Introduction
Accumulating evidence has demonstrated that nerve
terminals are vulnerable to various stimuli, including traumatic,
toxic and disease-associated neurodegenerative factors (1). As one of these pathological stimuli,
ischemia/hypoxia has been extensively investigated in the central
nervous system (CNS), including stroke-induced brain damage
(2). However, few studies have
focused on the stimulation of ischemia/hypoxia on the peripheral
nervous system (PNS), although the PNS can be highly susceptible to
reductions in blood supply and oxygen levels during surgical and
pathological conditions (3,4) and
can cause neurological complications, ranging from mild functional
loss to complete limb paralysis and permanent functional deficits
(5). Hypoxia is considered to be
the key pathological stimulus in CNS ischemia (6) and also induces functional changes at
the neuromuscular junction (7,8). The
mechanisms underlying nerve terminal vulnerability to
hypoxia-reperfusion injury in situations, including trauma, remain
to be fully elucidated (9) and
require further investigation. The majority of current in
vivo animal models of hypoxia-reperfusion rely on the
experimental application of high-pressure tourniquets, which may
induce considerable mechanical stress and potential crush injury to
the underlying nerves, making it difficult to conclusively
distinguish the effects of ischemia-reperfusion injury from the
effects of mechanical trauma (10).
During ischemia/hypoxia-induced damage to the
CNS/PNS or other organs, there are two major processes, necrosis
and apoptosis, leading to neural cell death. In the core ischemic
area, the severe restriction of blood flow leads to necrotic cell
death. However, in the peripheral ischemic area, where collateral
blood flow can buffer the degree of damage, cell apoptosis
predominantly develops (11,12).
Evidence suggests that activation of apoptotic pathways occurs in
the peripheral cells of the ischemic area, in a caspase-dependent
and caspase-independent manner (13,14).
Therefore, the apoptotic cascades during the
ischemia/hypoxia-induced damage to CNS/PNS are reversible and are a
major target of therapeutic interventions (15,16).
A variety of transcription factors have been
reported to be involved in regulating the genes responsible for the
metabolic responses to hypoxia-ischemia (17,18).
A key component among these factors is hypoxia-inducible factor 1
(HIF-1), existing as a heterodimer which is composed of a
constitutively expressed HIF-1β subunit and an oxygen sensitive
HIF-1α subunit. Following activation by hypoxia, the HIF-1α/HIF-1β
dimmer (19) binds to a conserved
DNA consensus on the promoters of its target genes, known as the
hypoxia-responsive element (20,21),
and induces a group of gene products that are crucial for hypoxic
adaptation (22). An
anti-apoptotic and a pro-apoptotic effect have been confirmed for
HIF-1 and/or hypoxia in hypoxia-ischemia. Severe or prolonged
hypoxia induces apoptosis, at least in part, whereas activated
HIF-1α, along with other molecules, protects neural cells from
apoptosis (23). HIF-1α can
prevent apoptosis by activating the phosphoinositide 3-kinase
(PI3K)/Akt pathway (24) or by
promoting the expression of survivin (25) in acute hypoxia, or by increasing
the expression of glycolytic enzymes, p21 and erythropoietin to
antagonize the effect of the hypoxia- and/or hypoglycaemia-induced
expression of tumor suppressor gene p53 (26) or BNIP3 (27). However, other molecules involved in
the pro- or anti-apoptotic effects of HIF-1α in hypoxia-ischemia
damage in the CNS/PNS require investigation.
In the present study, in order to investigate the
role of HIF-1α and Ras homolog gene family, member A (RhoA) in
hypoxic-ischemic damage to the CNS/PNS, PC12 neuroblastoma cells
were examined following hypoxia treatment. The expression levels of
HIF-1α and RhoA, which belongs to the Rho small GTPase family and
is reported to be implicated in hypoxia adaptation were assessed.
Subsequently, gain-of-function and loss-of-function strategies were
adopted to manipulate the expression levels of HIF-1α or RhoA in
PC12 cells, and the expression of hypoxia-induced RhoA and cell
apoptosis were determined again.
Materials and methods
Reagents, cell culture and treatment
The PC12 rat pheochromocytoma cell line was provided
by the National Platform of Experimental Cell Resources for
Sci-Tech (Beijing, China) and were cultured in F12K medium
(Invitrogen Life Technologies, Carlsbad, CA, USA) containing 15%
fetal calf serum (Invitrogen Life Technologies) at 37ºC under 5%
CO2. F12K medium supplemented with 2% fetal bovine serum
was used for the cell maintenance. For hypoxia treatment, the cells
at 85% confluence were placed in a hypoxia incubator infused with a
gas mixture of 5% CO2 and nitrogen to obtain a 3% oxygen
concentration, at 37ºC for 0, 2, 4, 6, 12, 24 or 48 h. The oxygen
concentration was monitored continuously (Forma 3130; Thermo Fisher
Scientific, Rockford, IL, USA). To overexpress HIF-1α in the PC12
cells, the wild-type HIF-1α coding sequence was amplified with Pfu
DNA polymerase (Promega, Madison, WI, USA) and cloned into a
pcDNA3.1 (+) vector (Invitrogen life Technologies), with
HindIII and BamHI (New England Biolabs, Ipswich, MA,
USA) as restriction endonucleases. The HIF-1α-pcDNA3.1 (+) or
control CAT-pcDNA3.1 (+) plasmids were then transfected,
respectively, into PC12 cells using Lipofectamine 2000 (Invitrogen
Life Technologies), and 25 or 50 nM small interfering (si) RNA-RhoA
or siRNA-Control (Sangon Biotech, Co., Ltd., Shanghai, China) were
transfected into the cells using Lipofectamine 2000 to knockdown
the expression of RhoA.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total cellular RNA was isolated from the cells using
a PureLink® RNA Mini kit (Invitrogen Life Technologies),
according to the manufacturer's instructions. The mRNA expression
levels of HIF-1α and RhoA were quantified by RT-qPCR using a Takara
One Step RT-PCR kit (Takara Biotechnology, Co., Ltd., Dalian,
China) and paired primers as follows: HIF-1α, forward
5′-aaccataacaaaaccatcca-3′, and reverse 5′-tattgaagatgacatgaaag-3′;
RhoA, forward 5′-gtggcagatatcgaggtgga-3′ and reverse
5′-aatcttcctgcccagctgtg-3′ and β-actin, forward
5′-tgtccaccttccagcagatgt-3′ and reverse 5′-gtaacagtccgcctaga-3′
(Sangon Biotech, Co., Ltd.). Equal quantities of mRNA sample (1
µl) were used for quantitative analysis. The RT-qPCR was
performed as follows: 65ºC for 5 min, 42ºC for 40 min and 95ºC for
10 sec for the reverse transcription step then 95ºC for 5 sec and
60ºC for 10 sec (40 cycles) for the PCR reaction. The mRNA samples
were amplified using primer/probe sets specific for the genes of
interest on a Lightcycler 480 II (Roche Diagnostics, Mannheim,
Germany). Relative quantification was determined using the ΔΔCt
method (28), with tubulin as a
reference gene (28).
