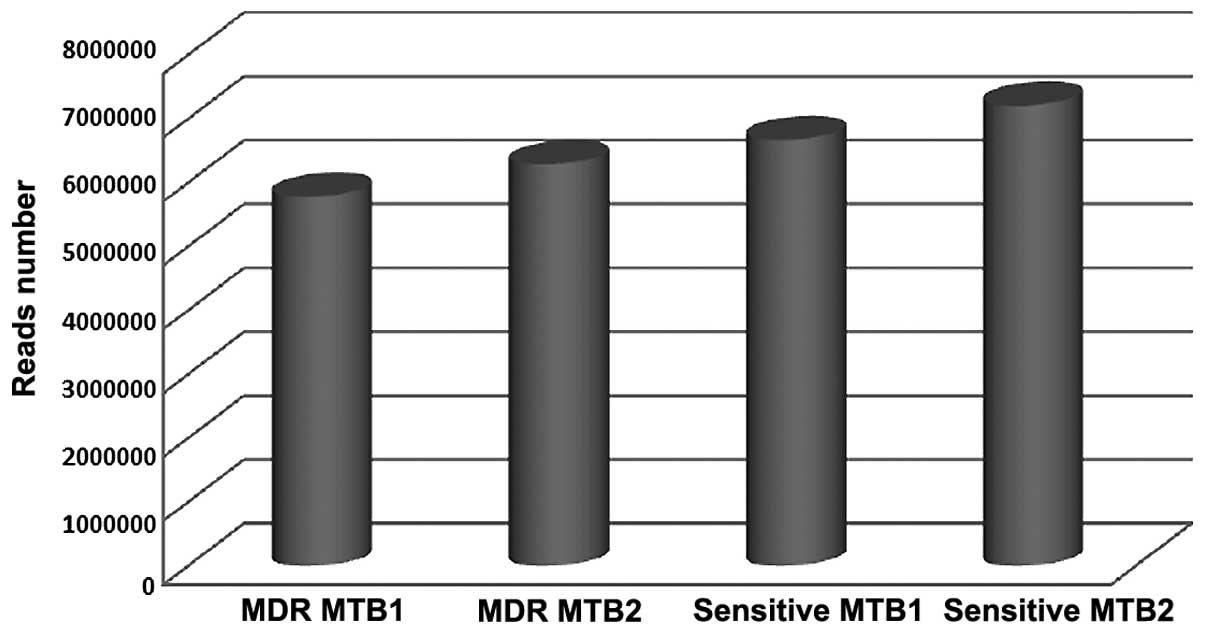
A total of 5, 785 and 195, and 6, 290 and 595
qualified Illumina reads were obtained from the two MDR MTB
strains; and 6, 673 and 665, and 7, 210 and 217 qualified Illumina
reads were obtained from the two sensitive MTB strains,
respectively (Table I and Fig. 1). According to the statistical
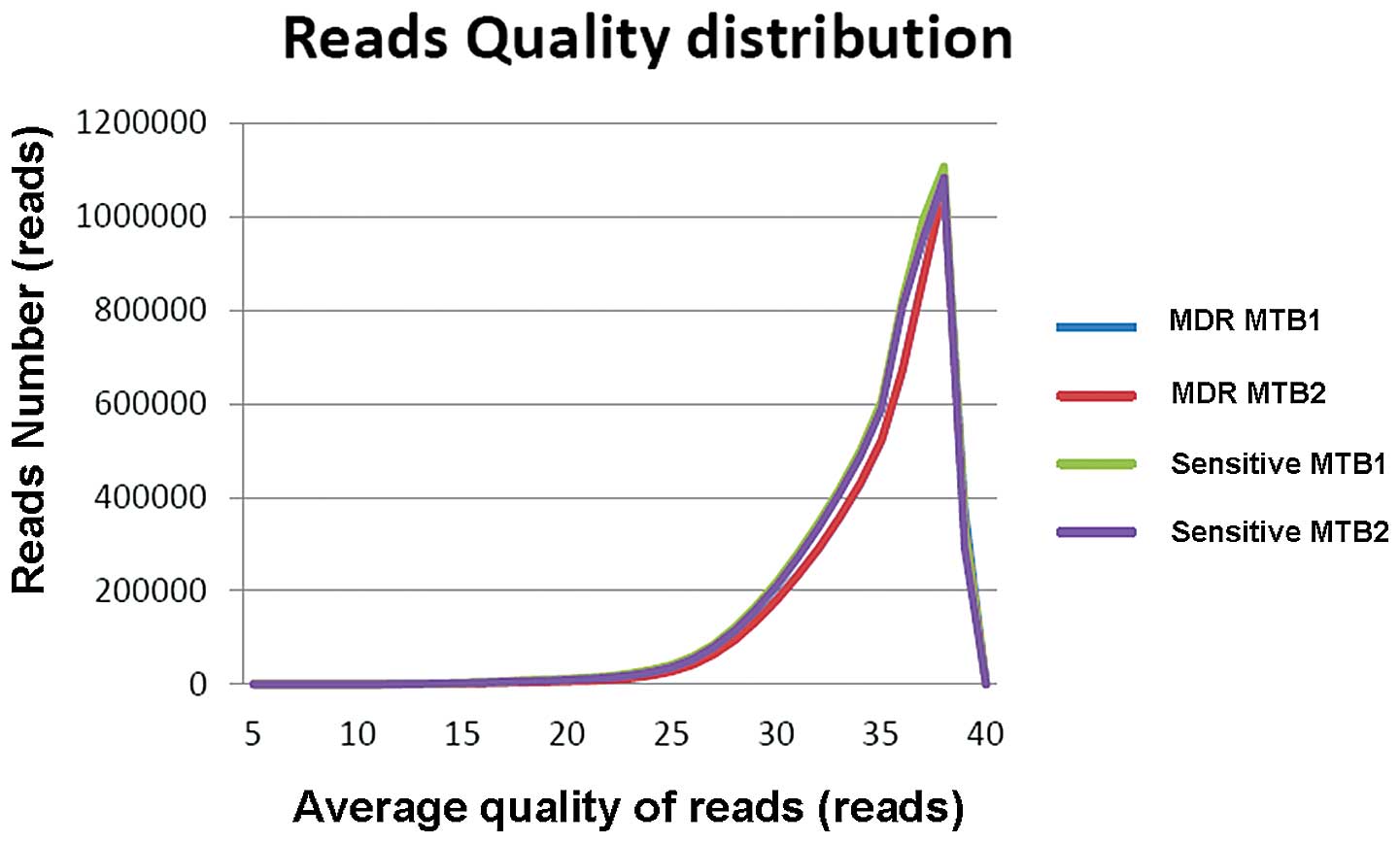
results, the average quality of >99% of the reads was >20 in
each sample, indicating the quality of the sequencing was suitable
(Table II and Fig. 2).
 | Table ICapacities of Illumina sequencing of
MTB strains. |
Table I
Capacities of Illumina sequencing of
MTB strains.
| Strain | Number of reads |
|---|
| MDR MTB1 | 5, 785, 195 |
| MDR MTB2 | 6, 290, 595 |
| Sensitive MTB1 | 6, 673, 665 |
| Sensitive MTB2 | 7, 210, 217 |
| Table IIQuality of Illumina sequencing of MTB
strains. |
Table II
Quality of Illumina sequencing of MTB
strains.
| Strain | Q20 (%) | Q30 (%) |
|---|
| MDR MTB1 | 99.56 | 91.50 |
| MDR MTB 2 | 99.65 | 91.93 |
| Sensitive MTB1 | 99.32 | 90.11 |
| Sensitive MTB2 | 99.45 | 90.62 |
Data pre-processing of NGS
The adapter sequence (3′ adapter:
AGATCGGAAGAGCACACGTCT) was filtered from the NGS raw data (Fig. 3) using the fastx_cliper program.
