Introduction
Since there are few effective treatments available
for acute stroke, further research is required to identify novel
strategies with which to treat this condition (1). One such strategy is ischemic
postconditioning, which may be induced by repeated cycles of
transient reperfusion and re-occlusion of the artery, which is
applied immediately after reperfusion (2–4).
There are two types of ischemic postconditioning: Early and delayed
ischemic postconditioning (5).
Rapid postconditioning is induced immediately or a few minutes
after reperfusion. Delayed post-conditioning may be induced up to 2
days after stroke, however at present 3–6 h is recognized to induce
robust neuroprotection after ischemic injury. It has previously
been reported that both forms of ischemic postconditioning are able
to improve neurological function and reduce infarct volume
following focal ischemic injury (6). However, as ischemia is unpredictable,
ischemic postconditioning, which may be implemented following a
stroke, requires further research and increased clinical attention.
Therefore, it may be important to identify additional models of
ischemic postconditioning. However, to date, there have been no
reports on the effects of the combination of early and delayed
ischemic postconditioning, and whether this may provide an
increased neuroprotective effect, compared with one type of
ischemic postconditioning alone.
Brain-derived neurotrophic factor (BDNF) is a member
of the neurotrophin family, which is hypothesized to be one of the
most important neurotrophic factors (NTFs) in the central and
peripheral nervous systems (7,8).
Numerous studies have indicated that NTFs, particularly BDNF, are
associated with neuronal development and differentiation, as well
as with improvement in neurological function following brain injury
(9,10). The cAMP response element-binding
protein (CREB) is a transcription factor of BDNF (11). In addition, extracellular
signal-regulated kinases 1/2 (ERK1/2) is an upstream
phosphorylating enzyme of BDNF (11,12).
Therefore, all of these proteins are important for the survival and
development of neuronal cells.
The present study aimed to investigate whether early
ischemic postconditioning, in combination with delayed ischemic
postconditioning, may protect against neuronal injury and
behavioral deficits following focal brain ischemia. Furthermore, in
order to explore the mechanisms underlying these neuroprotective
effects, the effects of ischemic postconditioning on the
disturbance of cerebral blood flow (CBF) and brain edema were
evaluated. The current study also investigated the role of the
ERK1/2-CREB-BDNF pathway in the neuroprotective effects of
combinative postconditioning.
Materials and methods
Animals, surgical procedures and ischemic
postconditioning
The experimental protocol of the present study was
approved by the ethical committee of the Animal Care and
Experimental Committee of the School of Medicine of Shanghai Jiao
Tong University (Shanghai, China). A total of 136 age-matched adult
male Sprague-Dawley rats (weight, 270–300 g; age, 7–8 weeks), were
housed in a temperature- and light-controlled environment with a
14/10-h light/dark cycle. The rats were purchased from the Animal
Center of the School of Medicine of Shanghai Jiao Tong University
(Shanghai, China). The rats were randomly divided into five groups:
Sham-operated (sham; n=24), middle cerebral artery occlusion (MCAO;
n=30), early ischemic postconditioning (IPe; n=28), delayed
ischemic postconditioning (IPd; n=28) and combinative ischemic
postconditioning treatment (IPc; n=26) groups. Transient cerebral
ischemia was induced using MCAO, as previously described (13). Briefly, the rats were anesthetized
by inhalation of 5% isoflurane (Ohio Medical Corporation, Gurnee,
IL, USA), and were treated with 2% isoflurane during surgery and
early reperfusion. A heating blanket was applied to the rats, in
order to maintain a rectal temperature of 37.0±0.5°C throughout the
experiments. A middle neck incision was made to expose the left
common carotid artery (CCA). The external carotid artery was
ligated using a bipolar coagulator (Hkpromed, Beijing, China). A
nylon filament (diameter 0.24–0.28 mm) was gently inserted into the
left internal carotid artery, until resistance was felt.
Subsequently, the CBF dropped sharply, indicating that the ischemic
model was successfully established. Reperfusion was induced by
removing the filament after 90 min of ischemia. Sham-operated
animals were subjected to the same anesthetic, incision and
exposure of the vessels, without arterial occlusion. Ischemic
postconditioning was established by repeatedly occluding and
releasing the left CCA using aneurysm clips (cat. no. FE 681 K;
Aesculap, Inc., Center Valley, PA, USA). In order to explore the
combinative effects of early and delayed postconditioning, the two
types of postconditioning were combined. The protocol for early
plus delayed ischemic postconditioning is shown in Fig. 1A. Both types of postconditioning
consisted of 10 cycles of 10-sec reperfusion/10-sec occlusion;
early postconditioning was conducted immediately following
reperfusion, and late postconditioning took place 3 h after
reperfusion.
