Introduction
Prostate cancer (PCa) is the most common tumor in
males in the USA, with an estimated 240,890 new cases and 33,720
mortali-ties related to PCa in 2011 (1). However, few risk factors have been
identified beyond age and family history (2,3).
Despite a proven link between cigarette smoking and a number of
types of tumors (4), the
association between cigarette smoking and PCa remains unresolved.
Observational case-control studies investigating the association
between cigarette smoking and PCa have produced inconclusive
results (5). Epidemiological
studies suggest that the results of investigations into the
correlation between cigarette smoking and the incidence of PCa may
differ from the results of those measuring PCa-related mortality
(6). An observational cohort study
confirmed that while smoking does not appear to be associated with
PCa incidence, there is increased mortality due to PCa among heavy
smokers (7). Furthermore,
cigarette smokers have a higher risk of developing advanced stage
and high-grade PCa, as compared with non-smokers (8). Hence, cigarette smoking is a
potential contributing factor to PCa progression.
Tumor progression is a multistep process that
includes cell migration, invasion and metastasis, to which
alterations in the expression of extracellular matrix (ECM) and
cell adhesion molecule (CAM)-related genes contribute (9). During PCa progression, tumor cell
migration and invasion occur following the degradation of the ECM,
which is composed of collagen, proteoglycans, fibronectin, laminin
and other glycoproteins (9–13).
Tumor metastasis involves the detachment of cancer cells from the
primary tumor, disruption of the basement membrane and invasion of
the surrounding stroma by tumor cells. Cancer cells enter the
vascular and lymphatic systems and distal sites, including the
liver, lungs and brain, where they extravasate, proliferate and
undergo angiogenesis (9,14–16).
ECM proteins such as matrix metalloproteinases (MMPs) are
associated with tumor growth, cancer progression and metastasis
(17–23).
Tobacco smoke contains at least 3,500 chemical
compounds, many of which are toxic, carcinogenic and/or mutagenic.
It consists of a particulate solid phase (tar) and a gaseous phase,
containing volatile organic compounds (VOCs), free radicals and
other volatile and semi-volatile compounds (24,25).
Exposing prostate cancer cells to cigarette smoke promotes cell
proliferation and the secretion of vascular endothelial growth
factor (VEGF), a potent angiogenic factor (26). Angiogenesis is the process of new
blood vessel formation, which is fundamental to the progression,
invasion, and metastasis of numerous cancers, including PCa
(27).
Interaction between the ECM and CAMs helps determine
tissue shape, structure and cellular function. ECM and CAMs are
involved in a range of disorders and diseases. ECM- and CAM-related
gene expression on cell surfaces is involved in pathophysiological
processes, such as wound healing, inflammation, and disease
(9,28–34).
Levels of ECM and CAM-related gene expression vary. For example,
studies have shown that an increase in the expression of
ECM-related genes, including MMP2 and MMP9, promotes cell invasion
and the progression of PCa (35–37).
The transition from healthy prostatic intraepithelial neoplasia to
invasive PCa is associated with a decrease in the expression of
MMP7 and the cell matrix adhesion protein, integrin β 4 (ITGB4)
(38). The loss of cell adhesion
molecules, such laminin α 3 (LAMA3), is associated with high
Gleason scores, high preoperative prostate-specific antigen (PSA)
levels and more advanced stages of PCa (39).
Cigarette smoke enhances prostate cancer cell
proliferation and VEGF secretion (26). The present study examined the
direct effects of cigarette smoke on cell migration, and the
expression of ECM- and CAM-related genes in PC3 cells.
Materials and methods
Preparation of SM
3R4F cigarettes (Reference Cigarette Program,
University of Kentucky Research Institute; Lexington, KY, USA)
contained 11.0 mg/cigarette (mg/cig) total particulate matter
(TPM), 9.4 mg/cig tar, 0.73 mg/cig nicotine, and 12 mg/cig carbon
monoxide (CO). SM was generated by collecting whole smoke from 20
reference cigarettes into 100 ml of cell culture media. Briefly,
cigarette smoke from 20 cigarettes was bubbled into cell culture
medium. The medium was then filtered, and was designated as SM.
Fresh SM was used in the specified final concentrations.
Cell culture
PC3 cells [American Type Culture Collection (ATCC),
Manassas, VA, USA] were plated onto a 12-well tissue culture plate
and cultured in ATCC-formatted F12K medium (cat. no. CRL-1435 and
cat. no. 30-2004; ATCC, Rockville, MD, USA; respectively)
supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals,
Norcross, GA, USA). Cells were then incubated at 37°C, at 5%
CO2.
siRNA transfection
Small interfering RNA for MMP2 (MMP2 siRNA; cat. no.
sc-29398; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA)
designed to knock down human MMP2 expression was resuspended to a
final 10 µΜ concentration in a buffer containing 10µM
Tris-HCl, 20 mM NaCl, and 1 mM EDTA at pH 8.0. For a control,
scrambled siRNA (cat. no. sc-37007, Santa Cruz Biotechnology, Inc.)
was prepared and used in the same condition. siRNA transfection was
performed according to the manufacturer's instructions. In brief,
PC3 cells were washed with siRNA transfection medium (Santa Cruz
Biotechnology, Inc.) and incubated at 37°C in 200 µl
transfection medium containing 0.8 µM of MMP2 siRNA or
scrambled siRNA (siCTL). Following 24 h of transfection, MMP2
expression was assessed using western blot analysis and a wound
healing assay was performed.
Wound healing assay
A scratch-wound assay was performed as previously
described by Rodriguez et al (40) in order to assess cell migration in
the presence of SM. Following incubation, when cells had reached
~100% confluence, they were washed with serum-free F12K medium, and
replenished with ATCC-formatted medium containing 0.5% FBS. The
cells were cultured for 24 h. Subsequently, a sterile 20 ml pipette
tip was used to scratch the monolayer of cells in two perpendicular
straight lines through the center of the wells. Wells were gently
washed with serum-free culture, medium replenished with the medium
containing 0.5% FBS and treated with 0 (control), 0.2, 0.5, 1 or 2%
SM in cell culture medium. Cells were cultured for 24 h, after
which, cells that had migrated into the gaps were counted using a
microscope (Diaphot 300; Nikon Corporation, Tokyo, Japan).