The low quality reads were removed using the fastq_quality_filter
program (http://seqanswers.com/forums/showthread.php?goto=nextoldest&t=24679),
to ensure the quality score of at least 95% of the bases was
>20. Clustering was then performed and identical base sequences
were recorded as one tag. The length of the mature miRNA sequences
was between 18 and 30 nt, which were further analyzed (Table III). The tags are shown in
Fig. 4.
| Table IIIData pre-processing of next
generation sequencing. |
Table III
Data pre-processing of next
generation sequencing.
| Strain | Trim 3′ adapter n
(%) | Filter low quality
n (%) | Collapse tags
(n) | 18–30 nt reads n
(%) | 18–30 nt tags
(n) |
|---|
| MDR MTB1 | 5, 445, 431
(94.13) | 4, 665, 152
(80.64) | 2, 494, 091 | 2, 583, 186
(44.65) | 1, 438, 171 |
| MDR MTB2 | 5, 378, 966
(85.51) | 4, 661, 476
(74.10) | 2, 497, 989 | 2, 829, 821
(44.98) | 1, 552, 250 |
| Sensitive MTB1 | 6, 231, 203
(93.37) | 5, 263, 218
(78.87) | 1, 880, 645 | 2, 541, 061
(38.08) | 1, 056, 595 |
| Sensitive MTB2 | 5, 992, 842
(83.12) | 5, 120, 235
(71.01) | 1, 858, 932 | 2, 766, 439
(38.37) | 1, 125, 144 |
Analysis of small RNA
The tags were mapped to the genome sequence of the
Mycobacterium tuberculosis strain CDC1551 in PubMed, using
match software Bowtie version 0.12.7. The genome-mapped tags and
reads, which were calculated based on 18–30 nt reads or tags, are
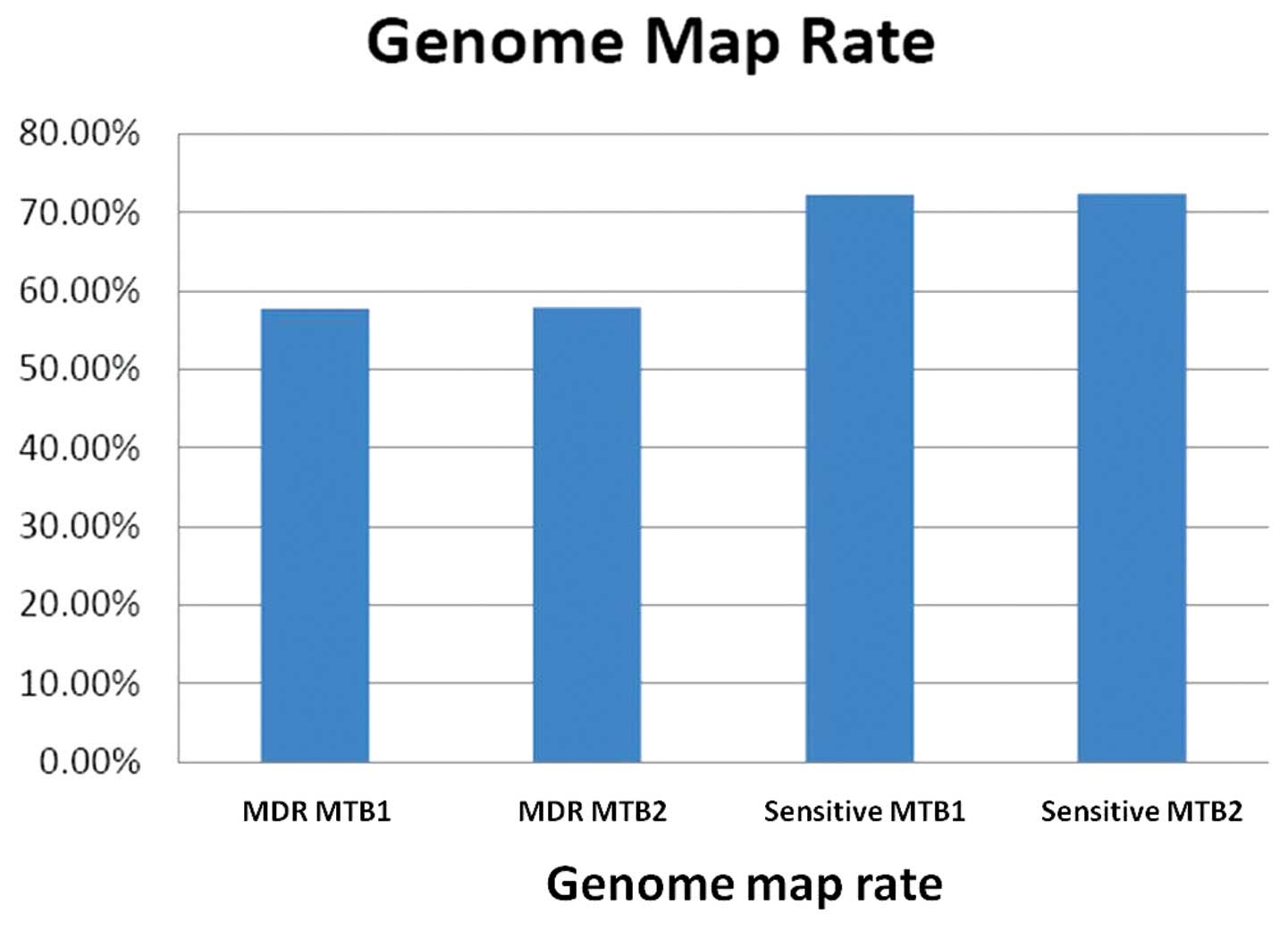
shown in Table IV. The genome map
rates of the samples are shown in Fig.
5. The RNA family database, Rfam (version 11.0; http://rfam.xfam.org/) was used to analyze the variety
of RNAs in the samples. The Rfam reads and tags are shown in
Tables V and VI.
| Table IVGenome mapped tags and reads. |
Table IV
Genome mapped tags and reads.
