Introduction
Colorectal cancer (CRC) is one of the most common
types of malignant cancer and is the third leading cause of
cancer-associated mortality worldwide (1). As is common with all cancers, the
overwhelming cause of fatality in patients with CRC is metastasis,
a complex series of multiple sequential steps during which primary
cancer cells acquire an invasive phenotype enabling them to
translocate from the primary tumor to a distant organ and form a
secondary tumor (2). Tumor
invasion and metastasis are parts of a complicated process in which
the tumor grows, then detaches from the primary site and
metastasizes to a distant organ. Aberrations in protein-coding
genes have been widely accepted to be critical in the pathology of
CRC, including oncogenes and tumor suppressor genes (3). The underlying molecular mechanism of
metastasis is yet to be elucidated. Thus, there is a growing need
to understand the molecular pathogenesis underlying CRC. Over the
last decade, microRNAs (miRNAs) have emerged as key players in
carcinogenesis. Aberrant expression of miRNAs has been demonstrated
to be critical in the initiation and progression of several types
of human cancer through post-transcriptional regulation of gene
expression (4–6).
miRNAs are emerging as regulators of metastasis by
acting on multiple signaling pathways (7). miRNAs are noncoding RNAs of 18–24
nucleotides that inhibit translation or induce mRNA decay through
binding to the 3′-untranslated regions (3′-UTRs) of their target
RNAs (8,9), influencing processes such as tumor
cell proliferation, differentiation, apoptosis and invasion
(4,7,10).
Recently, a series of studies has revealed that miRNAs regulate
various genes that are pivotal in the process of tumor progression
(11,12). It has specifically been shown that
miRNAs are important in the development of CRC. In CRC miRNAs
exhibit oncogenic or tumor suppressive roles by directly regulating
oncogenes or tumor-suppressor genes (13,14).
Several studies have indicated the role of miRNAs in regulating
tumor invasion and metastasis in CRC. For example, miR-133a
represses tumor growth and metastasis in CRC by targeting LIM and
SH3 protein 1 (LASP1) and inhibiting the mitogen-activated protein
kinase pathway (15).
Additionally, miR-132 inhibits CRC invasion and metastasis via
directly targeting ZEB2 (16).
miR-103/107 promotes metastasis of CRC by targeting the metastasis
suppressors Death-associated protein kinase (DAPK) and Kruppel-like
factor 4 (KLF4) (17). However,
miR-18a has instead been reported to suppress cell proliferation in
T24 bladder cancer cells (18).
Recently, the diagnostic value of circulating miR-18a in the plasma
was reported for pancreatic cancer (19). Therefore, miR-18a may exhibit
different roles in various types of cancer through different
targeted genes. However, the role of miR-18a in CRC progression and
metastasis remains largely unclear.
The present study aimed to examine the role of
miR-18a in the progression of CRC, and, to clarify the molecular
mechanism by which miR-18a promotes malignant tumor progression.
Efforts to elucidate the association between miR-18a and TBPL1 are
also included in the present study. In addition, the present study
aimed to provide a novel miRNA and target gene for cancer
therapy.
Materials and methods
Cell lines and clinical specimens
Human SW620 and HCT116 CRC cell lines were obtained
from the Cell Bank of the Type Culture Collection (Shanghai,
China). All cell lines were cultured in Dulbecco's modified Eagle's
medium (DMEM) supplemented with 10% fetal bovine serum (Gibco Life
Technologies, Carlsbad, CA, USA) in a humidified 37°C incubator
supplemented with 5% CO2. The medium was changed every
2–3 days, and the cells were trypsinized (Mingzhu Chemical Co.,
Shanghai, China) when 80–90% confluence was reached.
Twenty-five CRC tissue samples and adjacent normal
colon tissue samples were collected directly after surgical
resection at the First Affiliated Hospital of Zhengzhou University
(Zhengzhou, China). The NCM460 normal colonic epithelial cell lines
were purchased from the American Type Culture Collection (Manassas,
VA, USA). All patients had a histological diagnosis of CRC.
Following resection, all specimens were immediately frozen in
liquid nitrogen and stored at −80°C until RNA extraction. Approval
for this study was obtained from the Ethics Review Committee of the
Institutional Review Board of the First Affiliated Hospital of
Zhengzhou University and informed consent was obtained from each
patient.