RNA isolation
Isolation of total RNA was performed using
TRIzol® Reagent (cat. no. 15596-026; Invitrogen Life
Technologies, Carlsbad, CA, USA) according to the manufacturer's
instructions. Cells were seeded on 6-well plates and treated with
SM or F12K medium supplemented with 0.5% FBS. Subsequently,
chloroform (0.2 ml; Sigma-Aldrich, St. Louis, MO, USA) was added to
the wells. Samples were incubated at room temperature for 3 min,
and centrifuged at 12,000 x g at 4°C for 15 min. Subsequently,
isopropanol (0.5 ml; Thermo Fisher Scientific, Waltham, MA, USA)
was added to the supernatant. Following incubation at room
temperature for 10 min, samples were centrifuged at 12,000 x g at
4°C for 10 min. The pellets were washed with 75% ethanol, dissolved
in RNAse-free water (Thermo Fisher Scientific) and incubated at
60°C for 10 min.
Gene expression profiling
Cells were treated with 0.5% SM for 24 h.
Subsequently, total RNA was extracted using TRIzol and an RNeasy
mini kit (cat. no. 74104; Qiagen, Valencia, CA, USA). RNA integrity
was assessed using the bioanalyzer 'Agilent 2200 Tape Station'
(Agilent Technologies, Oxford, UK). The expression of 84 CAM- and
ECM-related genes were profiled using an RT2 Profiler
Polymerase Chain Reaction (PCR) Array for human extracellular
matrix and adhesion molecules, according to the manufacturer's
instructions (cat. no. PAHS-013A; SABiosciences, Qiagen). The gene
expression of 25 µg RNA per plate was measured. RNA was
converted into cDNA using a reverse transcription cocktail (cat.
no. 330401, Qiagen) at 42°C for 15 min. cDNA was then mixed with 2
x SABioscience RT PCR Master Mix (cat. no. 330520, Qiagen) and
subjected to PCR amplification using ABI 7300 and ABI 7500
platforms (AB Applied Biosystems, Foster City, CA, USA).
Quantitative (q)PCR primers and DNA oligos were purchased from Real
Time Primers, LLC (Elkins Park, PA, USA) and Integrated DNA
Technologies (Coralville, IA, USA), respectively. Threshold cycle
(Ct) was used to calculate changes in gene expression. Calculation
of Ct values and statistical analyses were performed using
web-based applications from SA Bioscience (Qiagen). Ct values were
normalized against those of actin and GAPDH. Ct values were
converted to linear values using the equation [2^
(−Ct)]. P-values were calculated using Student's t-test and a 95%
confidence interval (CI). Changes in gene expression were expressed
as fold change (FC) and fold regulation (FR). The PCR analysis was
conducted using web-based applications for RT2 Profiler PCR Array
Data Analysis version 3.5 (http://www.sabiosciences.com/RTPCR.php; Qiagen).
qPCR analysis
PCR reactions were performed using Power SYBR Green
PCR Master Mix (cat. no. 4309155; AB Applied Biosystems,
Warrington, UK) containing RT enzyme mix (cat. no. 4389988, AB
Applied Biosystems). RNA samples were subjected to qPCR
amplification using the ABI 7300 cycler (AB Applied Biosystems).
Using quantitative primers, the thermal cycler conditions were as
follows: 48°C for 30 min and 95°C for 10 min, followed by 42–52
cycles at 95°C for 15 sec and 60°C for 1 min.
Statistical analysis
Ct value calculations and statistical analyses were
performed using a web-based application (SA Bioscience) as
described above. Statistical significance was determined using
Student's t-test. Data represent the mean ± standard deviation.
P<0.05 was considered to indicate a statistically significant
difference.
Results
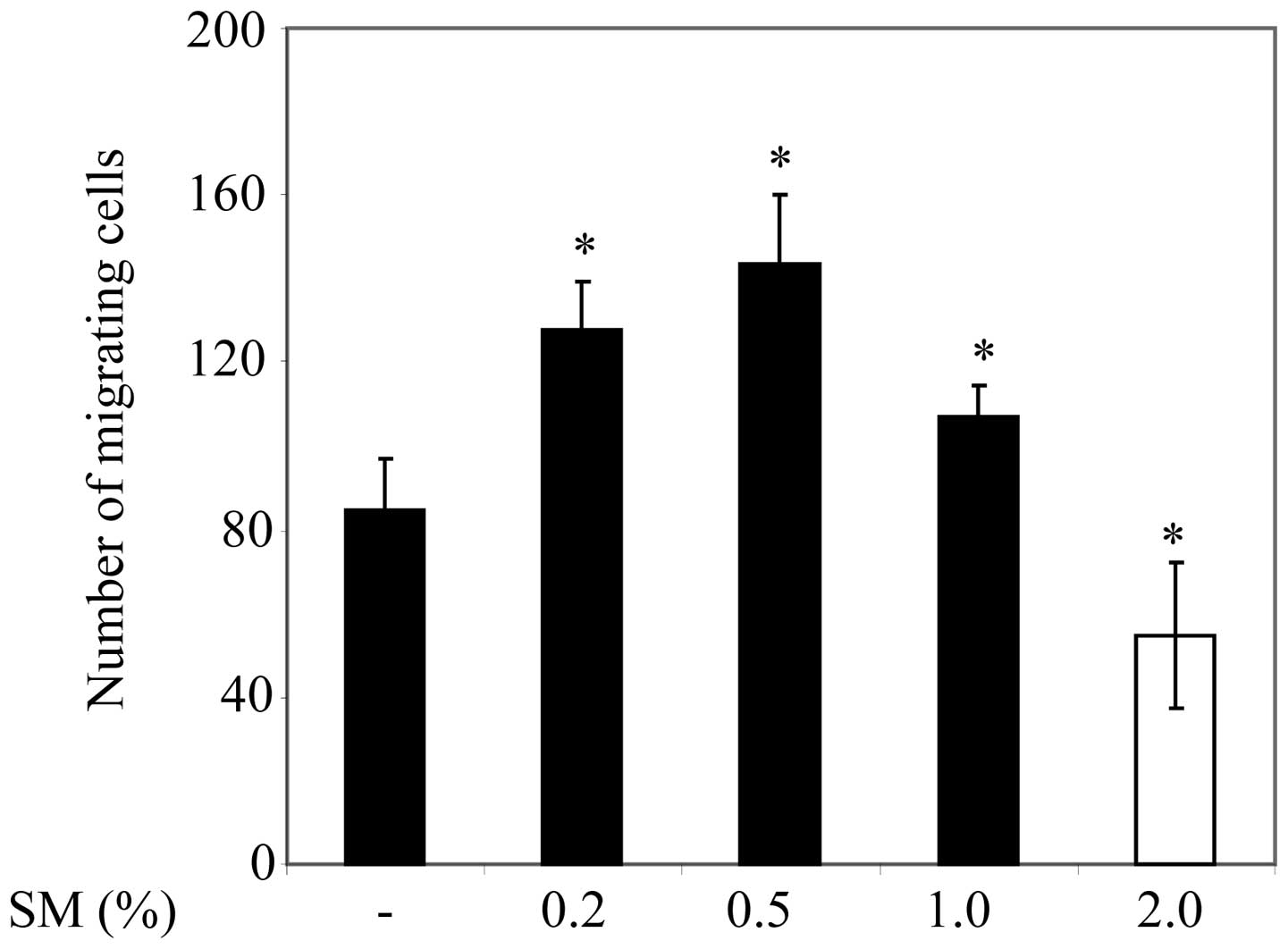
SM-mediated migration of PC3 cells
In order to examine the influence of cigarette smoke
on PC3 cell migration, the cells were cultured on 12-well plates.