| Strain | Genome-mapped tags
[n (%)] | Genome-mapped reads
[n (%)] |
|---|
| MDR MTB1 | 517, 409
(35.98) | 1, 490,704
(57.71) |
| MDR MTB2 | 554, 600
(35.73) | 1, 637, 876
(57.88) |
| Sensitive MTB1 | 557, 934
(52.80) | 1, 836, 536
(72.27) |
| Sensitive MTB2 | 590, 434
(52.48) | 2, 002, 245
(72.38) |
| Table VReads from Rfam analysis. |
Table V
Reads from Rfam analysis.
| Data | MDR MTB1 | MDR MTB2 | Sensitive MTB1 | Sensitive MTB2 |
|---|
| rRNA | 161, 738 | 176, 797 | 208, 101 | 226, 316 |
| Other RNA | 25, 089 | 27, 441 | 26, 246 | 28, 405 |
| tRNA | 23, 992 | 26, 474 | 80, 832 | 88, 883 |
| lncRNA | 98 | 83 | 136 | 130 |
| sRNA | 632 | 680 | 2, 411 | 2, 643 |
| snRNA | 309 | 292 | 367 | 417 |
| Genes | 710, 982 | 779, 884 | 932, 552 | 1, 016, 764 |
| Total reads | 922, 840 | 1, 011, 651 | 1, 250, 645 | 1, 363, 588 |
| Percentage | 61.91 | 61.77 | 68.10 | 68.10 |
| Table VITags from Rfam analysis. |
Table VI
Tags from Rfam analysis.
| Data | MDR MTB1 | MDR MTB 2 | Sensitive MTB1 | Sensitive MTB2 |
|---|
| rRNA | 10, 425 | 10, 708 | 10, 270 | 10, 387 |
| Other RNA | 3, 965 | 4, 127 | 4, 259 | 4, 425 |
| tRNA | 1, 963 | 2, 050 | 2, 310 | 2, 434 |
| lncRNA | 55 | 44 | 61 | 66 |
| sRNA | 264 | 256 | 365 | 370 |
| snRNA | 228 | 216 | 223 | 257 |
| Genes | 410, 107 | 440, 968 | 450, 926 | 478, 639 |
| Total reads | 427, 007 | 458, 369 | 468, 414 | 496, 578 |
| Percentage | 82.53 | 82.65 | 83.96 | 84.10 |
Analysis of miRNAs
Mirdeep (version 2; http://www.mdc-berlin.de/8551903/en/research/research_teams/systems_biology_of_gene_regulatory_elements/projects/miRDeep)
was used to predict the miRNAs in the samples. The data are shown
in Table VII. The target genes
of miRNAs were predicted using Miranda software (http://www.miranda-im.org/; score >150; energy
<−15).