Western blot analysis
To determine protein expression levels,
5×105 PC12 cells were collected and lyzed using a
cytoplasmic protein extraction kit (TP-001; ZmTech Scientific,
Inc., San Jose, CA, USA) and supplemented with a protease inhibitor
cocktail (Roche Diagnostics) according to the manufacturer's
instructions. All protein samples were quantified using Bradford
protein assay reagent (Bio-Rad Laboratories, Inc., Hercules, CA,
USA) and each sample with 30 µg (5 µl) was separated
on a 8–12% gradient SDS-PAGE gel, followed by being transferred
onto a polyvinylidene difluoride membrane and blocked in 5% skimmed
milk. Rabbit polyclonal antibodies against caspase 3 (1:1,000; cat.
no. ab2302; Abcam, Cambridge, UK), Poly ADP ribose polymerase
(PARP; 1:500; cat. no. ab137653; Abcam), cytochrome c
(1:1,000; cat. no. 4272; Cell Signaling Technology Inc., Danvers,
MA, USA), HIF-1α (1:1,000; cat. no. sc-10790; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), HIF-1β (1:500; cat. no.
C15A11, Cell Signaling Technology Inc.), RhoA (1:1,000; cat. no.
sc-179, Santa Cruz Biotechnology, Inc.), Rho-associated kinase
(Rock) 1 (1:500; cat. no. sc-5560, Santa Cruz Biotechnology, Inc.),
Rock 2 (1:1,000; cat. no. sc-5561, Santa Cruz Biotechnology, Inc.),
RhoB (1:1,000; cat. no. sc-180, Santa Cruz Biotechnology, Inc.) and
β-actin (1:500; cat. no. sc-130656, Santa Cruz Biotechnology, Inc.)
were used to quantify the protein level of each molecule via an
incubation at 4ºC overnight. Goat anti-rabbit IgG conjugated to
horseradish peroxidase (1:500; cat. no. 31212, Pierce
Biotechnology, Inc., Rockford, IL, USA) and an enhanced
chemiluminescence detection system (Super Signal West Femto; Pierce
Biotechnology, Inc.) were used for detection and quantification.
Prior to each inoculation, samples were washed four times with 1X
phosphate-buffered saline with Tween-20.
Apoptosis and caspase 3 assays
The apoptosis of the PC12 cells was examined using
an Annexin V-FITC Apoptosis Detection kit (Sigma-Aldrich, St.
Louis, MO, USA). Briefly, 4×105 cells were stained with
Annexin V-fluorescein isothiocyanate (FITC; 5 µl Annexin
V-FITC conjugate in 1 ml suspended cells) and propidium iodide (10
µl propidium iodide solution in 1 ml suspended cells), and
detected using a FACScan flow cytometer (Bio-Rad Laboratories,
Inc.) to analyze the levels of cellular apoptosis. The results were
calculated using CellQuest™ Pro software (Bio-Rad Laboratories,
Inc.) and expressed as the percentage of apoptotic cells in the
total cells. The activity of caspase 3 was examined using a Caspase
3 Activity Assay kit (Cell Signaling Technology, Inc.), according
to the manufacturer's instructions, with the activity expressed as
the relative fluorescence intensity of 7-amino-4-methylcou-marin
(AMC), compared with the control.
Statistical analysis
Statistical analyses were performed using SPSS 16.0
software (SPSS, Inc., Chicago, IL, USA). Comparison of the mRNA and
protein expression levels of HIF-1α and RhoA, the percentage of
apoptotic cells and the activity of caspase 3 between the two
groups were analyzed using Student's t-test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Hypoxia induces the apoptosis of PC12
cells
In the present study, flow cytometric analysis was
used to measure the level of apoptosis of cells of the PC12 rat
pheochromocytoma cell line, induced by hypoxia. When the cells were
exposed to hypoxia for 12 h, 24 h or 48 h, significantly more PC12
cells underwent apoptosis, compared with the cells under normoxic
conditions (P<0.05 or P<0.01; Fig. 1A). Procaspase 3 undergoes cleavage
and activation in apoptosis (29),
and hypoxia treatment also induced higher levels of caspase 3
activity in the present study, which was evaluated by measuring the
fluorescence intensity of AMC, compared with the control PC12 cells
(P<0.01 or P<0.001; Fig.
1B). The cleavage of procaspase 3 in the PC12 cells following
hypoxia treatment was also examined (Fig. 1C). As shown in Fig. 1D, a higher level of procaspase 3
was cleaved into its 17 and 12 kDa subunits in the soluble protein
in the PC12 cells, compared with the control (P<0.01). To
further determine whether PC12 cell death by hypoxia was mediated
by caspase-3 activation, the cleavage of poly(ADP-ribose)
polymerase (PARP) by activated caspase 3 was examined. The results
demonstrated that, following exposure to hypoxia for 48 h, PARP
cleavage was significantly promoted (P<0.05; Fig. 1C and E). The present study also
determined the release of cytochrome c, which is
translocated between the mitochondria and the cytosol where it
assists in activating caspases. As shown in Fig. 1C and F, hypoxia also significantly
enhanced the release of cytochrome c (P<0.05).