Cells were treated with 0 (negative control), 0.2, 0.5, 1 or 2% SM,
and cells that had migrated into the gap were subsequently counted.
PC3 cell migration was significantly greater following 0.2, 0.5,
and 1% SM treatment compared with the migration of 0% SM-treated
cells or control counterparts (Fig.
1). It is notable that PC3 cell migration was significantly
lower following 1% SM treatment compared with 0.2 and 0.5% SM
treatment. Furthermore, PC3 cell migration was significantly lower
in 2% SM-treated cells compared with that of the control cells.
Since 0.5% SM induced the greatest increase in cell migration
compared with control cells, 0.5% SM was used for the remaining
experiments in the present study.
SM-modulated changes in the expression of
CAM- and ECM-related genes
ECM- and CAM-related genes are involved in multiple
processes that contribute to tumor progression (9). In order to investigate the effect of
SM on the expression of ECM- and of CAM-related genes, PC3 cells
were incubated in a starvation medium (0.5% FBS) for 24 h and then
treated with 0.5% SM or 0% SM (negative control) for a further 24
h. Of the 84 genes, the expression of 23 (27.38%) exhibited an FC
value of ≥2 (Table I). These genes
included: C-type lectin domain family 3; collagen type I α 1
(COL1A1); COL5A1; COL11A1; COL14A1; COL16A1; contactin 1;
connective tissue growth factor (CTGF); catenin, α 1; catenin Δ 2;
integrin β 2 (ITGB2); kallmann syndrome 1 sequence (KAL1); laminin
α 3 (LAMA3); matrix metallopeptidase 2 (MMP2); MMP3; MMP7; MMP12;
MMP13; selectin E; secreted protein acidic, cysteine-rich (SPARC);
transforming growth factor, β-induced; Thrombospondin 3 (THBS3);
and Versican (VCAN).
 | Table ISM-modulated changes in gene
expression of CAM- and ECM-related molecules. |
Table I
SM-modulated changes in gene
expression of CAM- and ECM-related molecules.
| Gene | FC | FR | Unigene | Accession |
|---|
| CLEC3B | 0.43 | −2.33 | Hs.476092 | NM_003278 |
| CNTN1 | 0.33 | −2.99 | Hs.143434 | NM_001843 |
| COL11A1 | 2.28 | 2.28 | Hs.523446 | NM_080629 |
| COL14A1 | 0.43 | −2.34 | Hs.409662 | NM_021110 |
| COL16A1 | 0.47 | −2.14 | Hs.368921 | NM_001856 |
| COL1A1 | 0.49 | −2.03 | Hs.172928 | NM_000088 |
| COL5A1 | 0.49 | −2.04 | Hs.210283 | NM_000093 |
| CTGF | 0.47 | −2.11 | Hs.591346 | NM_001901 |
| CTNNA1 | 0.10 | −10.65 | Hs.534797 | NM_001903 |
| CTNND2 | 2.16 | 2.16 | Hs.314543 | NM_001332 |
| ITGB2 | 0.43 | −2.34 | Hs.375957 | NM_000211 |
| KAL1 | 0.48 | −2.08 | Hs.521869 | NM_000216 |
| LAMA3 | 0.41 | −2.46 | Hs.436367 | NM_000227 |
| MMP2 | 2.70 | 2.70 | Hs.513617 | NM_004530 |
| MMP3 | 6.37 | 6.37 | Hs.375129 | NM_002422 |
| MMP7 | 0.41 | 2.44 | Hs.2256 | NM_002423 |
| MMP12 | 2.74 | 2.74 | Hs.1695 | NM_002426 |
| MMP13 | 0.47 | −2.14 | Hs.2936 | NM_002427 |
| SELE | 3.49 | 3.49 | Hs.89546 | NM_000450 |
| SPARC | 0.24 | −4.16 | Hs.111779 | NM_003118 |
| TGFBI | 0.34 | −2.92 | Hs.369397 | NM_000358 |
| THBS3 | 0.38 | −2.60 | Hs.169875 | NM_007112 |
| VCAN | 0.35 | −2.82 | Hs.643801 | NM_004385 |
Analysis of SM-modulated changes in the
expression of CAM and ECM-related genes
Genes with an FR of ≥2 (Table II) were sorted into subgroups.
CAMs were divided into the following subgroups: Transmembrane
molecules (TM); cell-to-cell adhesion molecules (CCA); cell matrix
adhesion molecules (CMA); and other adhesion molecules (OAM).