RNA isolation and RT-qPCR analysis
Total RNA from the tissues and cells was extracted
using TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA,
USA) according to the manufacturer's instructions. RNA quality and
concentration were determined using the Nanodrop 2000 system
(Thermo Fisher Scientific, Wilmington, NC, USA). To analyze gene
expression levels, 5 µg RNA was reverse transcribed to cDNA
using a PrimeScript RT Reagent kit (Takara Biotechnology Co., Ltd.,
Dalian, China). Mature miR-18a and U6 levels were quantified with
TaqMan miRNA assays (Applied Biosystems, Foster City, CA, USA).
TBPL1 and β-actin mRNA levels were determined by RT-qPCR using the
SYBR Green Master mix on the HT 7500 System (Applied Biosystems).
The PCR primers for TBPL1 were 5′-CCTCTTCCCACGGATGTGAT-3′ (sense)
and 5′-GAGTCCAATGTGCAGCAGT-3′ (reverse). The qPCR cycle was as
follows: 98°C for 2 min, 40 cycles of 95°C for 15 s and 60°C for 30
s. The final melting curve analysis (60–95°C) was included. The
relative expression levels of each gene were calculated and
normalized using the 2−ΔΔCt method relative to U6 or
β-actin. All of the reactions were run in triplicate.
Western blot analysis
Cells were lysed with radioimmuno-precipitation
assay lysis buffer containing protease inhibitors (Roche,
Indianapolis, IN, USA). Protein concentrations were determined
using a Pierce Bicinchoninic Acid Protein Assay kit (Thermo Fisher
Scientific, Logan, UT, USA). Equal quantities of proteins were
loaded and separated on 10% SDS-PAGE and then transferred to a
polyvinylidene difluoride membrane (Millipore, Billerica, MA, USA).
Non-specific protein interactions were blocked by incubation with
3% fat-free milk in Tris-buffered saline buffer (containing 150 mM
NaCl and 50 mM Tris-HCl, pH 7.6) at 4°C for 1 h. The membranes were
incubated with the following primary antibodies at 4°C overnight:
Rabbit anti-human TBPL1 polyclonal antibody (cat. no. ABIN1875056;
1/1,000; Antibodies Online, Atlanta, GA, USA) and mouse anti-human
GAPDH monoclonal antibody (sc-365062; 1/2,000; Santa Cruz
Biotechnology Inc., Santa Cruz, CA, USA). Unbound antibody was
removed by washing in TBS with Tween-20 (Sigma-Aldrich, St. Louis,
MO, USA) buffer (TBST) three times (10 min/wash). The membranes
were then incubated with horseradish peroxidase-conjugated
secondary antibodies (Zhongshan, Beijing, China) at 25°C for 1 h,
followed by washing with TBST buffer three times (10 min/wash). The
blots were developed with the enhanced chemiluminescence reagent
(Millipore). GAPDH was used as an internal control.
Oligonucleotide transfection
Cells were transfected with oligonucleotides using
Lipofectamine 2000 (Invitrogen Life Technologies) according to the
manufacturer's instructions. The miR-18a mimic, miR-18a inhibitor
and negative control (NC) oligonucleotides were purchased from
Ribobio (Guangzhou, China). The cells were plated in a six-well
plate one day prior to transfection. All cell transfections were
performed with the miR-18a mimic (miR-18a inhibitor) or NC (50
nmol/l). Transfections were conducted in triplicate. Cells were
observed by inverted microscopy (Nikon TE 300, Nikon Inc.,
Melville, NY, USA) 24 h after transfection. Oligonucleotide
expression was confirmed by RT-qPCR.
Methylthiazol tetrazolium (MTT)
assay
An MTT assay was performed to quantify CRC cell
viability following transfection with the miR-18a mimic, miR-18a
inhibitor or NC. Following incubating the cells for 36 h at 37°C in
a humidified 5% CO2 atmosphere, the cells were stained
with 100 µl sterile 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl
tetrazolium bromide (0.5 mg/ml; Sigma-Aldrich) for 4 h at 37°C. The
culture medium was then removed and formazan crystals in the cells
were solubilized using dimethyl sulfoxide (Sigma-Aldrich) with
shaking of the plate for 30 min. The absorbance was measured at 570
nm with a spectrophotometer (Bio-Rad Laboratories, Hercules, CA,
USA). All experiments were performed in triplicate.