| Table VIImiRNAs from Mirdeep analysis. |
Table VII
miRNAs from Mirdeep analysis.
| Data | MDR MTB1 | MDR MTB 2 | Sensitive MTB1 | Sensitive MTB2 |
|---|
| miRNA (n) | 62 | 62 | 95 | 112 |
| miRNA reads
(n) | 33, 051 | 36, 243 | 75, 015 | 81, 954 |
| Percentage | 2.22 | 2.21 | 4.08 | 4.09 |
| miRNA tags (n) | 2, 606 | 2, 756 | 3, 627 | 4, 024 |
| Percentage | 0.50 | 0.50 | 0.65 | 80.68 |
Discussion
There are increasing cases of MDR MTB, which do not
respond to the existing first-line anti-TB drugs, including
rifampicin, and isoniazid. In addition, extremely drug resistant
(XDR) TB strains do not respond to even the most effective
second-line anti-TB drugs (18–23).
Therefore, there is an urgent requirement to identify the drug
resistance mechanisms of MTB, and develop anti-TB drugs, which are
effective against MDR and XDR TB strains. Bacterial RNA polymerase
(RNAP) is the target for the anti-TB drug, rifampicin (24–26).
Therefore, RNAP is considered an attractive target for the
development of novel anti-TB drugs (27–30).
To identify the drug resistance mechanisms of MTB, and screen
anti-TB agents, a high-throughput in vitro transcription and
transcript regulation assay is required.
The demand for low-cost sequencing has driven the
development of high-throughput sequencing, also termed NGS;
however, NGS is only modestly accurate at absolute quantification.
Thousands or millions of sequences are concurrently produced in the
NGS process. Genome-wide computational analysis is increasingly
being used for novel discoveries in biomedical research. However,
as the quantities of sequence data increase exponentially, an
analysis bottle-neck remains (31).
The present study performed a miRNA sequence
analysis of sensitive MTB and MDR MTB using NGS. A total of 5, 785
and 195, and 6, 290 and 595 qualified Illumina reads were obtained
from two MDR MTB strains, and 6, 673 and 665, and 7, 210 and 217
qualified Illumina reads were obtained from two sensitive MTB
strains. The quality of the data was considered to be high, with
the average quality of >99% of reads being >20. Data
pre-processing of NGS was performed using the fastx_cliper program,
and low quality reads were removed using the fastq_quality_filter
program. Clustering was performed, and the mature miRNA sequences
between 18 and 30 nt, were mapped to the genome sequence of the
Mycobacterium tuberculosis strains. The target genes of the
miRNAs were then predicted using Miranda software (score >150;
energy <−15).
Rfam is a collection of multiple sequence alignments
and covariance models, representing non-coding RNA families. Rfam
is available online (http://rfam.sanger.ac.uk/; http://rfam.janelia.org/), and allows users to search
a query sequence against a library of covariance models, and view
multiple sequence alignments and family annotation. The database
can also be downloaded in flatfile form and searched locally, using
the INFERNAL package (http://infernal.wustl.edu/). The first analysis by
Rfam contains 25 families, which annotate >50,000 non-coding RNA
genes in the taxonomic divisions of the EMBL nucleotide database
(http://www.ebi.ac.uk/embl).
The overall de novo assembly of miRNA
sequence data generated 62 and 62, and 95 and 112 miRNAs with a
length of 18–30 bp from the MDR MTB and sensitive MTB strains,
respectively. Comparative miRNA analysis identified 142
differentially expressed miRNAs in the MDR MTB strain, compared
with the sensitive MTB strain, and included 48 upregulated and 94
downregulated genes. There were 108 miRNAs expressed only in MDR
MTB.
miRNAs are short, non-coding RNAs, which bind to
complementary sequences in the 3′ untranslated regions of
protein-coding genes and regulate their expression (32,33).
Aberrant expression of miRNAs results in aberrant expression of
their target mRNAs. A single miRNA regulates the expression of
numerous genes (34), and several
miRNAs may concurrently regulate the expression of a single gene,
the function of which is pivotal in a given tissue (35). The recognition of mRNA by miRNA
depends on the 'seed region' of a miRNA, comprising 2–8 nt of a
mature molecule (17).