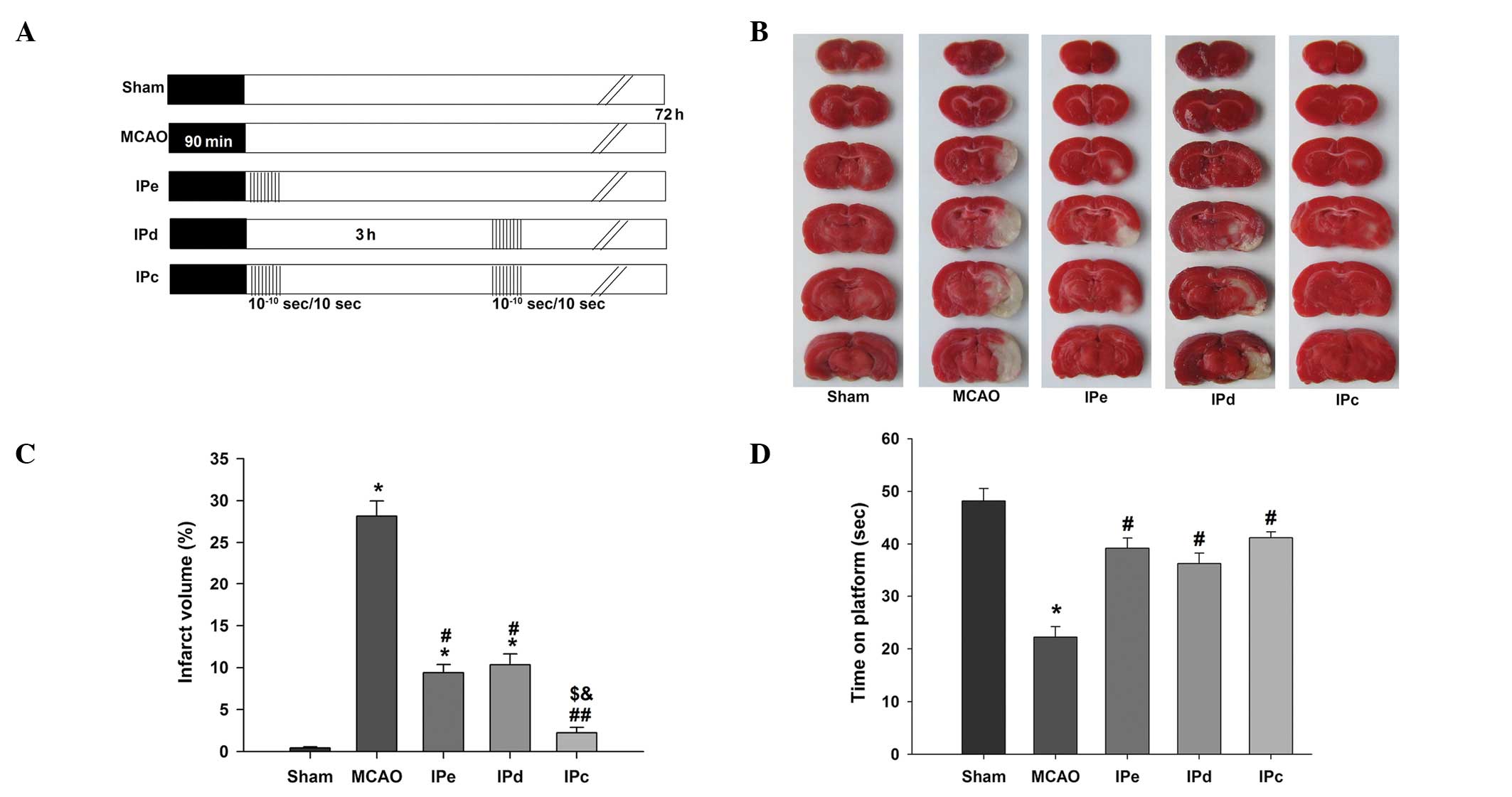 | Figure 1Combination of IPe and IPd reduced
infarct volumes and neurological deficit in MCAO rats. (A) Protocol
for IPc (n=26), in which postconditioning was conducted by
occluding and releasing the ipsilateral common carotid artery. IPe
(=28) was performed by conducting 10 cycles of 10/10-sec
reperfusion/re-occlusion immediately after MCAO. IPd (n=28) was
performed 3 h after MCAO. Ischemic injury was induced by MCAO
(MCAO, n=30). The sham group (n=24) was used as a control. (B)
Representative infarct volume from each group, demonstrated by
staining with 2,3,5-triphenyl tetrazolium chloride. (C) Average
infarct volume in rats treated with postconditioning. (D) Ischemic
postconditioning improved motor function, as evaluated by the
rotarod test. Data are presented as the mean ± standard error of
the mean. *P<0.05, compared with the sham group;
#P<0.05 and ##P<0.01, compared with the
MCAO group; $P<0.05, compared with the IPe group; and
&P<0.05, compared with the IPd group. MCAO,
middle cerebral artery occlusion; IPe, early ischemic
postcondi-tioning; IPd, delayed ischemic postconditioning; IPC,
combined IPe and IPd. |
CBF measurements
A laser Doppler flow meter (PeriFlux System 5000;
Perimed AG, Järfälla, Sweden) was used to measure CBF throughout
the experiments, and was connected to a standard laser Doppler
monitor (PeriFlux 5000 Laser Doppler Perfusion Monitor and PeriFlux
5000 main unit; Perimed AG). The holder for the Doppler probe was
fixed to the left side of the skull, in the region of the ischemic
penumbra (2 mm lateral and 2 mm posterior to the bregma). The
baseline value was regarded as the recorded CBF prior to occlusion.
All CBF values were expressed as a percentage relative to the
baseline.
Neurological behavioral assessment
A fully computerized electronically controlled
rotarod treadmill for rats (Accuscan Instruments, Inc., Columbus,
OH, USA) was used to investigate motor function. Each rat was
placed in a neutral position on a cylinder with a diameter of 3.75
inches. The speed of the rod accelerated linearly between 0 and 24
rpm within 60 sec. The time that rats maintained on the rotarod was
recorded automatically. The maximum score given to an animal was
fixed at 60 sec. All rats were given three trials and the average
score from these three trials was calculated.
Infarct volume measurement
The rats were euthanized 3 days after surgery using
CO2 and the brain tissue was harvested after
transcardial perfusion with cold phosphate-buffered saline (PBS).
Following 3 min of freezing, the brains were cut into six 2-mm
coronal sections. The tissue sections were then immediately stained
with 2% 2,3,5-triphenyl tetrazolium chloride (TTC) in PBS at 37°C
for 20 min. Images of the stained sections were captured using a
digital camera (Nikon E5100; Nikon Corporation, Tokyo, Japan), and
were then quantified using ImageJ version 1.37c software (National
Institutes of Health, Bethesda, MA, USA). The infarct volume was
the sum of the measured infarct areas of evenly sliced (2-mm) brain
sections, according to Simpson's rule (14).
Brain edema measurement
The cerebral hemispheres, cerebellum and brain stem
were separated. The wet weight was regarded as the immediate
weight, and the dry weight was regarded as the weight after drying
at 104°C for 48 h. The water content percentage was determined
using the following formula: [(wet weight-dry weight)/wet weight] ×
100.
In situ labeling of DNA fragmentation
using TUNEL staining
In order to determine the level of apoptosis in the
penumbral area, TUNEL staining was performed on each tissue
section, according to the manufacturer's instructions (Roche
Diagnostics GmbH, Mannheim, Germany). Briefly, the slides were
washed with PBS at room temperature for 30 min and incubated with
0.1% Triton X-100 in 0.1% sodium citrate solution at 4°C for 2 min.