Cell invasion assay
Cell invasion was determined using 24-well Transwell
chambers coated with Matrigel (BD Biosciences, Bedford, MD, USA).
The cells (5×104 cells suspended in 500 µl of
serum-free medium) were added to the upper chamber of the inserts,
which were coated with a Matrigel mix; fetal bovine serum (500
µl) was added to the bottom chamber as a chemoattractant.
After 24 h, the non-invading cells on the upper surface were
removed and the cells that had invaded the bottom side of the
membrane were fixed with methanol, stained with 0.1% crystal violet
(Shanghai Chemical Reagent Company, Shanghai, China), air dried and
subjected to digital image acquisition Olympus BX51 fluorescence
microscope (Olympus Optical Co., Tokyo, Japan). The number of
invasive cells was evaluated in five independent fields under a
microscope. The mean of triplicate assays for each experimental
condition was determined.
Wound healing assay
After transfection with the miR-18a mimic, miR-18a
inhibitor or NC (5.0×105 cells per well), the cell lines
were seeded into 6-well plates (Corning, Corning, NY, USA) for 24 h
and allowed to grow to 90% confluence. Linear scratch wounds were
then created (in triplicate) on the confluent monolayer using a
pipette tip and the cells were cultured in the same medium without
FBS. Immediately after wounding (time 0) and at 48 h, images were
captured using a digital camera mounted on a light microscope
(Axiophot; Carl Zeiss, Oberkochen, Germany). The width of the wound
gap was measured using NIH Image J 1.61 analysis (National
Institutes of Health, Bethesda, MA, USA) and normalized to the
width at time 0 for four independent experiments.
Vector construction and dual-luciferase
reporter assay
The potential miR-18a targets were predicted and
analyzed using three publicly available algorithms: PicTar
(http://www.pictar.org/cgi-bin/PicTar_vertebrate.cgi),
TargetScan (http://www.targetscan.org/) and microRNA.org (http://www.microrna.org/microrna/home.do). The number
of false-positive results was decreased by accepting only putative
target genes that were predicted by at least two programs.
The 1.77 kb 3′-untranslated region (UTR) of human
TBPL1 was amplified from a human cDNA library using PCR and cloned
into a pGL3 Vector (Promega Corporation, Madison, WI, USA) to
generate the Wt-pGL3-TBPL1-3′-UTR vector (WT vector). The following
primers were used: Forward: 5′-CTCTCTAGACTCAAAAGAAAACTGGACCAAC-3′;
and reverse: 5′-CTCTCTAGATGCACATTCAATTGAAAAA-3′. Site-directed
mutagenesis of the miR-18a target site in the TBPL1 3′-UTR was
performed using mutation primers to generate the
Mut-pGL3-TBPL1-3′-UTR vector (Mut vector). After cells were plated
in a 12-well plate at ~80% confluence, the experiments were
conducted in two parts. Firstly, cells were co-transfected with the
WT vector and 30 nM miRNA NC, miR-18a mimic or miR-18a inhibitor.
In addition, cells were co-transfected with miR-18a mimic and the
pGL3 vector, WT vector or Mut vector. Each sample was also
co-transfected with 0.05 µg phRL-null plasmid expressing
Renilla luciferase (Promega Corporation) as an internal
control for transfection efficiency. Cell transfection was
performed using Lipofectamine 2000 (Invitrogen Life Technologies)
according to the manufacturer's instructions. A luciferase assay
was performed 48 h after transfection using the Dual Luciferase
Reporter Assay system (Promega Corporation), and each experiment
was repeated in triplicate.
Statistical analysis
Data are expressed as the mean ± standard deviation
from at least three independent experiments. The significance
between groups was analyzed using Student's t-test. All statistical
analyses were performed using SPSS 13.0 (SPSS, Inc., Chicago, IL,
USA). Spearman's correlation test or the χ2 test were
also conducted. P<0.05 was considered to indicate a
statistically significant difference.