ECM-related molecules were divided into the following subgroups:
Basement membrane constituents (BMC); collagens and extracellular
matrix structural constituents (CEC); extracellular matrix
proteases (ECP); extracellular matrix protease inhibitors (ECPI);
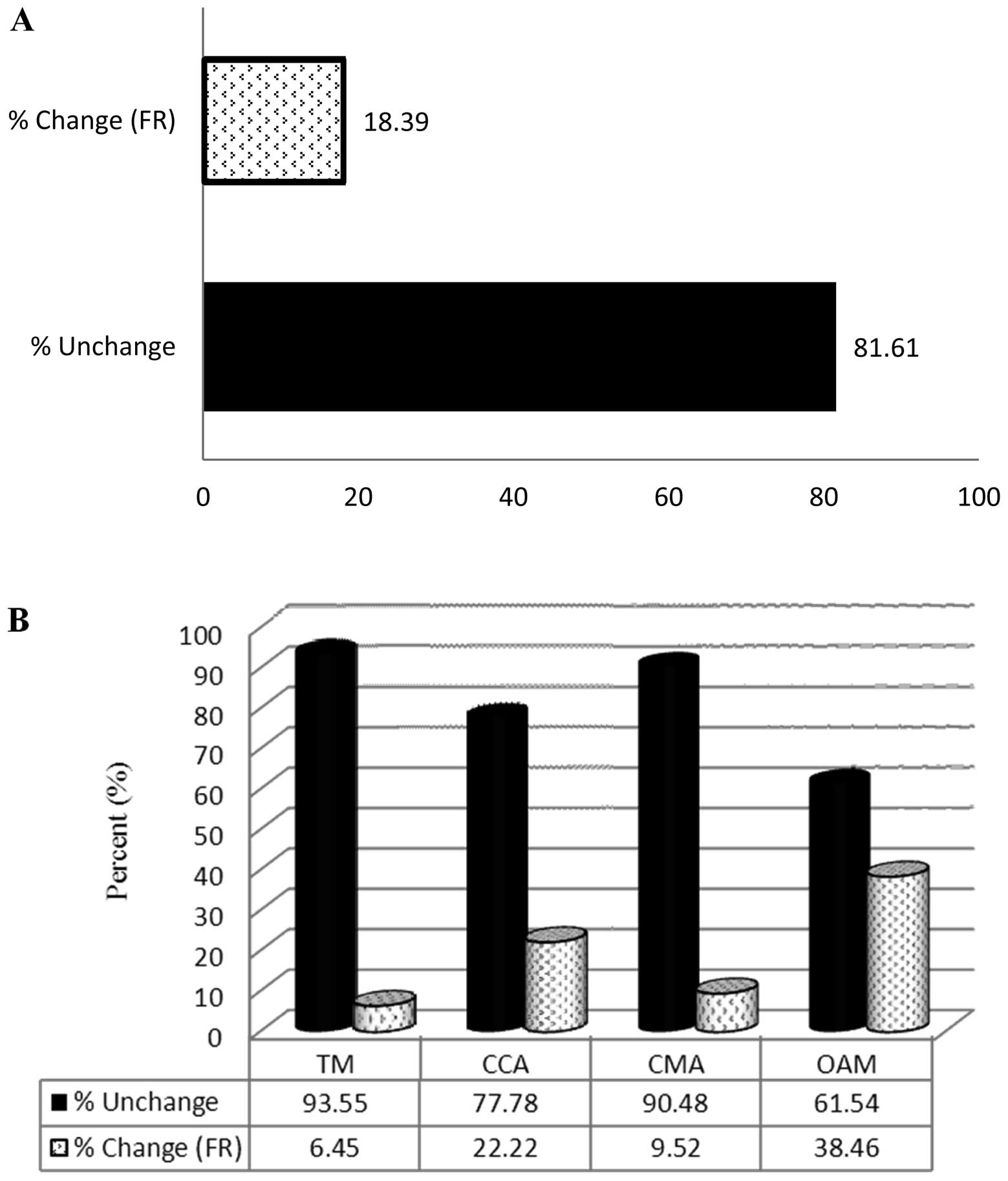
and other ECM (OECM). SM altered the expression of 18.39% (16/87)
of the CAM-related genes (Fig.
2A). The FR of 6.45% (2/31) of TM, 22.22% (2/9) of CCA, 9.52%
(2/21) of CMA and 38.46% (10/26) of OMA genes was altered by ≥2 in
the SM group compared with the control group (Fig. 2B). SM treatment altered the
expression of 33.92% (19/56) of ECM-related genes (Fig. 3A). The FR of 22.22% (2/9) of BMC,
42.85% (6/14) of CEC, 27.77% (5/18) of ECP, 25% (1/4) of ECPI and
45.45% (5/11) of OECM genes was altered in the SM-treated group
compared with the control group (Fig.
3B).
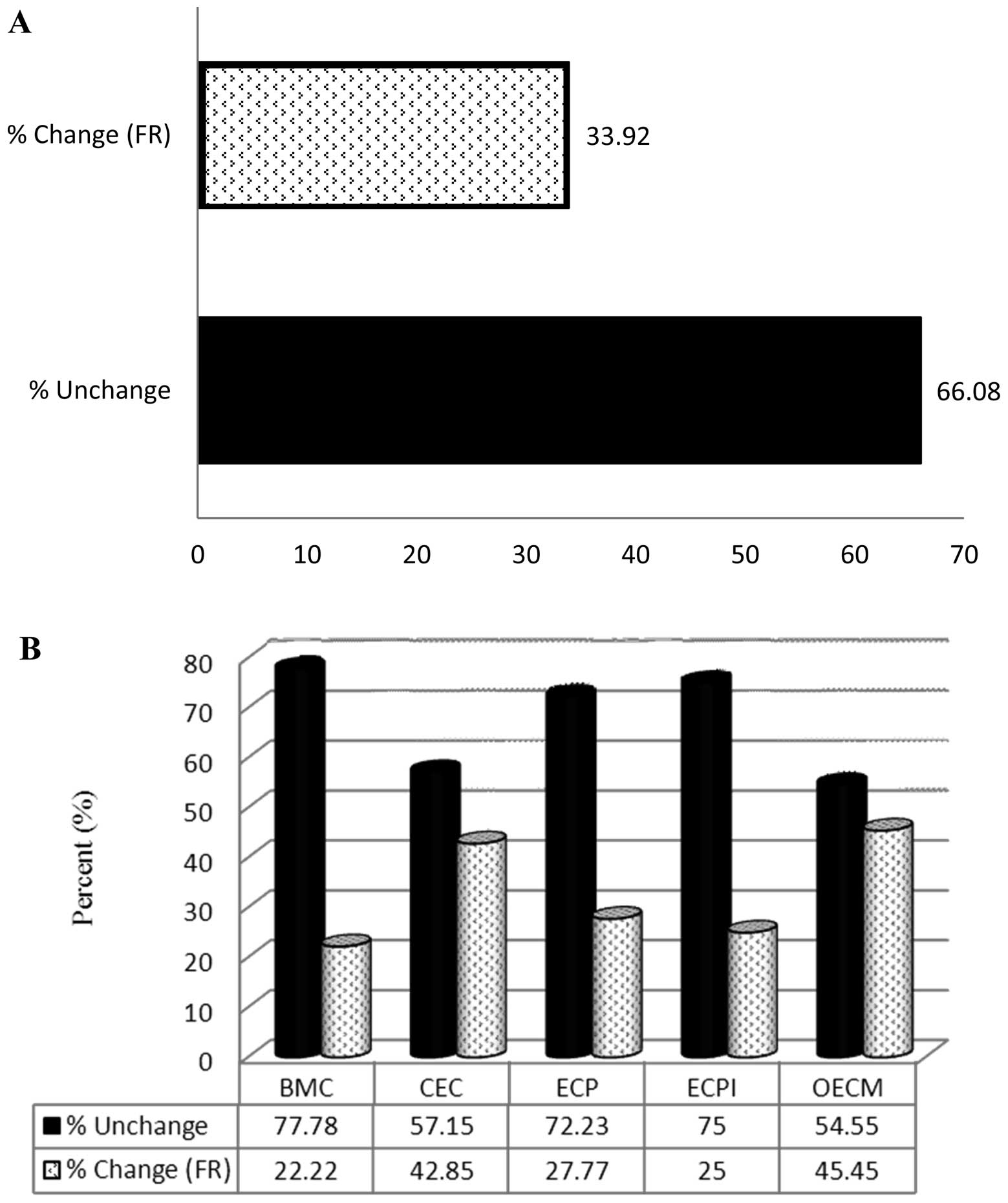 | Figure 3Effect of SM on the expression of
ECM-related genes assessed using an RT2 Profiler PCR
Array. (A) Percentage of ECM-related genes in which the FR was ≥2.0
following SM treatment, and the percentage of those in which
expression was not affected by SM. (B) Percentage of genes from
each ECM-related subgroup in which the FR was ≥2.0 following SM
treatment, and the percentage in which expression was not affected.