Approximately 50 µl TUNEL mixture solution was applied to
each slide at 37°C for 1 h. All of the sections were overlaid with
prolonged mounting reagent and DAPI (Invitrogen Life Technologies,
Carlsbad, CA, USA), and were covered with coverslips. The Nikon
ECLIPSE Ti fluorescence microscope (Nikon Corporation) and CoolSNAP
photometrics camera (Nikon Corporation) were used to visualize and
capture the images. TUNEL-positive cells, indicated by bright green
fluorescence, were quantified using the NIS-Elements BR diagnostic
software version 3.2 (Nikon Corporation). The number of
TUNEL-positive cells was counted from three random 1×1
mm2 areas.
Immunofluorescence staining
Brain sections (10 µm) were incubated with
10% normal goat serum/0.1% Triton-X 100 in PBS blocking solution at
room temperature for 1 h. Slides were then incubated with the
following corresponding primary antibodies: Polyclonal rabbit
anti-rat BDNF (1:200; cat. no. ab6201; Epitomics, Inc., Burlingame,
CA, USA), monoclonal anti-neuron-specific nuclear protein (NeuN;
1:500; cat. no. MAB377B; EMD Millipore, Bedford, MA, USA) and
monoclonal anti-glial fibrillary acidic protein (GFAP; 1:200; cat.
no. G3893; Sigma-Aldrich, St. Louis, MO, USA), overnight at 4°C.
Following incubation, slides were then washed and incubated for 1 h
with the following secondary antibodies: Alexa Fluor 488 goat
anti-rabbit Immunoglobulin G (1:300; cat. no. A31267; Invitrogen
Life Technologies, Camarillo, CA, USA) and Alexa Fluor 568 goat
anti-rat (1:500; cat. no. A10042; Invitrogen Life Technologies).
DAPI was used to stain the nuclei (Sigma-Aldrich). All of the
slides were visualized under a Nikon ECLIPSE Ti fluorescence
microscope, loaded with a CoolSNAP photometrics camera at 400×
magnification.
Western blot analysis
The expression levels of CREB-ERK pathway-associated
proteins were detected by western blot analysis. Protein extraction
reagents (Sigma-Aldrich) were applied for total protein isolation
and purification. Protein samples (~50 µg) were separated by
11.5% SDS-PAGE (Sigma-Aldrich). Polypeptides were
electrophoretically transferred onto an Immobilon polyvinylidene
fluoride membrane (EMD Millipore) at 300 mA for 1 h on ice. The
membranes were then blocked with 5% non-fat milk in Tris-buffered
saline with Tween (TBST). After blocking, the membranes were rinsed
with PBS and incubated with the following antibodies: Anti-BDNF
(1:500; Epitomics, Inc.), anti-phosphorylated (p) ERK1/2 (1:200;
cat. no. 14227s; Cell Signaling Technology, Inc., Danvers, MA,
USA), anti-p-CREB (1:600; cat. no. 8212s; Cell Signaling
Technology, Inc.) and anti-β-actin (1:1,000; cat. no. A2022;
Sigma-Aldrich) overnight at 4°C. The membranes were then washed
with TBS and were treated with horseradish peroxidase-conjugated
secondary antibodies (1:800; cat. no. sc-2448; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) using 1% non-fat milk in TBST
for 2 h at room temperature. The blots were visual-ized using the
ChemiDoc™ XRS Imaging system (Bio-Rad Laboratories, Inc., Hercules,
CA, USA), and the band densities were quantified using the Multi
Gauge Software of Science Lab 2006 (Fujifilm Corporation, Tokyo,
Japan). Six animals were analyzed from each group for the western
blot analyses.
Statistical analysis
Data are expressed as the mean ± standard error of
the mean. The neurological scores were analyzed using the
Mann-Whitney non-parametric test. Statistical analysis was
performed using analysis of variance, followed by Dunnett's post
hoc test. P<0.05 (95% confidence interval) was considered to
indicate a statistically significant difference. SPSS version 15.0
software (SPSS, Inc., Chicago, IL, USA) was used to conduct
statistical analyses.
Results
Combinative ischemic postconditioning
further reduces infarct volume, compared with early or late
postconditioning alone, and attenuates behavioral deficits in MCAO
rats
In order to determine the effects of the combination
of early and delayed ischemic postconditioning on brain ischemic
injury, the infarct size due to ischemic insult was measured.
Representative images of TTC-stained brain sections from rats
subjected to MCAO are shown in Fig.
1B. The rats in the sham group exhibited no cerebral infarcts.
In the MCAO group, large infarcts developed, which were
predominantly located in the cerebral cortex and striatum. The
normalized infarct volume in the MCAO group was 28.16±1.79%. Early
and delayed ischemic postconditioning reduced the infarct volume,
and the normalized infarct volumes in these groups were 9.38±0.96
and 10.33±1.31%, respectively. The combination of early and delayed
ischemic postconditioning further reduced the infarct volume to
2.22±0.65%, which was approximately 22% lower than that of the MCAO
group (P<0.01), 7% lower than that of the IPe group (P<0.05)
and 8% lower than that of the IPd group (P<0.05, Fig. 1C).
The rotarod test was performed in order to test the
neurological function of rats following ischemic injury. The
results indicated that early, delayed and combinative ischemic
postconditioning significantly attenuated the neurological
deficits, compared with the MCAO group (P<0.05, Fig. 1D). However, no significant
differences were detected among the three ischemic postconditioning
groups.
Combinative ischemic postconditioning
stabilizes changes in CBF and attenuates brain edema
During ischemia, CBF was decreased to equivalent
extents (51.64±3.08% of baseline) in all groups, apart from the
sham group. Following reperfusion, a short period of hyperperfusion
(168.67±6.11%), which lasted ~50 min, followed by a long period of
hypoperfusion, was observed in the MCAO group (Fig. 2A). Although early postconditioning
did not shorten the time of hyperperfusion, the level of
hyperperfusion was reduce to 138.67±3.28% in the IPe group
(P<0.05 compared with the MCAO and IPd groups) and 132.87±3.31%
in the IPc group (P<0.05 compared with the MCAO and IPd groups).
Following ~3 h of hypoperfusion, delayed postconditioning increased
the CBF value to 119.67±1.17% in the IPd group (P<0.05 compared
with the MCAO and IPe groups), and 122.17±2.74% in the IPc group
(P<0.05 compared with the MCAO and IPe groups). These results
suggest that combinative ischemic postconditioning significantly
attenuated early hyperperfusion following ischemic reperfusion, as
well as the subsequent hypoperfusion.