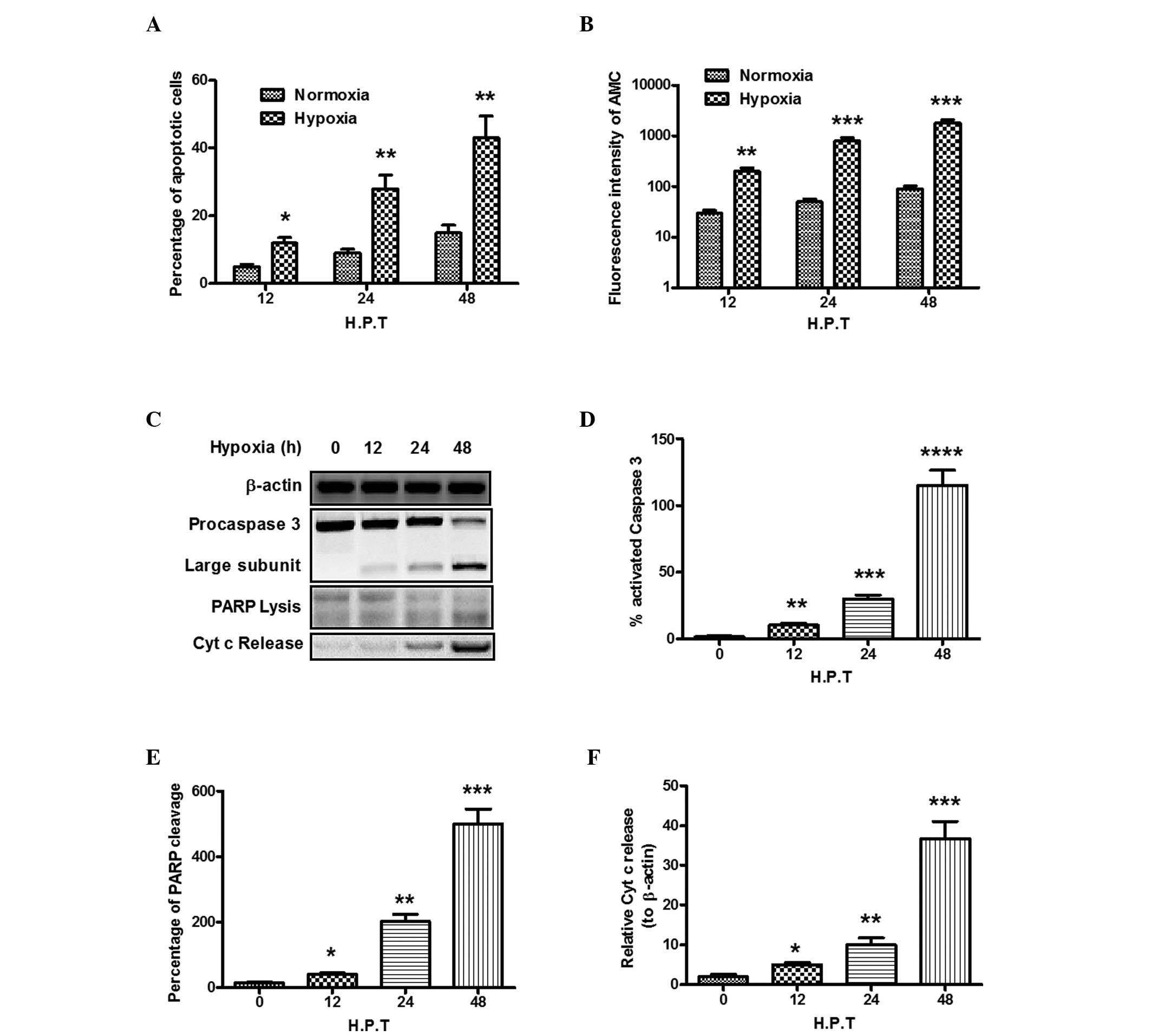 | Figure 1Hypoxia induces the apoptosis of PC12
cells. The PC12 cells were exposed to hypoxic or normoxic
conditions for 12, 24 or 48 h. (A) 4×105 cells were
stained with Annexin V-fluorescein isothiocyanate and propidium
iodide and detected using a FACS can flow cytometer to analyze
cellular apoptosis. Results are expressed as the percentage of
apoptotic cells to total cells. (B) Caspase 3 activity in the PC12
cells. Cell lysates were added to assay plates containing
Ac-DEVD-AMC substrate solution, and plates were incubated at 37ºC
in the dark. Relative fluorescent units were determined at 2 h. (C)
Western blot analysis of caspase 3 activation, PARP cleavage and
cytochrome c release in PC12 cells under hypoxic conditions.
(D) Percentage of activated caspase 3 to pro-caspase 3; (E)
Percentage of lyzed PARP to intact PARP; (F) Percentage of
cytochrome c release, relative to β-actin. All experiments
were performed in triplicate and data are presented as the mean ±
standard error of the mean. *P<0.05,
**P<0.01, ***P<0.001 and
****P<0.0001, compared with the normoxic control.
AMC, 7-amino-4-methylcoumarin; PARP, poly(ADP-ribose) polymerase;
Cyt c, cytochrome c; H.P.T, hours post-treatment. |
HIF-1α antagonizes hypoxia-induced
apoptosis of PC12 cells
To determine whether HIF-1α alters hypoxia-induced
apoptosis in PC12 cells, an plasmid overexpressing the HIF-1α gene
was transfected into PC12 cells, and the mRNA and protein levels of
HIF-1α and HIF-1β were evaluated using RT-qPCR and western blot
analysis. As shown in Fig. 2A, the
expression of HIF-1α was over four times higher in the
HIF-1α-overexpressing plasmid-transfected cells (P<0.01),
compared with the control plasmid-transfeted cells. The protein
level of HIF was also increased in the HIF-1α
overexpression-plasmid-transfected cells (P<0.01 or P<0.001;
Fig. 2B and C), whereas the
expression of HIF-1β was not regulated by HIF-1α overexpression
(Fig. 2B and D). Subsequently, to
determine whether HIF-1α alters hypoxia-induced apoptosis, the
present study re-evaluated the activity of caspase 3 by measuring
the AMC fluorescence intensity of the PC12 cells with or without
HIF-1α transfection. As shown in Fig.
2E, HIF-1α overexpression inhibited caspase 3 activity from 24
h post-transfection, and less AMC fluorescence intensity was
observed in the HIF-1α-overexpressed PC12 cells (P<0.05) 24 or
48 h post-transfection). In addition, flow cytometric analysis
revealed that the overexpression of HIF-1α inhibited the
hypoxia-induced apoptosis of the PC12 cells (P<0.05) 24 or 48 h
post-transfection; Fig. 2F). Taken
together, these findings indicated that the overexpression of
HIF-1α attenuated hypoxia-induced PC12 cell apoptosis.