BMC, basement membrane constituent; ECM, extracellular matrix; CEC,
collagens and ECM structural constituents; ECP, extracellular
proteases; ECPI, ECP inhibitors; OEM, other ECM molecules; SM,
cigarette smoke medium; FR, fold regulation. |
| Table IISM-modulated changes in gene
expression of CAMs and ECM-related molecules using qPCR
analysis. |
Table II
SM-modulated changes in gene
expression of CAMs and ECM-related molecules using qPCR
analysis.
| Gene | FC | 95% CI | P-value | FR |
|---|
| COL5A1 | 0.25 | (0.08, 0.42) | 0.048875 | −2.17 |
| CTGF | 0.31 | (0.21, 0.41) | 0.000417 | −3.24 |
| ITGB2 | 0.36 | (0.30, 0.42) | 0.000338 | −2.74 |
| KAL1 | 0.26 | (0.07, 0.45) | 0.049642 | −3.91 |
| LAMA3 | 0.24 | (0.15, 0.33) | 0.001575 | −4.19 |
| MMP2 | 1.28 | (1.08, 1.48) | 0.042917 | 1.28 |
| MMP7 | 0.44 | (0.27, 0.61) | 0.027884 | −2.26 |
| MMP12 | 3.47 | (1.74, 5.20) | 0.009610 | 3.47 |
| MMP13 | 0.48 | (0.37, 0.59) | 0.007994 | −2.26 |
| SPARC | 0.15 | (0.11, 0.19) | 0.001258 | −6.59 |
| THBS3 | 0.14 | (0.09, 0.19) | 0.004143 | −7.35 |
| VCAN | 0.78 | (0.69, 0.87) | 0.015940 | −1.28 |
Validation of SM-modulated changes in
gene expression
SM-treated and control PC3 cells were harvested. RNA
was then extracted, purified and subjected to qPCR analysis using
the primer sets provided in Table
III. Ct values were normalized to those of GAPDH. Statistically
significant differences in gene expression in SM-treated compared
with control samples were determined using Student's t-test, and
calculated with a 95% confidence interval. SM significantly
increased MMP2 and MMP12 expression, and significantly decreased
COL5A1, CTGF, ITGB2, KAL1, LAMA3, MMP7, MMP13, SPARC, THBS3 and
VCAN expression in PC3 cells compared with that in control cells.
The data suggest there may be a correlation between SM-induced
migration (Fig. 1) and changes in
the expression of ECM- and CAM-related genes. These changes in gene
expression may be associated with interactions between CAM- and
ECM-related molecules, which may lead to SM-enhanced migration of
PC3 cells.
| Table IIIList of quantitative polymerase chain
reaction primers. |
Table III
List of quantitative polymerase chain
reaction primers.