The majority of previous studies on miRNA
aberrations were based on the analysis of the expression of
canonical, reference miRNAs, as the analysis of isomiRs requires
the use of more robust technologies, including NGS, and extensive
analysis of the obtained data. Therefore, the present study used
NGS to identify all the miRNA isoforms, which may be expressed in
sensitive MTB or MDR MTB, the aberrances of which potentially
underlie the initiation and progression of drug resistance. The aim
of the present study was to identify novel, previously unknown
isomiRs, and the results revealed the expression profiles of
canonical miRNAs and their newly identified isoforms.
In conclusion, the present study demonstrated that
miRNAs may serve as an invaluable resource for revealing the
molecular basis of the regulation of expression associated with the
mechanism of drug resistance in MTB. The differential expression of
miRNAs between sensitive and MDR MTB was identified by NGS, and
identified miRNAs that may be associated with the drug resistance
of TB. However, the mechanism by which miRNA regulates the
expression of mRNA at the transla tional level in TB remains to be
elucidated. The present study analyzed the differences between the
miRNA expression profiles of MDR MTB and drug-sensitive TB, in
order to identify novel mRNA transcripts associated with drug
resistance in TB.
Acknowledgments
The present study was funded by the projects of the
National Key Program of Mega Infectious Disease (grant nos.
2012ZX10005010-001, 2012ZX10005010-003 and 2013ZX10003002-001). The
funding bodies had no role in the study design, data collection and
analysis, manuscript preparation or decision to publish.
References
|
1
|
World Health Organization (WHO): Global
Tuberculosis Control Report 2010. WHO; Geneva: 2010
|
|
2
|
US Agency for International Development:
Report to congress: Health-related research and development
activities at USAID-an update on the five-year strategy, 2006–2010.
US Agency for International Development. 182:1788–1790. 2009.
|
|
3
|
Tare P, China A and Nagaraja V: Distinct
and contrasting transcription initiation patterns at Mycobacterium
tuberculosis promoters. PLoS One. 7:e439002012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jia Y and Patel SS: Kinetic mechanism of
transcription initiation by bacteriophage T7 RNA polymerase.
Biochemistry. 36:4223–4232. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Martínez-Antonio A, Salgado H, Gama-Castro
S, et al: Environmental conditions and transcriptional regulation
in Escherichia coli: A physiological integrative approach.
Biotechnol Bioeng. 84:743–749. 2003. View Article : Google Scholar
|
|
6
|
Holley CL and Topkara VK: An introduction
to small non-coding RNAs: miRNA and snoRNA. Cardiovas Drugs Ther.
25:151–159. 2011. View Article : Google Scholar
|
|
7
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Baroni D and Arrigo P: MicroRNA target and
gene validation in viruses and bacteria. Methods Mol Biol.
1107:223–231. 2014. View Article : Google Scholar
|
|
9
|
Chinese Medical Association: TB volume of
clinical diagnosis and treatment guidelines. People's Medical
Publishing House; Beijing: 2005
|
|
10
|
World Medical Association: World Medical
Association Declaration of Helsinki: Ethical principles for medical
research involving human subjects. JAMA. 310:2191–2194. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huard RC, Lazzarini LC, Butler WR, van
Soolingen D and Ho JL: PCR-based method to differentiate the
subspecies of the Mycobacterium tuberculosis complex on the basis
of genomic deletions. J Clin Microbiol. 41:1637–1650. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bao X, Lian L, Xu D, et al: Rapid species
identification of 391 clinical Mycobacterium isolates from Anhui
province by multi-locus PCR. Chinese Journal of Zoonoses.
28:659–663. 2012.
|
|
13
|
Kozomara A and Griffiths-Jones S: miRBase:
Integrating microRNA annotation and deep-sequencing data. Nucleic
Acids Res. 39:D152–D157. 2011. View Article : Google Scholar :
|
|
14
|
Martin M: Cutadapt removes adaptor
sequences from high-throughput sequencing reads. EMBnetjournal.