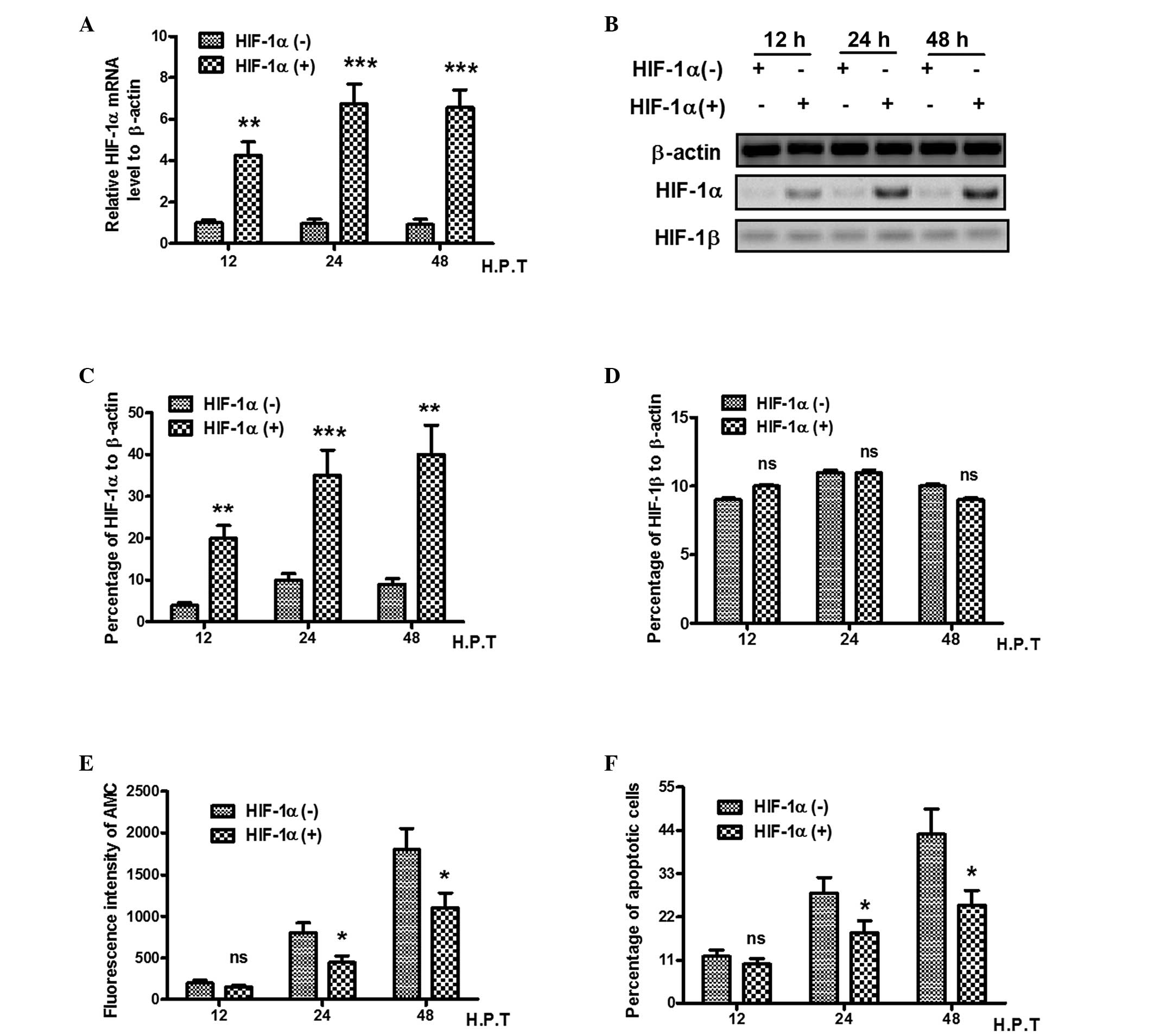 | Figure 2HIF-1α antagonizes the
hypoxia-induced apoptosis of PC12 cells. The PC12 cells were
transfected with a pcDNA3.1-HIF-1α or control pcDNA3.1-CAT-plasmid
for 12, 24 and 48 h. (A) Relative mRNA expression of HIF-1α in
HIF-1α-overexpressed PC12 cells. (B) Western blot analysis of
HIF-1α and HIF-1β in HIF-1α-overexpressed PC12 cells. Relative
expression of (C) HIF-1α and (D) HIF-1β as percentage of β-actin.
(E) Caspase 3 activity, determined by Acetyl-DEVD-AMC substrate
lyzation analysis, in HIF-1α-overexpressed PC12 cells under hypoxic
conditions. (F) Apoptosis in HIF-1α-overexpressed PC12 cells under
hypoxic conditions, revealed using a FACScan flow cytometer. All
experiments were performed in triplicate and data are presented as
the mean ± standard error of the mean. *P<0.05 and
**P<0.01, compared with the HIF-1α (-) group, ns, no
significance; HIF, hypoxia-inducible factor; AMC,
7-amido-4-methylcoumari; H.P.T, hours post-treatment. |
Hypoxia induces RhoA in PC12 cells
RhoA has been reported to be activated by chronic
hypoxia in the lungs (30,31), as two of the key downstream
effectors of RhoA, Rock 1 and Rock 2, have been confirmed to be
involved in the lung response to hypoxia (32). In present study, to determine
possible role of RhoA, Rock 1, Rock 2 and RhoB in hypoxia-induced
PC12 cell apoptosis, the present study examined the expression
levels of the three molecules in PC12 cells. As shown in Fig. 3A and B, the expression of RhoA was
markedly upregulated by >10-fold, between 2 and 6 h post-hypoxia
(P<0.001). The expression of Rock 1 was also significantly
upregulated in the PC12 cells from 2 h post-hypoxia exposure, with
a ~30-fold elevation in the expression of Rock 1 following hypoxia
stimulation (P<0.01 or P<0.001; Fig 3C). Notably, the expression levels of
Rock 2 appeared to change minimally on hypoxia stimulation, and
began to increase 4 h post-hypoxia exposure, with an elevation of
only three-fold (Fig. 3D). RhoB,
an inhibitor of Rho activity, was unresponsive to hypoxia
stimulation, with no significant change between 2 and 6 h
post-hypoxia exposure (Fig. 3E).
Therefore, RhoA signaling was significantly promoted by hypoxia in
the PC12 cells, and RhoA became the focus of subsequent experiments
to investigate levels of expression and to investigate the
molecular mechanisms used by the Rho proteins to regulate hypoxic
responses.