| Gene | Forward primer | Reverse primer |
|---|
| COL5A1 |
5′-TGTATTTCCCCTGACCTTCA-3′ |
5′-ACCTTTAATCCATCGGGAAG-3′ |
| CTGF |
5′-TTTGGCCCAGACCCAACTAT-3′ |
5′-GTGCAGCCAGAAAGCTAAA-3′ |
| ITGB2 |
5′-ACAAGCTGGCTGAAAACAAC-3′ |
5′-GCAGAAGGAGTCGTAGGTGA-3′ |
| KAL1 |
5′-CAGCAAACACTTCCGTTCTT-3′ |
5′-GCTTCTTCTTTGTTGGGACA-3′ |
| LAMA3 |
5′-CTGCCAGTGCATCTGAATCT-3′ |
5′-TTTCTGACCATGCTCTTTGC-3′ |
| MMP2 |
5′-TTGACGGTAAGGACGGACTC-3′ |
5′-ACTTGCAGTACTCCCCATCG-3′ |
| MMP7 |
5′-AGCCAAACTCAAGGAGATGC-3′ |
5′-GCCAATCATGATGTCAGCAG-3′ |
| MMP12 |
5′-ACACATTTCGCCTCTCTGCT-3′ |
5′-CCTTCAGCCAGAAGAACCTG-3′ |
| MMP13 |
5′-TGACCCTTCCTTATCCCTTG-3′ |
5′-ATACGGTTGGGAAGTTCTGG-3′ |
| SPARC |
5′-GCACGGACTGTCAGTTCTCT-3′ |
5′-AAGAACAACCGATTCACCAA-3′ |
| THBS3 |
5′-ACACAGTTCTCCTGCGACTC-3′ |
5′-ATGGACCCACCCAGAATAAT-3′ |
| VCAN |
5′-TTGCTGTGGAAGGAACTGAG-3′ |
5′-CATAGGTGGCAGAAGCAGAA-3′ |
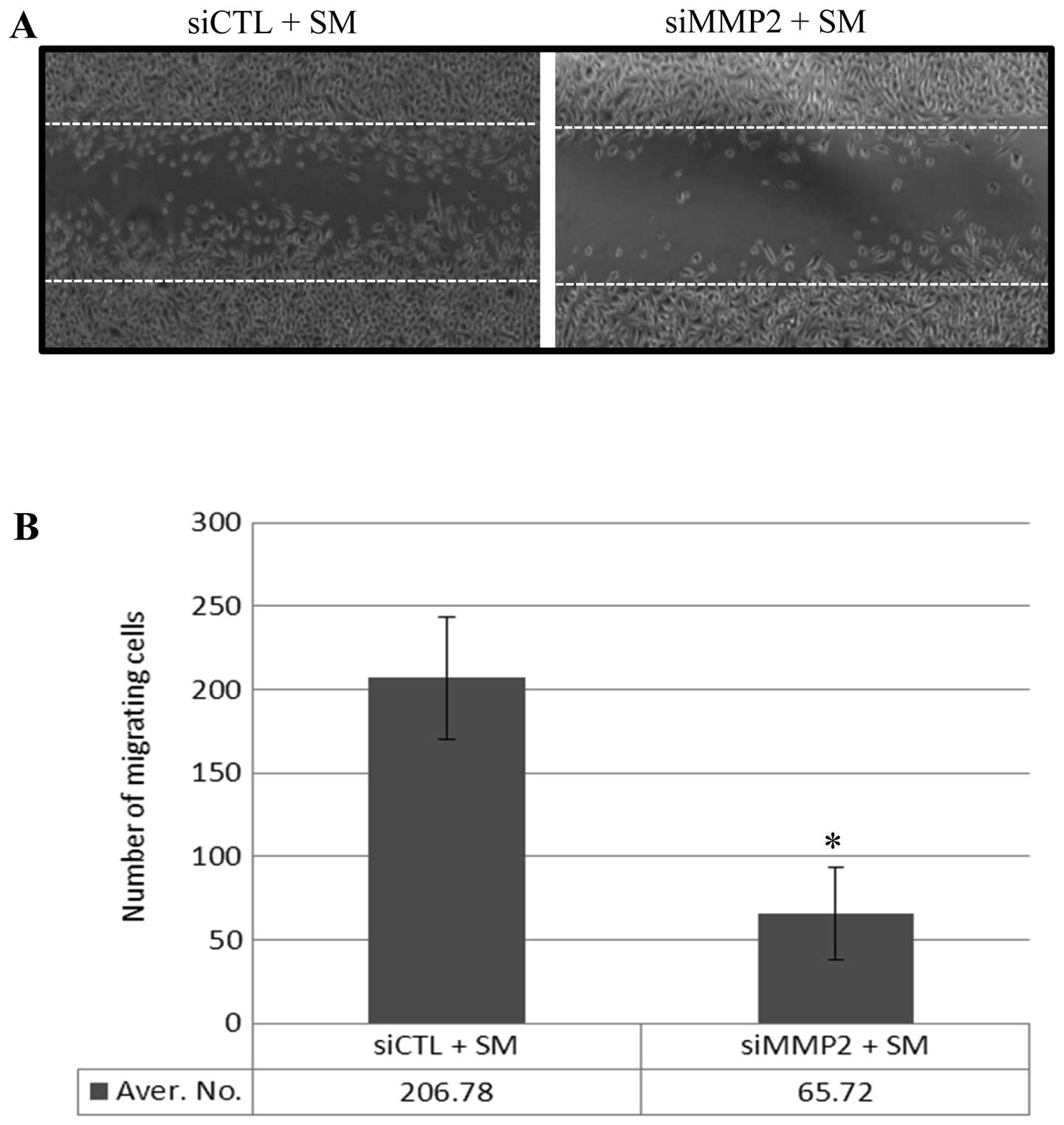
MMP2-modulated migration of SM-treated
PC3 cells
SM treatment led to an increase in the number of
migrating PC3 cells (Fig. 1) and
enhanced the expression of a number of MMPs, including MMP2
(Tables I and II). In order to confirm that the
increase in MMP2 expression contributed to enhanced PC3 cell
migration, PC3 cells were transfected either with a scrambled siRNA
(siCTL) or with MMP2 siRNA (siMMP2), which inhibited MMP2
expression. Following 24 h of transfection, a reduction in MMP2
expression was measured using western blot analysis (data not
shown). Following 24 h incubation in 0.5% FBS medium, the monolayer
was 'wounded' and treated with 0.5% SM for 12 h. Migration of the
siMMP2-transfected PC3 cells (with reduced MMP2 expression) was 68%
lower than that of siCTL transfected PC3 cells (Fig. 4A and B). These data suggest that
SM-enhanced MMP2 expression may contribute to the migration of PC3
cells.
Discussion
A recent observational cohort study demonstrated
that smoking is associated with the progression rather than the
incidence of PCa (7). However, to
the best of our knowledge the underlying molecular mechanism of PCa
progression is yet to be investigated. A previous study suggested
that cigarette smoke enhanced the growth of PCa cells and increased
secretion of VEGF, a potent angiogenic factor (26). In the present study, the effect of
cigarette smoke on PC3 cell migration was investigated, and the
expression of genes involved in tumor progression was measured. The
results suggested that cigarette smoke may increase PC3 cell
migration by altering the expression of a number of CAM- and
ECM-related genes.
Physiological interactions between CAMs and the ECM
define the shape, structure, and function of healthy tissues, which
are modulated by cellular activities, such as cell growth,
division, differentiation and migration (41,42).
However, ECM-CAM interactions are also involved in a range of
pathophysiological disorders, including cancer (9,28–34).
Changes in the expression of ECM- and CAM-related genes may alter
the interaction between these molecules, which may affect PCa
progression. Cigarette smoke has been shown to enhance PC3 cell
growth and to increase VEGF secretion (26). In the present study SM treatment
led to an increase in PC3 cell migration (Fig. 1). Therefore, exposure to cigarette
smoke may accelerate PCa progression. These results are consistent
with previous findings, which suggest that smokers exhibit a higher
risk of developing advanced stage and high-grade PCa, and report an
increased mortality among heavy smokers (7).