Results
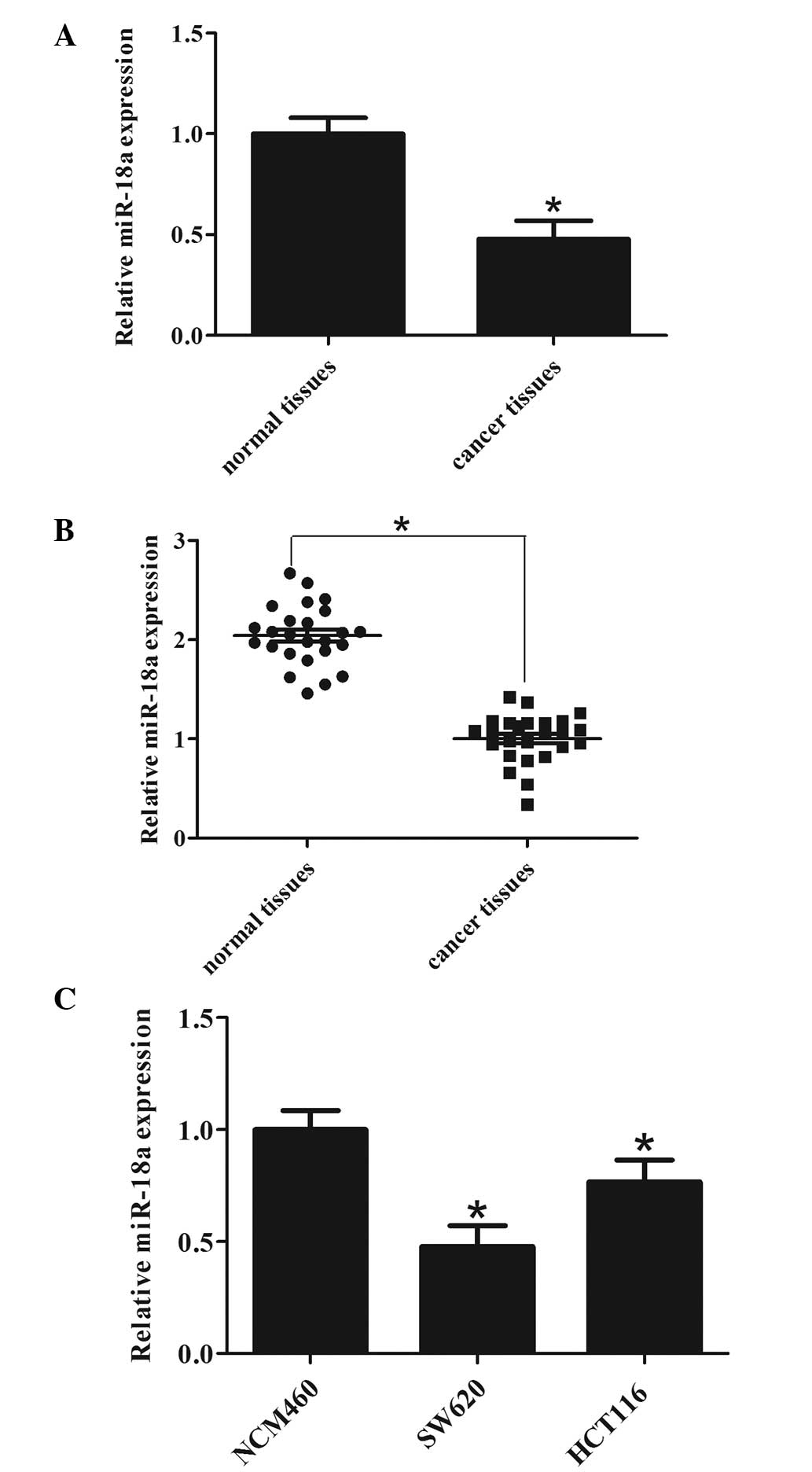
miR-18a is significantly downregulated in
CRC cell lines and CRC tissues
According to the miRNA array data from the patients
with CRC, miR-18a was found to be significantly downregulated in
CRC tissues compared with their matched normal tissues (Fig. 1A). To confirm this, miR-18a levels
were measured in the 25 CRC tissues by RT-qPCR. The results showed
that miR-18a was downregulated in cancer tissues compared with
normal controls (Fig. 1B). To
further confirm that this phenomenon is consistent and common in
CRC cell lines, the expression of miR-18a in two CRC cell lines and
a NCM460 normal colonic cell line was detected by RT-qPCR. As shown
in Fig. 1C, miR-18a was
downregulated in CRC cell lines compared with the NCM460 normal
colonic cell line. These data revealed that the expression of
miR-18a was universally downregulated in CRC cells and in patients
with CRC, indicating that increased miR-18a expression may
contribute to the malignant phenotype and development of the
tumor.