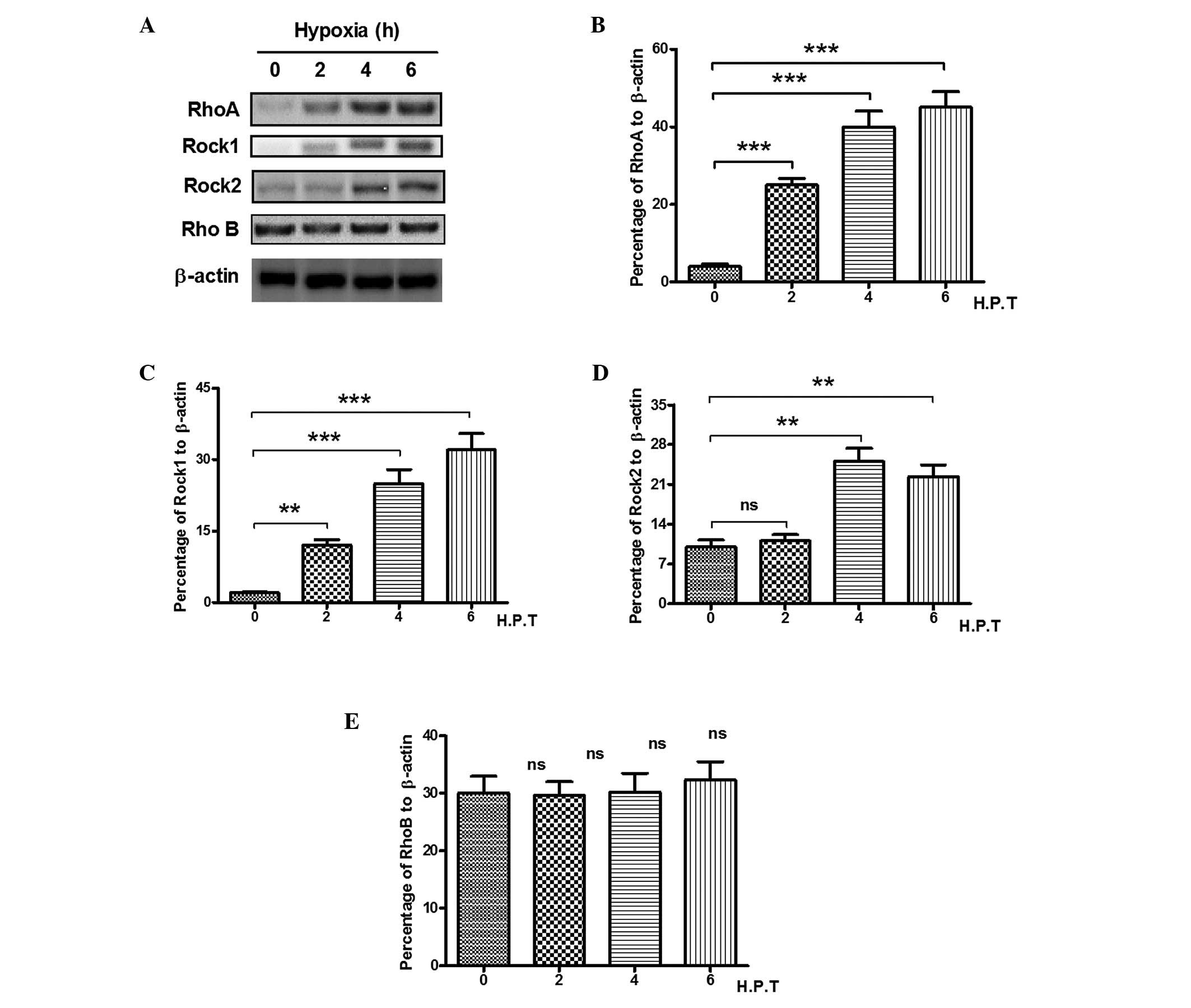 | Figure 3Hypoxia induces RhoA in PC12 cells.
The PC12 cells were exposed to hypoxic conditions for 0, 2, 4, 6
and 8 h and were analyzed for the expression of RhoA, Rock 1 and
Rock 2. (A) Western blot analysis of the expression levels of RhoA,
Rock1, Rock2 and RhoB in hypoxia-treated PC12 cells. Percentage of
(B) RhoA, (C) Rock1, (D) Rock2 or (E) RhoB to β-actin in the
hypoxia-treated PC12 cells. All experiments were performed in
triplicate and data are presented as the mean ± standard error of
the mean. **P<0.01 and ***P<0.001. Rho,
Ras homolog gene family, member A; Rock, Rho-associated kinase; ns,
no significance; H.P.T, hours post-treatment. |
Hypoxia-induced HIF-1α is
RhoA-dependent
The present study used RhoA specific siRNA to
investigate the effect of RhoA on the promotion of HIF-1α. First,
the mRNA and protein levels of RhoA were measured using RT-qPCR and
western blotting following the transfection of siRNAs specific for
RhoA (25 or 50 nM) into the PC12 cells. It was observed that 25 or
50 nM anti-RhoA siRNA significantly inhibited the mRNA and protein
expression levels of RhoA (Fig. 4A and
B). It was also demonstrated that the mRNA expression of HIF-1α
under hypoxia treatment also reduced following anti-RhoA siRNA
transfection in the PC12 cells between 12 and 24 h, which suggested
that hypoxia-induced HIF-1α was RhoA-dependent (P<0.05). The
present study also examined whether transfection with anti-RhoA
siRNA regulated the protein expression of HIF-1α. As shown in
Fig. 4D and E, compared with the
siRNA control-transfected PC12 cells, siRNA targeting RhoA
significantly inhibited hypoxia-induced the protein expression of
HIF-1α at 12, 24 h (P<0.05) and 48 h (P<0.01)
post-hypoxia.
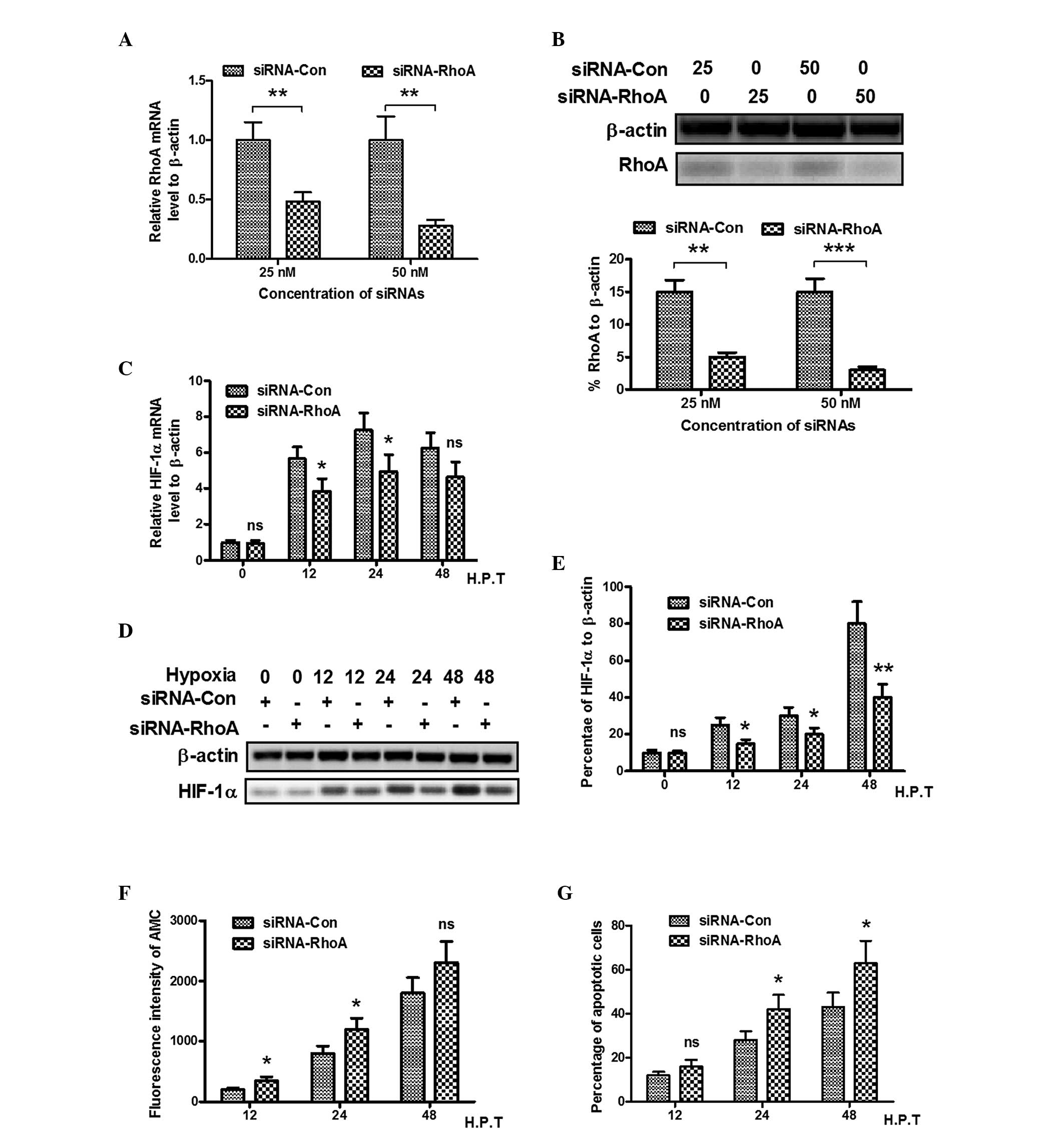 | Figure 4Hypoxia-induced HIF-1α is
RhoA-dependent. (A) Relative mRNA expression levels of RhoA in PC12
cells transfected with RhoA-targeted siRNA; results are expressed
relative to β-actin. (B) Western blot analysis of RhoA in PC12
cells following transfection with RhoA-targeted siRNA. (C) Relative
mRNA expression of HIF-1α in PC12 cells with RhoA knockdown,
results are expressed relative to β-actin. (D) Western blot
analysis of the induction of HIF-1α by hypoxia in PC12 cells
following RhoA knockdown (transfection with 50 nM siRNA-RhoA or
siRNA-Con for 12, 24 and 48 h). (E) Relative expression of HIF-1α
as percentage of β-actin. (F) Caspase 3 activity determined using
Acetyl-DEVD-AMC substrate lyzation analysis, in PC12 cells under
hypoxic conditions following RhoA knockdown. (G) Apoptotic cells
induced by hypoxia following RhoA knockdown. All experiments were
performed in triplicate and data are presented as the mean ±
standard error of the mean. *P<0.05,
**P<0.01 and ***P<0.001, compared with
the control. HIF, hypoxia-inducible factor; RhoA, Ras homolog gene
family, member A; ROCK, Rho-associated kinase; siRNA, small
interfering RNA; AMC, 7-amido-4-methylcoumari; Con, control; H.P.T,
hours post-treatment; ns, no significance. |
RhoA knockdown abrogates the antagonism
of HIF-1α in hypoxia-induced apoptosis of PC12 cells
As shown in Fig. 2,
HIF-1α was observed to antagonize hypoxia-induced apoptosis of the
PC12 cells, whereas the overexpression of HIF-1α in the PC12 cells