The results of the present investigation suggested
that PCa cell growth and migration was greater following 0.5% SM
treatment compared with that of PCa cells treated with 0.2, 1 and
2% SM (26; Fig. 1), which suggests
that 0.5% is an optimal concentration with which to enhance PCa
progression. This optimal SM concentration is estimated to contain
levels of TPMs, nicotine, and CO, which are equivalent to those
found in 1/100 smoke produced by one reference cigarette, according
to the Reference Cigarette Program. Cigarette smoke also contains
VOCs and reactive free radicals (24,26),
which can dysregulate healthy physiological processes, and promote
the proliferation, migration, invasion and metastasis of cancer
cells (25). Chronic exposure to
cigarette smoke, even at low doses, may pose a risk for advanced
stage and high-grade PCa in smokers and non-smokers.
Changes in the expression of ECM- and CAM-related
genes may alter the interactions of these moleules, influencing PCa
progression (43,44). An RT2 Profiler PCR array
analysis of PC3 cells suggested that 0.5% SM led to a ≥2-FC change
in the expression of 23 out of 84 ECM- and CAM-related genes in PC3
cells (Table I). This is
consistent with the results of the PCR analysis conducted in the
present study.
The amplitude and type of expression change in ECM-
and CAM-related genes varies between disease groups and disorders
(38). The transition from healthy
epithelial cells to PC3 cells is associated with a decrease in
ITGB4 and MMP7 expression, and the transition from prostatic
intraepithelial neoplasia to PCa is associated with a
downregulation in COL6A1 and ITGB2 expression (38). Loss of the cell adhesion molecule,
LAMA3, is associated with high Gleason scores, elevated
preoperative PSA levels and advanced stages of PCa (39). Reduced CTGF expression promotes
cell growth, migration and invasion of nasopharyngeal carcinoma
(45). Elevated COL11A1 expression
is detected malignant lesions of the stomach (46) and in sporadic colorectal tumors
(47). COL11A1 also promotes tumor
progression and is associated with a poor survival outcome in
patients with ovarian cancer (48). Among ECM-related genes, an increase
in MMP2 expression promotes cell invasion and PCa progression
(35–37), and an upregulation of MMP12 is
associated with the degradation and invasion of type I collagen in
PCa cells (49). In accordance
with previous reports (35–37,46–49),
the results of the present study suggested that SM treatment
enhances the expression of COL11A, MMP2 and MMP12 in PC3 cells
(Tables I and II). These finding suggested that
SM-mediated alteration of these molecules may be associated with
the progression of PCa.
To the best of our knowledge, the present study was
the first to examine the direct effects of cigarette smoke on PCa
progression. The results of the present study are consistent with
those of with epidemiological studies linking cigarette smoking to
PCa progression. CAM- and ECM-related genes may serve as biomarkers
for understanding PCa progression in smokers. In addition, the
present study suggested that MMP inhibitors may be useful
therapeutic treatments to prevent PCa progression.
Acknowledgments
This study was supported by grants from the Flight
Attendant Medical Research Institute (S.S) and National Institutes
of Health [grant no. 2R56HL09211 (S.T.)].
References
|
1
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: the impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chan JM, Jou RM and Carroll PR: The
relative impact and future burden of prostate cancer in the United
States. J Urol. 172:S13–S16. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huncharek M, Haddock KS, Reid R and
Kupelnick B: Smoking as a risk factor for prostate cancer: a
meta-analysis of 24 prospective cohort studies. Am J Public Health.
100:693–701. 2010. View Article : Google Scholar
|
|
4
|
Stewart SL, Cardinez CJ, Richardson LC, et
al: Surveillance for cancers associated with tobacco use - United
States, 1999–2004. MMWR Surveill Summ. 57:1–33. 2008.PubMed/NCBI
|
|
5
|
Colditz G: Consensus conference: smoking
and prostate cancer. Cancer Causes Control. 7:560–562. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hickey K, Do KA and Green A: Smoking and
prostate cancer. Epidemiol Rev. 23:115–125. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rohrmann S, Linseisen J, Allen N, et al:
Smoking and the risk of prostate cancer in the European Prospective
Investigation into Cancer and Nutrition. Br J Cancer. 108:708–714.
2013. View Article : Google Scholar :
|
|
8
|
Zu K and Giovannucci E: Smoking and
aggressive prostate cancer: a review of the epidemiologic evidence.
Cancer Causes Control. 20:1799–1810. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Roomi MW, Kalinovsky T, Rath M, et al:
Down-regulation of urokinase plasminogen activator and matrix
metalloproteinases and up-regulation of their inhibitors by a novel
nutrient mixture in human prostate cancer cell lines PC-3 and
DU-145. Oncol Rep. 26:1407–1413. 2011.PubMed/NCBI
|
|
10
|
Yurchenco PD and Schittny JC: Molecular
architecture of basement membranes. FASEB J. 4:1577–1590.
1990.PubMed/NCBI
|
|
11
|
Barsky SH, Siegal GP, Jannotta F, et al:
Loss of basement membrane components by invasive tumors but not by
their benign counterparts. Lab Invest. 49:140–147. 1983.PubMed/NCBI
|
|
12
|
Liotta LA, Tryggvason K, Garbisa S, et al:
Metastatic potential correlates with enzymatic degradation of
basement membrane collagen. Nature. 284:67–68. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liotta LA, Rao CN and Barsky SH: Tumor
invasion and the extracellular matrix. Lab Invest. 49:636–649.
1983.PubMed/NCBI
|
|
14
|
Egeblad M and Werb Z: New functions for
the matrix metalloproteinases in cancer progression. Nat Rev
Cancer. 2:161–174. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Folkman J: Role of angiogenesis in tumor
growth and metastasis. Semin Oncol. 29:15–18. 2002. View Article : Google Scholar
|
|
16
|
Chambers AF and Matrisian LM: Changing
views of the role of matrix metalloproteinases in metastasis. J
Natl Cancer Inst. 89:1260–1270. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nelson AR, Fingleton B, Rothenberg ML, et
al: Matrix metalloproteinases: biologic activity and clinical
implications. J Clin Oncol. 18:1135–1149. 2000.PubMed/NCBI
|
|
18
|
Bérubé M, Deschambeault A, Boucher M, et
al: MMP-2 expression in uveal melanoma: differential activation
status dictated by the cellular environment. Mol Vis. 11:1101–1111.