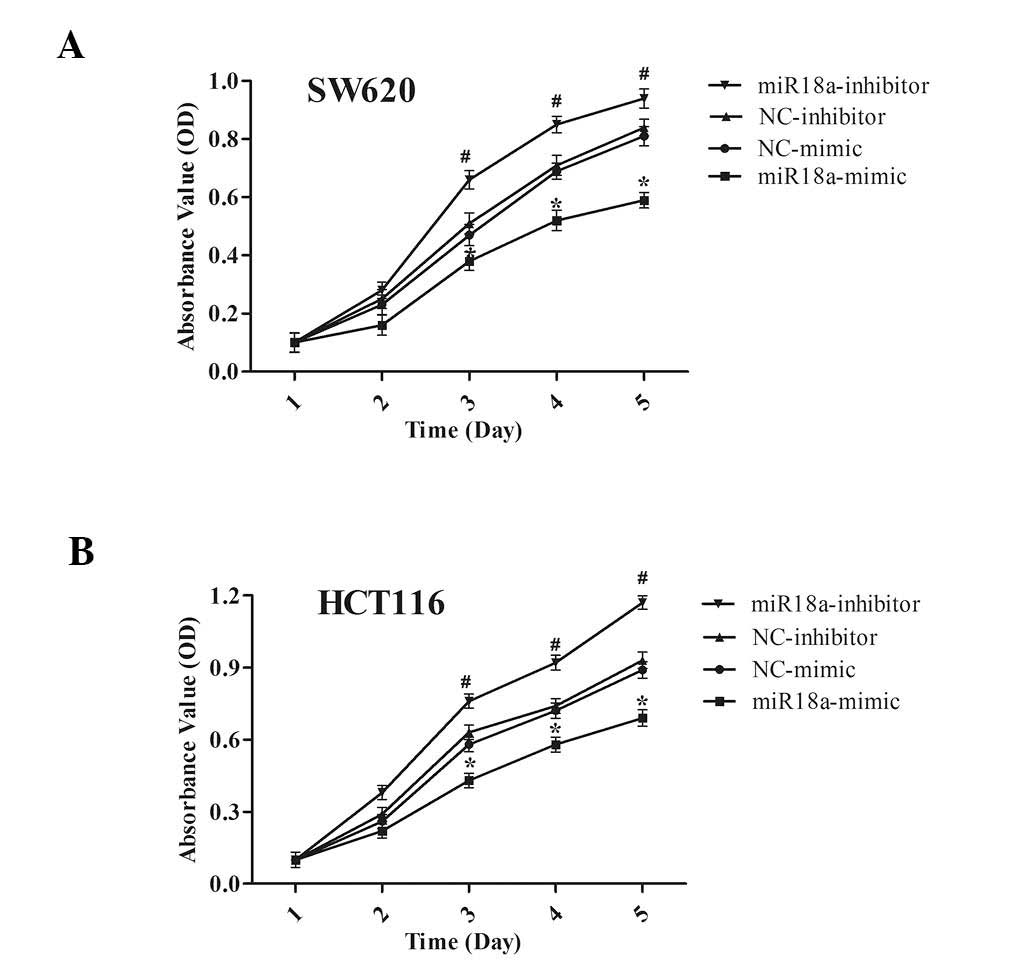
miR-18a inhibits CRC cell
proliferation
To investigate whether miR-18a is involved in the
progression of CRC, an MTT assay was conducted to determine the
effect of miR-18a on cell proliferation in SW620 and HCT116 cells
transiently transfected either with miR-18a mimic or inhibitor. It
was revealed that miR-18a inhibited cell proliferation in these two
cell lines. As shown in Fig. 2,
the increased expression of miR-18a significantly suppressed cell
proliferation in SW620 (Fig. 2A)
and HCT116 (Fig. 2B) cells
(P<0.05). Correspondingly, after transfection with miR-18a
inhibitors, cell proliferation was increased compared with the
control group. These findings demonstrate that miR-18a expression
significantly reduced the proliferation of SW620 and HCT116
cells.