was inhibited by RhoA knockdown by siRNA transfection. The present
study investigated whether RhoA knockdown can reverse this change.
Following co-transfection with anti-RhoA siRNA and the pcDNA-HIF-1α
plasmid, the PC12 cells were treated with hypoxia for 12, 24 and 48
h. The fluorescence intensity of AMC revealed that the transfection
with RhoA siRNA significantly improved hypoxia-induced caspase 3
activity with HIF-1α treatment (Fig.
4E). Flow cytometric analysis also indicated that more
apoptotic cells were formed in the RhoA siRNA transfected PC12
cells, compared with the control siRNA group (Fig. 4F). In conclusion, knockdown of RhoA
in the PC12 cells abrogated the antagonism of HIF-1α to
hypoxia-induced apoptosis.
Discussion
Hypoxic conditions regulate several metabolic
enzymes and transcription factors, which are involved in cancer,
ischemia and pulmonary diseases (33). HIF-1 is a transcription factor
induced by hypoxia, which possesses two subunits, HIF-1α and HIF-1β
(34). Several studies have
demonstrated that reactive oxygen species (ROS) generation is
necessary for the transcriptional response induced by hypoxia
(35,36). The addition of diphenylene
iodonium, an inhibitor of ROS, or the use of cells depleted in
mitochondria can eradicate the hypoxic induction of HIF-1α,
erythropoietin, glycolytic enzymes and vascular endothelial growth
factor (37). Analysis of the
events upstream of HIF-1α has suggested the importance of the
generation of ROS in hypoxia via mitochondria as a sensor of oxygen
levels, and that ROS may be involved in stabilizing HIF-1α
(36,38).
RhoA belongs to the Rho small GTPase family, within
the Ras-like protein superfamily, which also includes the Ras, Rab,
Arf and Ran families (39). Rho
molecules are highly conserved between eukaryotes and mammals
(40). Following activation, Rho
GTPases trigger a signaling cascade to direct a variety of cellular
responses (41–43). Rho GTPases have been reported to
contribute to the majority of steps of cancer initiation and
progression, including unlimited proliferation potential and
evasion from apoptosis, and several Rho GTPases are upregulated in
certain types of human tumor, including RhoA, RhoC, Rac1 (44,45).
The upregulated Rho GTPases exert their pro-oncogenic effects via
the stimulation of cell cycle progression and regulation of gene
transcription (46). Certain Rho
GTPases are considered to be able to regulate the release of
pro-angiogenic factors to promote neovascularisation (47). Interacting with the plasma
membrane, RhoA modulates downstream signaling pathways to regulate
cell cycle and survival (48,49).
In addition, RhoA is essential in cancer cell migration and
invasion (50–52). RhoA has been suggested to be
involved in several anti-apoptotic pathways in the suppression of
apoptosis. The expression of RhoA promotes activation of
extracellular signal-regulated kinase (ERK) and facilitates
glomerular epithelial cells survival (53). RhoA also upregulates the expression
of anti-apoptotic Bcl-2 in T cells (54), vascular smooth muscle cells
(55) and osteosarcoma cells
(56). In addition, Rho inhibition
induces p53 in human endothelial cells (57).
Apoptosis during CNS/PNS damage is reversible and is
regarded as a major target of therapeutic interventions (15,16).