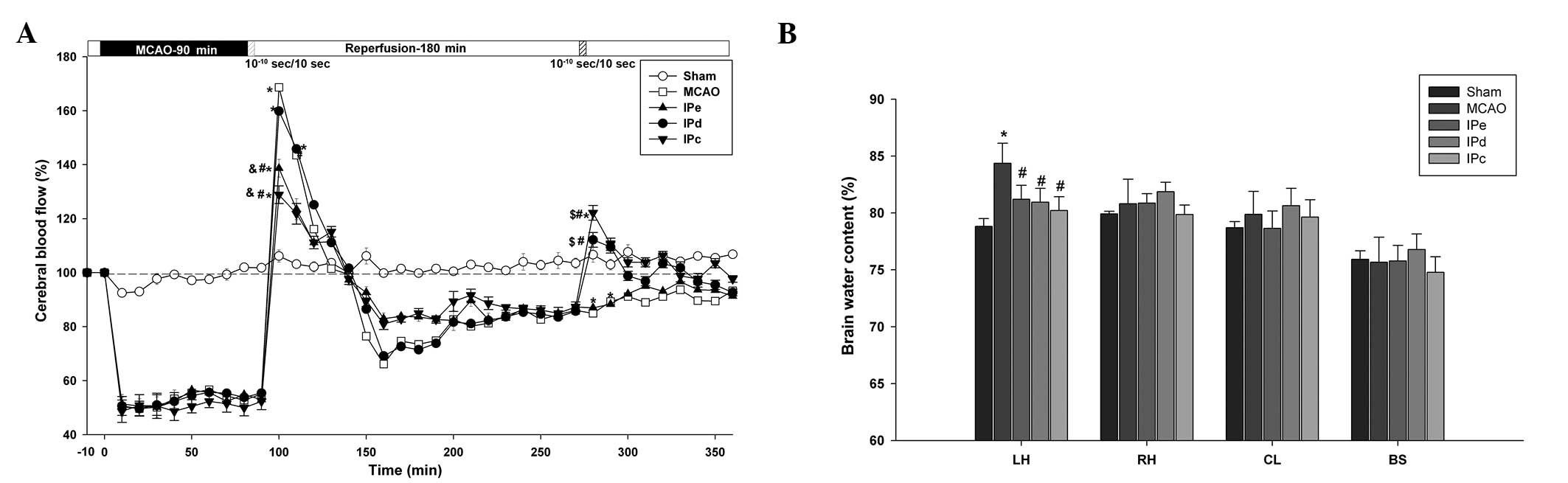 | Figure 2Combination of early and delayed
postconditioning stabilized CBF and attenuated brain edema. (A)
Changes in CBF in the MCA area following ischemia and ischemic
postconditioning. Data are presented as the mean ± standard error
of the mean; n=24. *P<0.05, compared with the sham
group; #P<0.05, compared with the MCAO group;
$P<0.05, compared with the IPe group; and
&P<0.05, compared with the IPd group. (B) Brain
water content of the LH, RH, CL and BS following ischemia and
ischemic postconditioning. Data are presented as the mean ±
standard error of the mean; n=6. *P<0.05, compared
with the sham group; and #P<0.05 compared with the
MCAO group. CBF, cerebral blood flow; MCAO, middle cerebral artery
occlusion; IPe, early ischemic postconditioning; IPd, delayed
ischemic postconditioning; IPc, combinative ischemic
postconditioning; LH, left hemisphere; RH, right hemisphere; CL,
cerebellum; BS, brain stem. |
The wet-dry method was used to assess the level of
brain edema 3 days after MCAO. The left brain hemisphere
(84.36±0.72%) contained more water than the right brain hemisphere
(80.81±0.88%) in the MCAO group. Early, delayed and combinative
ischemic postconditioning all significantly reduced the water
content of the left brain hemisphere (P<0.05), compared with the
MCAO group. However, no significant differences were detected among
the three ischemic postconditioning groups (Fig. 2B).
Combinative ischemic postconditioning
further inhibits apoptosis in the penumbral area compared with
early or late postconditioning alone
In pathological and anatomical terms, the penumbra
is an area surrounding a severely ischemic core. This area is where
pharmacological interventions are most likely to attenuate the
neurological injury and improve clinical outcome (Fig. 3A). TUNEL-positive staining was
absent in the sham-treated group (Fig.
3B). Numerous bright green dots, which indicate the presence of
TUNEL-positive cells, were observed in the MCAO group (Fig. 3C). There were few TUNEL-positive
cells observed among rats that were administered with early,
delayed and combinative ischemic postconditioning treatment
(Fig. 3D–F). Quantitative analysis
indicated that the number of TUNEL-positive cells in the MCAO group
was 409.78±45.23. Early and delayed ischemic postconditioning
significantly reduced the level of apoptosis, compared with the
MCAO group (P<0.05, Fig. 3G).
Combinative ischemic postconditioning further decreased the rate of
apoptosis following ischemia in the penumbral area (P<0.01,
compared with the MCAO group; P<0.05, compared with the IPe and
IPd groups; Fig. 3G).