2005.PubMed/NCBI
|
|
19
|
Garzetti G, Ciavattini A, Lucarini G, et
al: Tissue and serum metalloproteinase (MMP-2) expression in
advanced ovarian serous cystoadenocarcinomas: clinical and
prognostic implications. Anticancer Res. 15:2799–2804.
1995.PubMed/NCBI
|
|
20
|
Gohji K, Fujimoto N, Hara I, Fujii A,
Gotoh A, et al: Serum matrix metalloproteinase-2 and its density in
men with prostate cancer as a new predictor of disease extension.
Int J Cancer. 79:96–101. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lokeshwar BL: MMP inhibition in prostate
cancer. Ann NY Acad Sci. 878:271–289. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shariat SF, Karam JA and Roehrborn CG:
Blood biomarkers for prostate cancer detection and prognosis.
Future Oncol. 3:449–461. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Achbarou A, Kaiser S, Tremblay G, et al:
Urokinase overproduction results in increased skeletal metastasis
by prostate cancer cells in vivo. Cancer Res. 54:2372–2377.
1994.PubMed/NCBI
|
|
24
|
Pryor WA and Stone K: Oxidants in
cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and
peroxynitrite. Ann NY Acad Sci. 686:12–27. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hecht SS: Tobacco smoke carcinogens and
lung cancer. J Natl Cancer Inst. 91:1194–1210. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Birrane G, Li H, Yang S, et al: Cigarette
smoke induces nuclear translocation of heme oxygenase 1 (HO-1) in
prostate cancer cells: nuclear HO-1 promotes vascular endothelial
growth factor secretion. Int J Oncol. 42:1919–1928. 2013.PubMed/NCBI
|
|
27
|
Aragon-Ching JB: Active surveillance for
prostate cancer: has the time finally come? J Clin Oncol.
28:e265–e266. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dilly AK, Ekambaram P, Guo Y, et al:
Platelet-type 12-lipoxygenase induces MMP9 expression and cellular
invasion via activation of PI3K/Akt/NF-κB. Int J Cancer.
133:1784–1791. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tian TV, Tomavo N, Huot L, et al:
Identification of novel TMPRSS2:ERG mechanisms in prostate cancer
metastasis: involvement of MMP9 and PLXNA2. Oncogene. 24:2204–2214.
2014. View Article : Google Scholar
|
|
30
|
Delassus GS, Cho H, Hoang S and Eliceiri
GL: Many new down- and up-regulatory signaling pathways, from known
cancer progression suppressors to matrix metalloproteinases, differ
widely in cells of various cancers. J Cell Physiol. 224:549–558.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Verslegers M, Lemmens K, Van Hove I and
Moons L: Matrix metalloproteinase-2 and -9 as promising benefactors
in development, plasticity and repair of the nervous system. Prog
Neurobiol. 105:60–78. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jinka R, Kapoor R, Sistla PG, Raj TA and
Pande G: Alterations in cell-extracellular matrix interactions
during progression of cancers. Int J Cell Biol. 2012:2191962012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yoo SY and Kwon SM: Angiogenesis and its
therapeutic opportunities. Mediators Inflamm. 2013:1271702013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Grässel S and Bauer RJ: Collagen XVI in
health and disease. Matrix Biol. 32:64–73. 2013. View Article : Google Scholar
|
|
35
|
Goel HL, Li J, Kogan S and Languino LR:
Integrins in prostate cancer progression. Endocr Relat Cancer.
15:657–664. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vandewalle C, Van Roy F and Berx G: The
role of the ZEB family of transcription factors in development and
disease. Cell Mol Life Sci. 66:773–787. 2009. View Article : Google Scholar
|
|
37
|
Xiao LJ, Lin P, Lin F, et al: ADAM17
targets MMP-2 and MMP-9 via EGFR-MEK-ERK pathway activation to
promote prostate cancer cell invasion. Int J Oncol. 40:1714–1724.
2012.
|
|
38
|
Ashida S, Nakagawa H, Katagiri T, et al:
Molecular features of the transition from prostatic intraepithelial
neoplasia (PIN) to prostate cancer: genome-wide gene-expression
profiles of prostate cancers and PINs. Cancer Res. 64:5963–5972.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sathyanarayana UG, Padar A, Suzuki M, et
al: Aberrant promoter methylation of laminin-5-encoding genes in
prostate cancers and its relationship to clinicopathological
features. Clin Cancer Res. 9:6395–6400. 2003.PubMed/NCBI
|
|
40
|
Rodriguez LG, Wu X and Guan JL:
Wound-healing assay. Methods Mol Biol. 294:23–29. 2005.
|
|
41
|
Gumbiner BM: Cell adhesion: the molecular
basis of tissue architecture and morphogenesis. Cell. 84:345–357.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sheetz MP, Felsenfeld DP and Galbraith CG:
Cell migration: regulation of force on
extracellular-matrix-integrin complexes. Trends Cell Biol. 8:51–54.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhen Y, Ye Y, Yu X, et al: Reduced CTGF
expression promotes cell growth, migration, and invasion in
nasopharyngeal carcinoma. PLoS One. 8:e649762013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jinka R, Kapoor R, Sistla PG, Raj TA and
Pande G: Alterations in cell-extracellular matrix interactions
during progression of cancers. Int J Cell Biol. 2012:2191962012.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Okegawa T, Pong RC, Li Y and Hsieh JT: The
role of cell adhesion molecule in cancer progression and its
application in cancer therapy. Acta Biochim Pol. 51:445–457.
2004.PubMed/NCBI
|
|
46
|
Zhao Y, Zhou T, Li A, et al: A potential
role of collagens expression in distinguishing between premalignant
and malignant lesions in stomach. Anat Rec (Hoboken). 292:692–700.
2009. View Article : Google Scholar
|
|
47
|
Fischer H, Salahshor S, Stenling R, et al:
COL11A1 in FAP polyps and in sporadic colorectal tumors. BMC
Cancer. 1:172001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wu YH, Chang TH, Huang YF, et al: COL11A1
promotes tumor progression and predicts poor clinical outcome in
ovarian cancer. Oncogene. 33:3432–3440. 2014. View Article : Google Scholar
|
|
49
|
Nabha SM, dos Santos EB, Yamamoto HA, et
al: Bone marrow stromal cells enhance prostate cancer cell invasion
through type I collagen in an MMP-12 dependent manner. Int J
Cancer. 122:2482–2490. 2008. View Article : Google Scholar : PubMed/NCBI
|