In the present study, hypoxia was demonstrated to induce apoptosis
of PC12 cells. Hypoxia had a significant effect on PC12 cell
apoptosis (Fig. 1), including
caspase 3 activation and cytochrome c release. Secondly,
transfection of an expression plasmid into PC12 cells revealed that
the overexpression of HIF-1α inhibited hypoxia-induced PC12 cell
apoptosis (Fig. 2). To improve
understanding of the mechanisms involved in the upregulation of
RhoA, the present study investigated its expression in
hypoxia-induced PC12 cells. The promotion of HIF-1α and RhoA, which
is reported to be involved in hypoxia adaptation, was observed in
the neuroblastoma PC12 cells following exposure to hypoxia
(Fig. 3). By inducing the
overexpression and inhibition of RhoA, the present study
manipulated the level of HIF-1α in the PC12 cells, and revealed
that hypoxia induced RhoA upregulation and cell apoptosis. The
results further confirmed the protective role of HIF-1α and RhoA in
hypoxia-induced PC12 cell apoptosis, and that the upregulation of
RhoA by hypoxia was HIF-1α-dependent (Fig. 4).
In conclusion, the present study demonstrated that
HIF-1α upregulation protected PC12 neuroblastoma cells from
hypoxia-induced apoptosis in a RhoA-dependent manner, and the
promotion of HIF-1α and RhoA may be a valuable strategy for
therapeutic intervention for hypoxic-ischemic damage to the
CNS/PNS.
Acknowledgments
This study was supported by a postdoctoral grant
(grant no. QL2012035) from Qilu Hospital of Shandong University
(Jinan, China).
References
|
1
|
Wishart TM, Parson SH and Gillingwater TH:
Synaptic vulnerability in neurodegenerative disease. J Neuropathol
Exp Neurol. 65:733–739. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Whiteley W, Tseng MC and Sandercock P:
Blood biomarkers in the diagnosis of ischemic stroke: A systematic
review. Stroke. 39:2902–2909. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kam PC, Kavanagh R and Yoong FF: The
arterial tourniquet: Pathophysiological consequences and
anaesthetic implications. Anaesthesia. 56:534–545. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McEwen JA: Complications of and
improvements in pneumatic tourniquets used in surgery. Med Instrum.
15:253–257. 1981.PubMed/NCBI
|
|
5
|
Rorabeck CH: Tourniquet-induced nerve
ischemia: An experimental investigation. J Trauma. 20:280–286.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bickler PE and Donohoe PH: Adaptive
responses of vertebrate neurons to hypoxia. J Exp Biol.
205:3579–3586. 2002.PubMed/NCBI
|
|
7
|
Nishimura M: Factors influencing an
increase in spontaneous transmitter release by hypoxia at the mouse
neuromuscular junction. J Physiol. 372:303–313. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bukharaeva EA, Salakhutdinov RI, Vyskocil
F and Nikolsky EE: Spontaneous quantal and non-quantal release of
acetylcholine at mouse endplate during onset of hypoxia. Physiol
Res. 54:251–255. 2005.PubMed/NCBI
|
|
9
|
David G, Nguyen K and Barrett EF: Early
vulnerability to ischemia/reperfusion injury in motor terminals
innervating fast muscles of SOD1-G93A mice. Exp Neurol.
204:411–420. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hatzipantelis KP, Natsis K and Albani M:
Effect of acute limb ischaemia on neuromuscular function in rats.
Eur J Surg. 167:831–838. 2001. View Article : Google Scholar
|
|
11
|
Northington FJ, Ferriero DM, Flock DL and
Martin LJ: Delayed neurodegeneration in neonatal rat thalamus after
hypoxia-ischemia is apoptosis. J Neurosci. 21:1931–1938.
2001.PubMed/NCBI
|
|
12
|
Graham SH and Chen J: Programmed cell
death in cerebral ischemia. J Cereb Blood Flow Metab. 21:99–109.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Linnik MD, Zobrist RH and Hatfield MD:
Evidence supporting a role for programmed cell death in focal
cerebral ischemia in rats. Stroke. 24:2002–2008; discussion
2008–2009. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ferrer I and Planas AM: Signaling of cell
death and cell survival following focal cerebral ischemia: Life and
death struggle in the penumbra. J Neuropathol Exp Neurol.
62:329–339. 2003.PubMed/NCBI
|
|
15
|
Kato H and Kogure K: Biochemical and
molecular characteristics of the brain with developing cerebral
infarction. Cell Mol Neurobiol. 19:93–108. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Newcomb JD, Ajmo CJ, Sanberg CD, Sanberg
PR, Pennypacker KR and Willing AE: Timing of cord blood treatment
after experimental stroke determines therapeutic efficacy. Cell
Transplant. 15:213–223. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cummins EP and Taylor CT:
Hypoxia-responsive transcription factors. Pflugers Arch.
450:363–371. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Licausi F, Weits DA, Pant BD, Scheible WR,
Geigenberger P and van Dongen JT: Hypoxia responsive gene
expression is mediated by various subsets of transcription factors
and miRNAs that are determined by the actual oxygen availability.
New Phytol. 190:442–456. 2011. View Article : Google Scholar
|
|
19
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang GL, Jiang BH, Rue EA and Semenza GL:
Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS
heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci
USA. 92:5510–5514. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pouyssegur J, Dayan F and Mazure NM:
Hypoxia signalling in cancer and approaches to enforce tumour
regression. Nature. 441:437–443. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kaelin WJ and Ratcliffe PJ: Oxygen sensing
by metazoans: The central role of the HIF hydroxylase pathway. Mol
Cell. 30:393–402. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Piret JP, Mottet D, Raes M and Michiels C:
Is HIF-1alpha a pro- or an anti-apoptotic protein? Biochem
Pharmacol. 64:889–892. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Akakura N, Kobayashi M, Horiuchi I, Suzuki
A, Wang J, Chen J, Niizeki H, Kawamura KI, Hosokawa M and Asaka M:
Constitutive expression of hypoxia-inducible factor-1alpha renders
pancreatic cancer cells resistant to apoptosis induced by hypoxia
and nutrient deprivation. Cancer Res. 61:6548–6554. 2001.PubMed/NCBI
|
|
25
|
Zhang B, Yin CP, Zhao Q and Yue SW:
Upregulation of hif-1α by hypoxia protect neuroblastoma cells from
apoptosis by promoting survivin expression. Asian Pac J Cancer
Prev. 15:8251–8257. 2014. View Article : Google Scholar
|
|
26
|
Chen D, Li M, Luo J and Gu W: Direct
interactions between HIF-1 alpha and Mdm2 modulate p53 function. J
Biol Chem. 278:13595–13598. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kothari S, Cizeau J, McMillan-Ward E,
Israels SJ, Bailes M, Ens K, Kirshenbaum LA and Gibson SB: BNIP3
plays a role in hypoxic cell death in human epithelial cells that
is inhibited by growth factors EGF and IGF. Oncogene. 22:4734–4744.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2 (-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
29
|
Bailly K, Ridley AJ, Hall SM and Haworth
SG: RhoA activation by hypoxia in pulmonary arterial smooth muscle
cells is age and site specific. Circ Res. 94:1383–1391. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nicholson DW and Thornberry NA: Apoptosis.