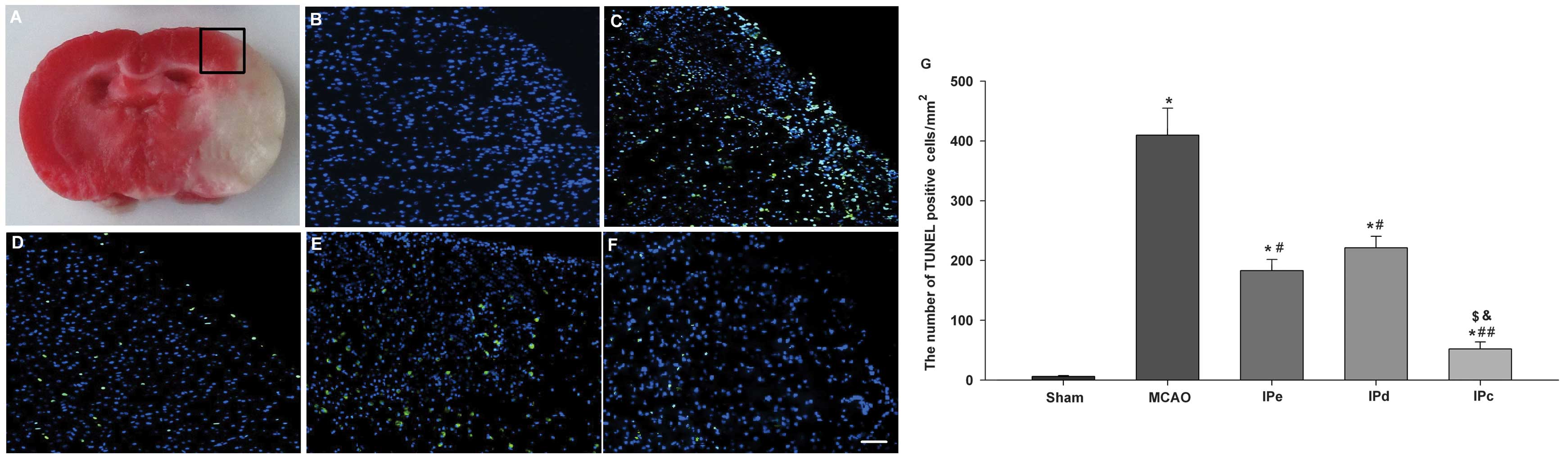 | Figure 3Combination of IPe and IPd inhibited
the levels of apoptosis in the penumbral area. (A) The black square
indicates the penumbral area. Immunofluorescence results of the
TUNEL assay in the (B) sham, (C) MCAO, (D) IPe, (E) IPd and (F) IPc
groups. Bright green dots indicate TUNEL-positive apoptotic cells.
Scale bar represents 50 µm. (G) Analysis of apoptotic cells
3 days after MCAO. TUNEL-positive cells were counted from three
random 1×1 mm2 areas. Data are presented as the mean ±
standard error of the mean; n=9 per group. *P<0.05,
compared with the sham group; #P<0.05 or
##P<0.01, compared with the MCAO group;
$P<0.05, compared with the IPe group; and
&P<0.05, compared with the IPd group. MCAO,
middle cerebral artery occlusion; IPe, early ischemic
postconditioning; IPd, delayed ischemic postconditioning; IPc,
combinative ischemic postconditioning. |
Combinative ischemic postconditioning
increases the expression levels of BDNF in the penumbral area
The present study sought to determine whether
ischemic postconditioning induces BDNF in the penumbral area, using
immunofluorescence staining. Neurons in the sham control group
exhibited BDNF-positive immunoreactivity, as assessed by BDNF+NeuN
double staining (Fig. 4A).
Astrocytes in this groups were also positive for BDNF, as assessed
by BDNF+GFAP double staining (Fig.
4F). BDNF immunoreactivity was significantly lower in the MCAO
group, compared with the sham group (Fig. 4B and G). Neither early nor delayed
ischemic postconditioning alone had a significant effect on
expression of BDNF (Fig. 4C, D, H and
I). However, BDNF immunoreactivity was higher in the
combinative ischemic postconditioning group, compared with that in
the MCAO, IPe and IPd groups (Fig. 4E
and G). Quantitative analysis also indicated that BDNF-positive
signals were higher in the IPc group, compared with those in the
MCAO (P<0.01), IPe (P<0.05) and IPd groups (P<0.05;
Fig. 4K).
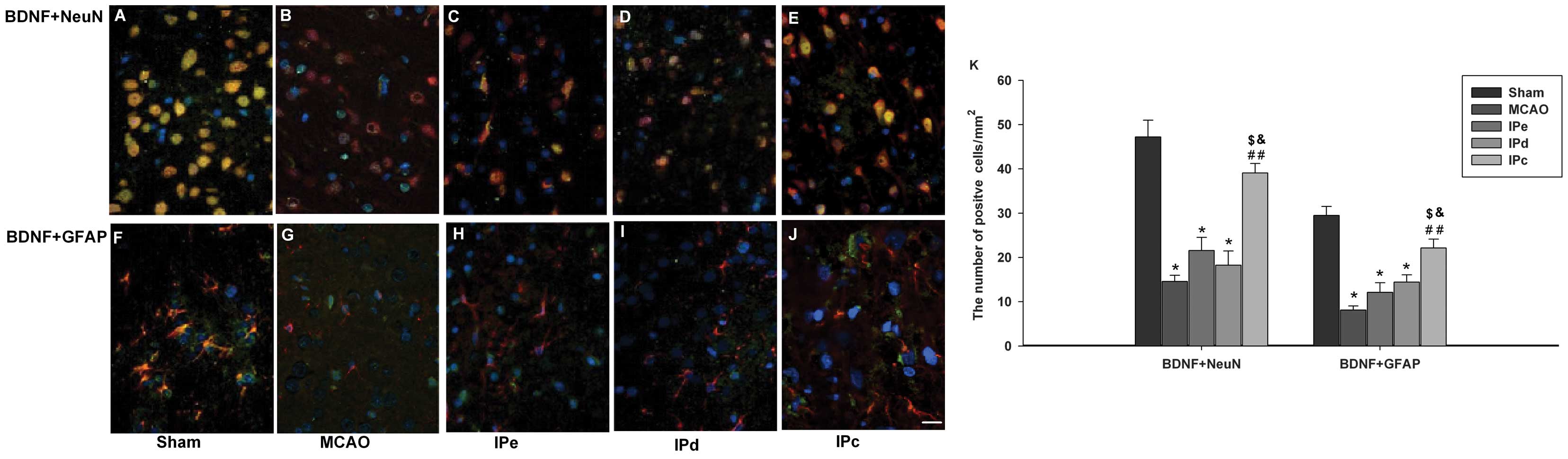 | Figure 4Combination of IPe and IPd increased
the expression of BDNF in the penumbral area. Coronal sections were
stained with the following specific antibodies 3 days after the
operation: BDNF (green), with either NeuN (red) or GFAP (red).