17:10–12. 2011.
|
|
15
|
Langmead B, Trapnell C, Pop M and Salzberg
SL: Ultrafast and memory-efficient alignment of short DNA sequences
to the human genome. Genome Biol. 10:R252009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wojcicka A, Swierniak M, Kornasiewicz O,
et al: Next generation sequencing reveals microRNA isoforms in
liver cirrhosis and hepatocellular carcinoma. Int J Biochem Cell
Biol. 53:208–217. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nielsen CB, Shomron N, Sandberg R,
Hornstein E, Kitzman J and Burge CB: Determinants of targeting by
endogenous and exogenous microRNAs and siRNAs. RNA. 13:1894–1910.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
World Health Organization (WHO): Global
tuberculosis report, 2012. WHO; Geneva: 2012
|
|
19
|
Dye C, Espinal MA, Watt CJ, Mbiaga C and
Williams BG: Worldwide incidence of multidrug-resistant
tuberculosis. J Infect Dis. 185:1197–1202. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Raviglione MC, Gupta R, Dye CM and Espinal
MA: The burden of drug-resistant tuberculosis and mechanisms for
its control. Ann NY Acad Sci. 953:88–97. 2001. View Article : Google Scholar
|
|
21
|
Zumia A and Grange JM: Multidrug-resistant
tuberculosis - can the tide be turned? Lancet Infect Dis.
1:199–202. 2001. View Article : Google Scholar
|
|
22
|
World Health Organization (WHO):
Anti-tuberculosis drug resistance in the world: The WHO/IUATLD
global project on anti-tuberculosis drug resistance suveillance:
Third global report. WHO; Geneva: 2003
|
|
23
|
Banerjee R, Rudra P, Prajapati RK,
Sengupta S and Mukhopadhyay J: Optimization of recombinant
Mycobacterium tuberculosis RNA polymerase expression and
purification. Tuberculosis (Edinb). 94:397–404. 2014. View Article : Google Scholar
|
|
24
|
Artsimovitch I, Vassylyeva MN, Svetlov D,
et al: Allosteric modulation of the RNA polymerase catalytic
reaction is an essential component of transcription control by
rifamycins. Cell. 122:351–363. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Campbell EA, Korzheva N, Mustaev A, et al:
Structural mechanism for rifampicin inhibition of bacterial RNA
polymerase. Cell. 104:901–912. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Feklistov A, Mekler V, Jiang Q, et al:
Rifamycins do not function by allosteric modulation of binding of
Mg2+ to the RNA polymerase active center. Proc Natl Acad
Sci USA. 105:14820–14825. 2008. View Article : Google Scholar
|
|
27
|
Chopra I: Bacterial RNA polymerase: A
promising target for the discovery of new antimicrobial agents.
Curr Opin Investig Drugs. 8:600–607. 2007.PubMed/NCBI
|
|
28
|
Chopra I, Hesse L and O'Neill AJ:
Exploiting current understanding of antibiotic action for discovery
of new drugs. Symp Ser Soc Appl Microbiol. (31): 4S–15S. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Darst SA: New inhibitors targeting
bacterial RNA polymerase. Trends Biochem Sci. 29:159–160. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Villain-Guillot P, Bastide L, Gualtieri M
and Leonetti JP: Progress in targeting bacterial transcription.
Drug Discov Today. 12:200–208. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang J, Chiodini R, Badr A and Zhang G:
The impact of next-generation sequencing on genomics. J Genet
Genomics. 38:95–109. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Filipowicz W, Bhattacharyya SN and
Sonenberg N: Mechanisms of post-transcriptional regulation by
microRNAs: Are the answers in sight? Nat Rev Genet. 9:102–114.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lim LP, Lau NC, Garrett-Engele P, et al:
Microarray analysis shows that some microRNAs downregulate large
numbers of target mRNAs. Nature. 433:769–773. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jazdzewski K, Boguslawska J, Jendrzejewski
J, et al: Thyroid hormone receptor beta (THRB) is a major target
gene for microRNAs deregulated in papillary thyroid carcinoma
(PTC). J Clin Endocrinol Metab. 96:E546–E553. 2011. View Article : Google Scholar
|