Life and death decisions. Science. 299:214–215. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dada LA, Novoa E, Lecuona E, Sun H and
Sznajder JI: Role of the small GTPase RhoA in the hypoxia-induced
decrease of plasma membrane Na, K-ATPase in A549 cells. J Cell Sci.
120:2214–2222. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Desbuards N, Antier D, Rochefort GY,
Apfeldorfer CS, Schenck E, Hanton G and Hyvelin JM: Dexfenfluramine
discontinuous treatment does not worsen hypoxia-induced pulmonary
vascular remodeling but activates RhoA/ROCK pathway: Consequences
on pulmonary hypertension. Eur J Pharmacol. 602:355–363. 2009.
View Article : Google Scholar
|
|
33
|
Semenza GL: HIF-1 and human disease: One
highly involved factor. Genes Dev. 14:1983–1991. 2000.PubMed/NCBI
|
|
34
|
Wang GL and Semenza GL: Purification and
characterization of hypoxia-inducible factor 1. J Biol Chem.
270:1230–1237. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li C and Jackson RM: Reactive species
mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol
Cell Physiol. 282:C227–C241. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chandel NS and Schumacker PT: Cellular
oxygen sensing by mitochondria: Old questions, new insight. J Appl
Physiol (1985). 88:1880–1889. 2000.
|
|
37
|
Chandel NS, Maltepe E, Goldwasser E,
Mathieu CE, Simon MC and Schumacker PT: Mitochondrial reactive
oxygen species trigger hypoxia-induced transcription. Proc Natl
Acad Sci USA. 95:11715–11720. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pearlstein DP, Ali MH, Mungai PT, Hynes
KL, Gewertz BL and Schumacker PT: Role of mitochondrial oxidant
generation in endothelial cell responses to hypoxia. Arterioscler
Thromb Vasc Biol. 22:566–573. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vega FM and Ridley AJ: Rho GTPases in
cancer cell biology. FEBS Lett. 582:2093–2101. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Boureux A, Vignal E, Faure S and Fort P:
Evolution of the Rho family of ras-like GTPases in eukaryotes. Mol
Biol Evol. 24:203–216. 2007. View Article : Google Scholar
|
|
41
|
Jaffe AB and Hall A: Rho GTPases:
Biochemistry and biology. Annu Rev Cell Dev Biol. 21:247–269. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bishop AL and Hall A: Rho GTPases and
their effector proteins. Biochem J. 48(Pt 2): 241–255. 2000.
View Article : Google Scholar
|
|
43
|
Wilkins A and Insall RH: Small GTPases in
Dictyostelium: Lessons from a social amoeba. Trends Genet.
17:41–48. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gomez del Pulgar T, Benitah SA, Valerón
PF, Espina C and Lacal JC: Rho GTPase expression in tumourigenesis:
Evidence for a significant link. Bioessays. 27:602–613. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gouw LG, Reading NS, Jenson SD, Lim MS and
Elenitoba-Johnson KS: Expression of the Rho-family GTPase gene RHOF
in lymphocyte subsets and malignant lymphomas. Br J Haematol.
129:531–533. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Benitah SA, Valerón PF, van Aelst L,
Marshall CJ and Lacal JC: Rho GTPases in human cancer: An
unresolved link to upstream and downstream transcriptional
regulation. Biochim Biophys Acta. 1705:121–132. 2004.PubMed/NCBI
|
|
47
|
Merajver SD and Usmani SZ: Multifaceted
role of Rho proteins in angiogenesis. J Mammary Gland Biol
Neoplasia. 10:291–298. 2005. View Article : Google Scholar
|
|
48
|
Wong WW, Dimitroulakos J, Minden MD and
Penn LZ: HMG-CoA reductase inhibitors and the malignant cell: The
statin family of drugs as triggers of tumor-specific apoptosis.
Leukemia. 16:508–519. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Graaf MR, Richel DJ, van Noorden CJ and
Guchelaar HJ: Effects of statins and farnesyltransferase inhibitors
on the development and progression of cancer. Cancer Treat Rev.
30:609–641. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Denoyelle C, Vasse M, Korner M, Mishal Z,
Ganné F, Vannier JP, Soria J and Soria C: Cerivastatin, an
inhibitor of HMG-CoA reductase, inhibits the signaling pathways
involved in the invasiveness and metastatic properties of highly
invasive breast cancer cell lines: An in vitro study.
Carcinogenesis. 22:1139–1148. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sawada K, Morishige K, Tahara M, Kawagishi
R, Ikebuchi Y, Tasaka K and Murata Y: Alendronate inhibits
lysophosphatidic acid-induced migration of human ovarian cancer
cells by attenuating the activation of rho. Cancer Res.
62:6015–6020. 2002.PubMed/NCBI
|
|
52
|
Kusama T, Mukai M, Tatsuta M, Nakamura H
and Inoue M: Inhibition of transendothelial migration and invasion
of human breast cancer cells by preventing geranylgeranylation of
Rho. Int J Oncol. 29:217–223. 2006.PubMed/NCBI
|
|
53
|
Bijian K, Takano T, Papillon J, Le Berre
L, Michaud JL, Kennedy CR and Cybulsky AV: Actin cytoskeleton
regulates extracellular matrix-dependent survival signals in
glomerular epithelial cells. Am J Physiol Renal Physiol.
289:F1313–F1323. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gómez J, Martínez C, Giry M, García A and
Rebollo A: Rho prevents apoptosis through Bcl-2 expression:
Implications for interleukin-2 receptor signal transduction. Eur J
Immunol. 27:2793–2799. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Blanco-Colio LM, Villa A, Ortego M,
Hernández-Presa MA, Pascual A, Plaza JJ and Egido J:
3-Hydroxy-3-methyl-glutaryl coenzyme A reductase inhibitors,
atorvastatin and simvastatin, induce apoptosis of vascular smooth
muscle cells by downregulation of Bcl-2 expression and Rho A
prenylation. Atherosclerosis. 161:17–26. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fromigué O, Haÿ E, Modrowski D, Bouvet S,
Jacquel A, Auberger P and Marie PJ: RhoA GTPase inactivation by
statins induces osteosarcoma cell apoptosis by inhibiting
p42/p44-MAPKs-Bcl-2 signaling independently of BMP-2 and cell
differentiation. Cell Death Differ. 13:1845–1856. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Li X, Liu L, Tupper JC, Bannerman DD, Winn
RK, Sebti SM, Hamilton AD and Harlan JM: Inhibition of protein
geranylgeranylation and RhoA/RhoA kinase pathway induces apoptosis
in human endothelial cells. J Biol Chem. 277:15309–15316. 2002.
View Article : Google Scholar : PubMed/NCBI
|