Mounting medium contained DAPI, which counterstained the nuclei
blue. Merged pictures, which co-expressed (A)-(E) NeuN and BDNF or
(F)-(J) GFAP and BDNF, were shown with yellow. Scale bar indicates
100 µm. (K) Quantitative analysis of BDNF-positive cells
were analyzed by ImageJ software. Data are presented as the mean ±
standard error of the mean; n=9 per group. *P<0.05,
compared with the sham group; ##P<0.01, compared with
the MCAO group; $P<0.05, compared with the IPe group;
and &P<0.05, compared with the IPd group. BDNF,
brain-derived neurotrophic factor; GFAP, glial acidic fibrillary
protein; NeuN, neuron-specific nuclear protein; MCAO, middle
cerebral artery occlusion; IPe, early ischemic postconditioning;
IPd, delayed ischemic postconditioning; IPc, combinative ischemic
postconditioning. |
Combinative ischemic postconditioning
upregulates BDNF protein expression levels by phosphorylating CREB
and ERK1/2
Western blot analyses indicated that combinative
ischemic postconditioning increased the protein expression levels
of BDNF in the penumbral area (Fig.
5A). Western blot analysis was also performed to investigate
whether combinative ischemic postconditioning results in the
phosphorylation of CREB and ERK1/2 within the cortex. No
significant differences in the protein expression of total CREB and
ERK1/2 were detected among the groups. However, administration of
combinative ischemic postconditioning significantly increased the
expression of p-CREB, compared with that of the MCAO (P<0.01),
IPe (P<0.05) and IPd groups (P<0.05; Fig. 5A and B). Similar results were
obtained for the expression of p-ERK1/2 (Fig. 5A and B), which is the upstream
phosphorylating enzyme of CREB.
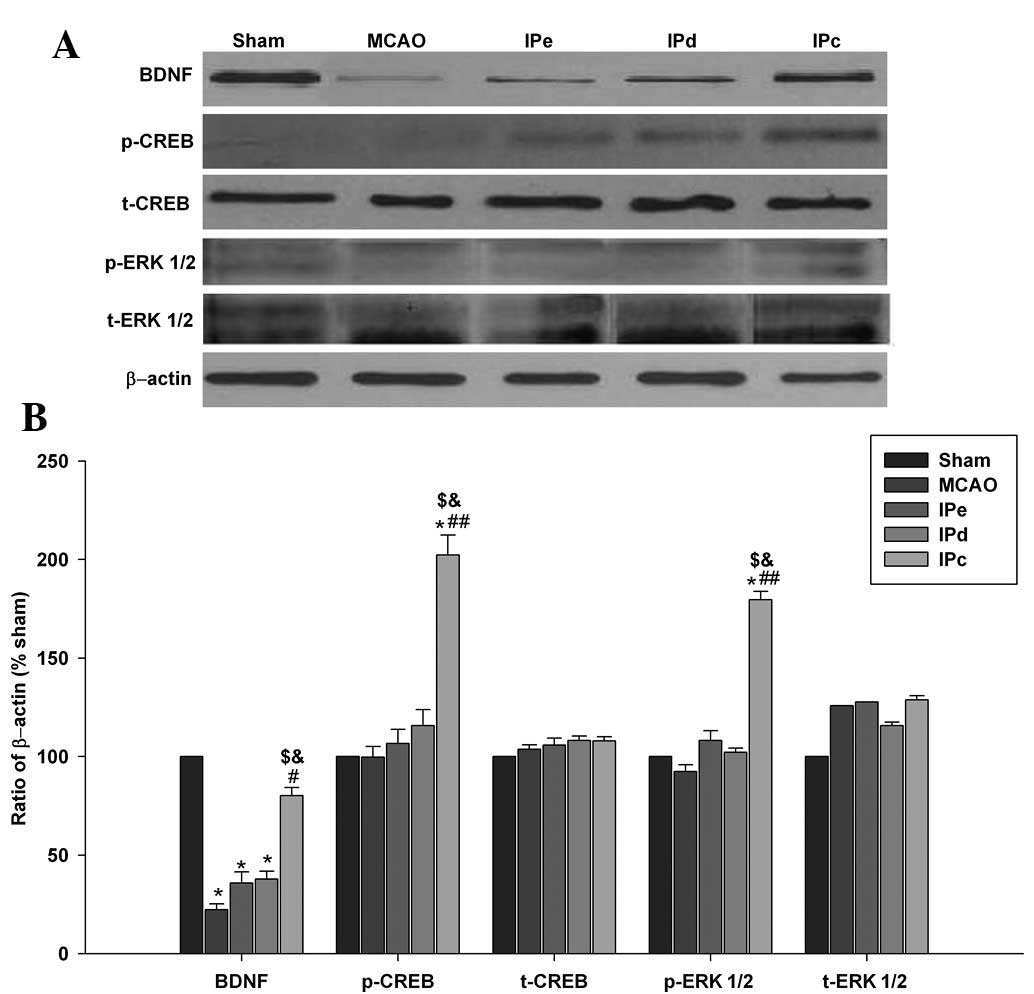 | Figure 5Combination of IPe and IPd upregulated
the protein expression levels of BDNF, p-CREB and p-ERK1/2. (A)
Protein expression levels of BDNF, p-CREB, t-CREB, p-ERK1/2 and
t-ERK1/2 in the sham, MCAO, IPe, IPd and IPc rats. (B) Optical
density analysis of BDNF, p-CREB, t-CREB, p-ERK1/2 and t-ERK1/2
expression. Data are presented as the mean ± standard error of the
mean; n=9 per group. *P<0.05, compared with the sham
group; #P<0.05 or ##P<0.01, compared
with the MCAO group; $P<0.05 compared with the IPe
group; and &P<0.05, compared with the IPd group.
BDNF, brain-derived neurotrophic factor; p, phosphorylated; t,
total; CREB, cAMP response element-binding protein; ERK 1/2,
extracellular signal-regulated kinases 1/2; MCAO, middle cerebral
artery occlusion; IPe, early ischemic postconditioning; IPd,
delayed ischemic postconditioning; IPc, combinative ischemic
postconditioning. |
Discussion
The results of the present study demonstrated that
the combination of early and delayed ischemic postconditioning
reduced infarct volume and stabilized CBF disturbance in rats,
following focal brain ischemia, compared with no treatment, as well
as with early or late postconditioning treatment alone. The
potential neuroprotective mechanisms underlying the protective
effect of combinative ischemic postconditioning against focal
ischemic injury may involve the reduction of neuronal apoptosis in
the penumbral area. Furthermore, it was demonstrated that
combinative ischemic postconditioning upregulated the expression of
BDNF, which aids the repair of neuronal injury in the central
nervous system. In addition, the majority of BDNF-positive cells
were detected by staining with either anti-NeuN or anti-GFAP.
Combinative ischemic postconditioning also activated ERK1/2 and
CREB in the cortex, following focal ischemia. These results
suggested that combinative ischemic postconditioning induces BDNF
expression in neurons and astrocytes, and protects against
neurological injury following brain ischemia, which may be mediated
by the phosphorylation of ERK1/2 and CREB.
It has been established that numerous
neuroprotectants, which are highly effective in animal models of
stroke, fail in clinical trials due to side effects, which
frequently result in premature termination of a trial (e.g.,
MK-801, ZK200755) (15–17). Previous studies have aimed to
overcome this limitation by developing methods that induce, mimic
or enhance endogenous protective responses, and do not interfere
with physiological neurotransmission (18–20).
Therefore, endogenous neuroprotection is a novel strategy that has
been increasingly focused on within stroke research (21).
Ischemic conditioning has been suggested as a useful
method with which to generate ischemic tolerance or endogenous
neuroprotection (20). There are
two types of ischemic conditioning: Ischemic preconditioning and
ischemic postconditioning. Ischemic preconditioning may be induced
by the application of transient episodes of nonlethal ischemia
prior to stroke (22). Ischemic
postconditioning is defined as numerous repeated cycles of brief
reperfusion and re-occlusion, which may protect against lethal
ischemic injury and subsequent reperfusion damage (23). Currently, both of these types of
ischemic conditioning are hypothesized to be possible therapeutic
strategies. However, the translational application of ischemic
preconditioning is hindered by the fact that it can only be applied
for strokes or ischemic injuries that may be predicted (24,25).
Therefore, ischemic postconditioning, which can be applied after
ischemia, may be a better approach.
Recently, it has been demonstrated that two types of
postconditioning, early ischemic postconditioning (26) and delayed ischemic postconditioning
(23), exhibit neuroprotective
effects, and may reduce neuronal injury following brain ischemia.
However, there are few reports regarding the neuroprotective
effects of a combination of these two types of postconditioning.
Therefore, the aim of the present study was to evaluate the effects
of combined early and delayed ischemic postconditioning. In the
present study, early ischemic postconditioning or delayed ischemic
postconditioning each reduced the infarct volume following brain
ischemia. However, combinative ischemic postconditioning further
reduced infarct volume (P<0.05), compared with either approach
alone. Disturbances to CBF occur throughout the period of
reperfusion. Following reperfusion, there is a short period of
hyperperfusion, followed by a longer period of hypoperfusion. The
results of the present study indicated that combinative ischemic
postconditioning may stabilize CBF disturbances during the early
hyperperfusion and later hypoperfusion periods.
BDNF is considered an important member of the
neurotrophin family, which contributes to the maintenance and
survival of neurons (27), and
stimulates the differentiation and growth of new neurons and
synapses (28). The primary role
of BDNF is associated with increased long-term potentiation and
neurogenesis (29). Numerous
studies have shown that BDNF decreases infarct volume and improves
neurological outcomes, when exogenously administered (30) or when overexpressed using genetic
methods in vivo (31) in
experimental models of stroke. However, the mechanisms underlying
the effects of ischemic postconditioning on the production of BDNF
remain unclear. The present study used immunofluorescence staining
and western blot analysis to detect BDNF expression in the brain
penumbral area following focal brain ischemia. The results
indicated that neither early nor delayed ischemic postconditioning
significantly increased the expression levels of BDNF. However,
combinative ischemic postconditioning upregulated the expression
levels of BDNF in neuronal cells and astrocytes.
Additional mechanisms of combinative ischemic
postconditioning were hypothesized to involve CREB, a transcription
factor of BDNF. Furthermore, ERK1/2, which is the upstream
phosphorylating enzyme of CREB, activates and phosphorylates CREB
at Ser133 (32), resulting in the
upregulation of pro-survival CREB target genes, including BDNF
(33,34). Therefore, in the present study
western blotting was used to detect the protein expression levels
of CREB and ERK1/2. The results demonstrated that no difference in
the expression of total CREB and ERK1/2 protein among the groups.
However, combinative ischemic postconditioning significantly
increased the protein expression levels of p-CREB and p-ERK1/2 in
the penumbral area following focal brain ischemia.
The results of the present study demonstrated that a
combination of early and delayed ischemic postconditioning had
stronger neuroprotective effects on focal brain ischemia, compared
with early or delayed ischemic postconditioning alone. This effect
may be associated with the stabilization of CBF disturbances and a
reduction in apoptosis in the penumbral area following focal brain
ischemia. Furthermore, it was indicated that combinative ischemic
postconditioning upregulated the expression of BDNF, in neurons and
astrocytes, and protected against neurological damage following
brain ischemic injury. These effects may be associated with
activation of ERK1/2 and CREB. Although there were some limitations
to the present study, such as the use of only one model of
combinative postconditioning and the fact that the neuroprotective
effects were examined using only a rat model, combinative ischemic
postconditioning appeared to alleviate or even prevent ischemic
brain injury. Therefore, combinative ischemic postconditioning has
the potential for future clinical application and requires further
investigation.
Acknowledgments
The present study was supported by the National
Nature Science Foundation (grant no. 81000498).
References
|
1
|
Blanco M and Castillo J: Stroke in 2012:
Major advances in the treatment of stroke. Nat Rev Neurol. 9:68–70.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu J, Xu Q, Wang H, et al:
Neuroprotection of ischemic post-conditioning by downregulating the
postsynaptic signaling mediated by kainate receptors. Stroke.
44:2031–2035. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sandu N and Schaller B: Postconditioning:
A new or old option after ischemic stroke? Expert Rev Cardiovasc
Ther. 8:479–482. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhao H: Ischemic postconditioning as a
novel avenue to protect against brain injury after stroke. J Cereb
Blood Flow Metab. 29:873–885. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Danielisova V, Burda J, Nemethova M, et
al: An effective combination of two different methods of
postconditioning. Neurochem Res. 37:2085–2091. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhao H, Ren C, Chen X and Shen J: From
rapid to delayed and remote postconditioning: The evolving concept
of ischemic postconditioning in brain ischemia. Curr Drug Targets.
13:173–187. 2012. View Article : Google Scholar :
|
|
7
|
Dauncey MJ: Recent advances in nutrition,
genes and brain health. Proc Nutr Soc. 71:581–591. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu B, Nagappan G, Guan X, et al:
BDNF-based synaptic repair as a disease-modifying strategy for
neurodegenerative diseases. Nat Rev Neurosci. 14:401–416. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Melo CV, Okumoto S, Gomes JR, et al:
Spatiotemporal resolution of BDNF neuroprotection against glutamate
excitotoxicity in cultured hippocampal neurons. Neuroscience.
237:66–86. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Singh M and Su C: Progesterone,
brain-derived neurotrophic factor and neuroprotection.
Neuroscience. 239:84–91. 2013. View Article : Google Scholar :
|
|
11
|
Alboni S, Tascedda F, Corsini D, et al:
Stress induces altered CRE/CREB pathway activity and BDNF
expression in the hippocampus of glucocorticoid receptor-impaired
mice. Neuropharmacology. 60:1337–1346. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pardon MC, Roberts RE, Marsden CA, et al:
Social threat and novel cage stress-induced sustained
extracellular-regulated kinase1/2 (ERK1/2) phosphorylation but
differential modulation of brain-derived neurotrophic factor (BDNF)
expression in the hippocampus of NMRI mice. Neuroscience.
132:561–574. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yifeng M, Bin W, Weiqiao Z, et al:
Neuroprotective effect of sophocarpine against transient focal
cerebral ischemia via down-regulation of the acid-sensing ion
channel 1 in rats. Brain Res. 1382:245–251. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thiele H, Paetsch I, Schnackenburg B, et
al: Improved accuracy of quantitative assessment of left
ventricular volume and ejection fraction by geometric models with
steady-state free precession. J Cardiovasc Magn Reson. 4:327–339.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Diener HC, Lees KR, Lyden P, et al: SAINT
I and II Investigators: NXY-059 for the treatment of acute stroke:
Pooled analysis of the SAINT I and II Trials. Stroke. 39:1751–1758.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Furlan AJ: Challenges in acute ischemic
stroke clinical trials. Curr Cardiol Rep. 14:761–766. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shuaib A, Lees KR, Lyden P, et al: SAINT
II Trial Investigators: NXY-059 for the treatment of acute ischemic
stroke. N Engl J Med. 357:562–571. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ducruet AF, Zacharia BE, Sosunov SA, et
al: Complement inhibition promotes endogenous neurogenesis and
sustained anti-inflammatory neuroprotection following reperfused
stroke. PloS One. 7:e386642012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Iadecola C and Anrather J: Stroke research
at a crossroad: Asking the brain for directions. Nat Neurosci.
14:1363–1368. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ding ZM, Wu B, Zhang WQ, et al:
Neuroprotective effects of ischemic preconditioning and
postconditioning on global brain ischemia in rats through the same
effect on inhibition of apoptosis. Int J Mol Sci. 13:6089–6101.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Y, Reis C, Applegate R, et al:
Ischemic conditioning-induced endogenous brain protection:
Applications pre-, per- or post-stroke. Exp Neurol. 15:123–125.
2015.
|
|
22
|
Liu XQ, Sheng R and Qin Z: The
neuroprotective mechanism of brain ischemic preconditioning. Acta
Pharmacol Sin. 30:1071–1080. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ren C, Gao X, Niu G, et al: Delayed
postconditioning protects against focal ischemic brain injury in
rats. PloS One. 3:e38512008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang JY, Shen J, Gao Q, et al: Ischemic
postconditioning protects against global cerebral
ischemia/reperfusion-induced injury in rats. Stroke. 39:983–990.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xing B, Chen H, Zhang M, et al: Ischemic
postconditioning inhibits apoptosis after focal cerebral
ischemia/reperfusion injury in the rat. Stroke. 39:2362–2369. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu C, Weaver J and Liu KJ: Early
conditioning with oxygen oscillation: Neuroprotection by
intermittent normobaric hyperoxia after transient focal cerebral
ischemia in rats. Stroke. 43:220–226. 2012. View Article : Google Scholar
|
|
27
|
Stahl K, Mylonakou MN, Skare Ø, et al
Cytoprotective effects of growth factors: BDNF more potent than
GDNF in an organotypic culture model of Parkinson's disease. Brain
Res. 1378:105–118. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Baldelli P, Novara M, Carabelli V, et al:
BDNF up-regulates evoked GABAergic transmission in developing
hippocampus by potentiating presynaptic N− and P/Q-type
Ca2+ channels signalling. Eur J Neurosci. 16:2297–2310.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Babu H, Ramirez-Rodriguez G, Fabel K, et
al: Synaptic network activity induces neuronal differentiation of
adult hippocampal precursor cells through BDNF signaling. Front
Neurosci. 3(49)2009.
|
|
30
|
Hyacinth HI, Gee BE, Adamkiewicz TV, et
al: Plasma BDNF and PDGF-AA levels are associated with high TCD
velocity and stroke in children with sickle cell anemia. Cytokine.
60:302–308. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chang DJ, Lee N, Choi C, et al:
Therapeutic effect of BDNF-overexpressing human neural stem cells
(HB1.F3.BDNF) in a rodent model of middle cerebral artery
occlusion. Cell Transplant. 22:1441–1452. 2013. View Article : Google Scholar
|
|
32
|
Cao G, Zhu J, Zhong Q, et al: Distinct
roles of methamphetamine in modulating spatial memory
consolidation, retrieval, reconsolidation and the accompanying
changes of ERK and CREB activation in hippocampus and prefrontal
cortex. Neuropharmacology. 67:144–154. 2013. View Article : Google Scholar :
|
|
33
|
Chen DY, Bambah-Mukku D, Pollonini G and
Alberini CM: Glucocorticoid receptors recruit the CaMKIIα-BDNF-CREB
pathways to mediate memory consolidation. Nat Neurosci.
15:1707–1714. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lesiak A, Pelz C, Ando H, et al: A
genome-wide screen of CREB occupancy identifies the RhoA inhibitors
Par6C and Rnd3 as regulators of BDNF-induced synaptogenesis. PloS
One. 8:e646582013. View Article : Google Scholar : PubMed/NCBI
|