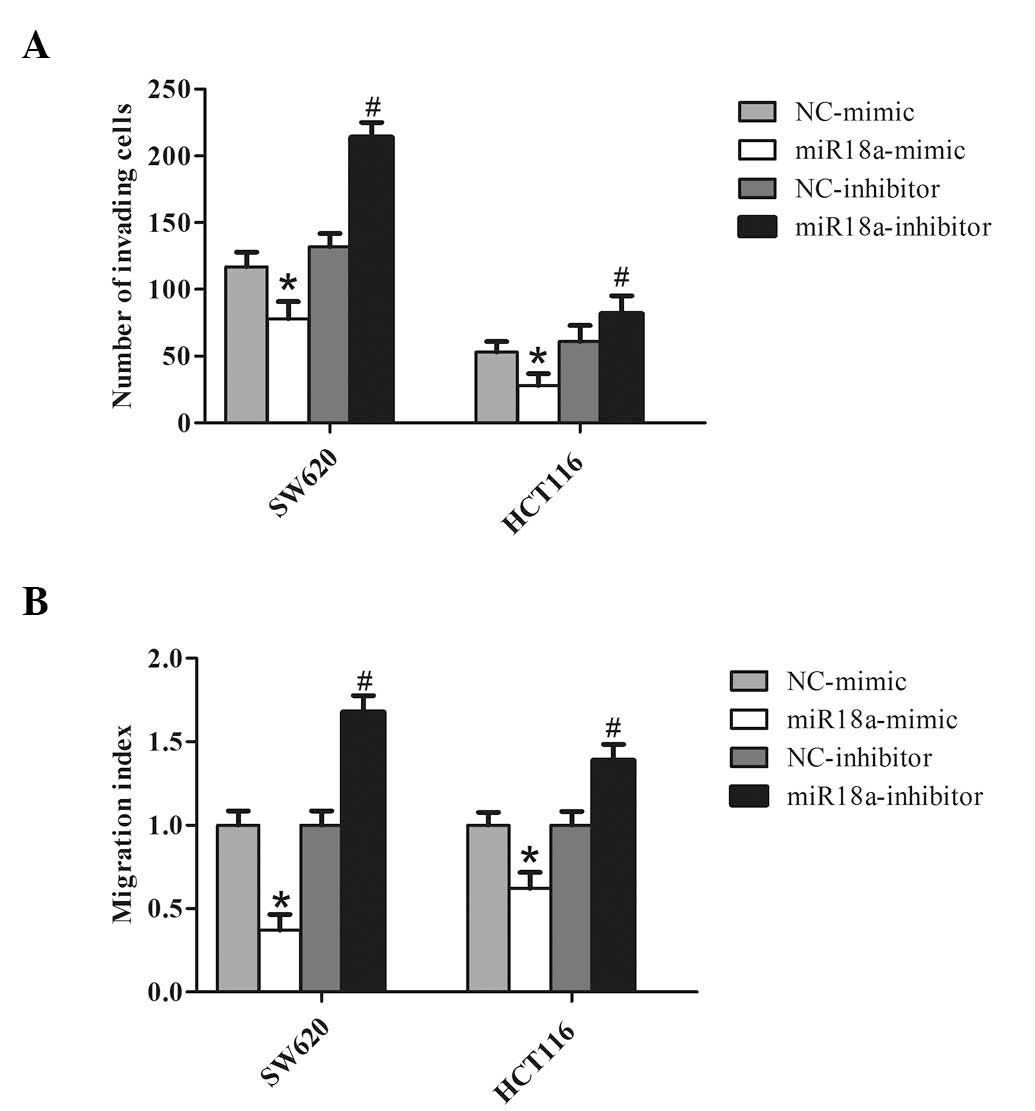
miR-18a suppresses CRC cell invasion and
migration in vitro
In light of the findings described previously, it
was hypothesized that miR-18a could inhibit cell invasion by CRC.
Two cell lines, SW620 and HCT116, were transfected with the miR-18a
mimic to overexpress miR-18a, or transfected with the miR-18a
inhibitor to knockdown endogenous miR-18a. As expected,
overexpression of miR-18a decreased CRC cell invasion. By contrast,
knockdown of miR-18a significantly increased the invasiveness of
the CRC cells (Fig. 3A).
Similarly, the wound healing assay demonstrated that overexpression
of miR-18a markedly reduced the migration of CRC cells, and
knockdown of miR-18a in CRC cells increased wound healing (Fig. 3B). These observations suggest that
miR-18a significantly promotes in vitro migration and
invasion of CRC cells.
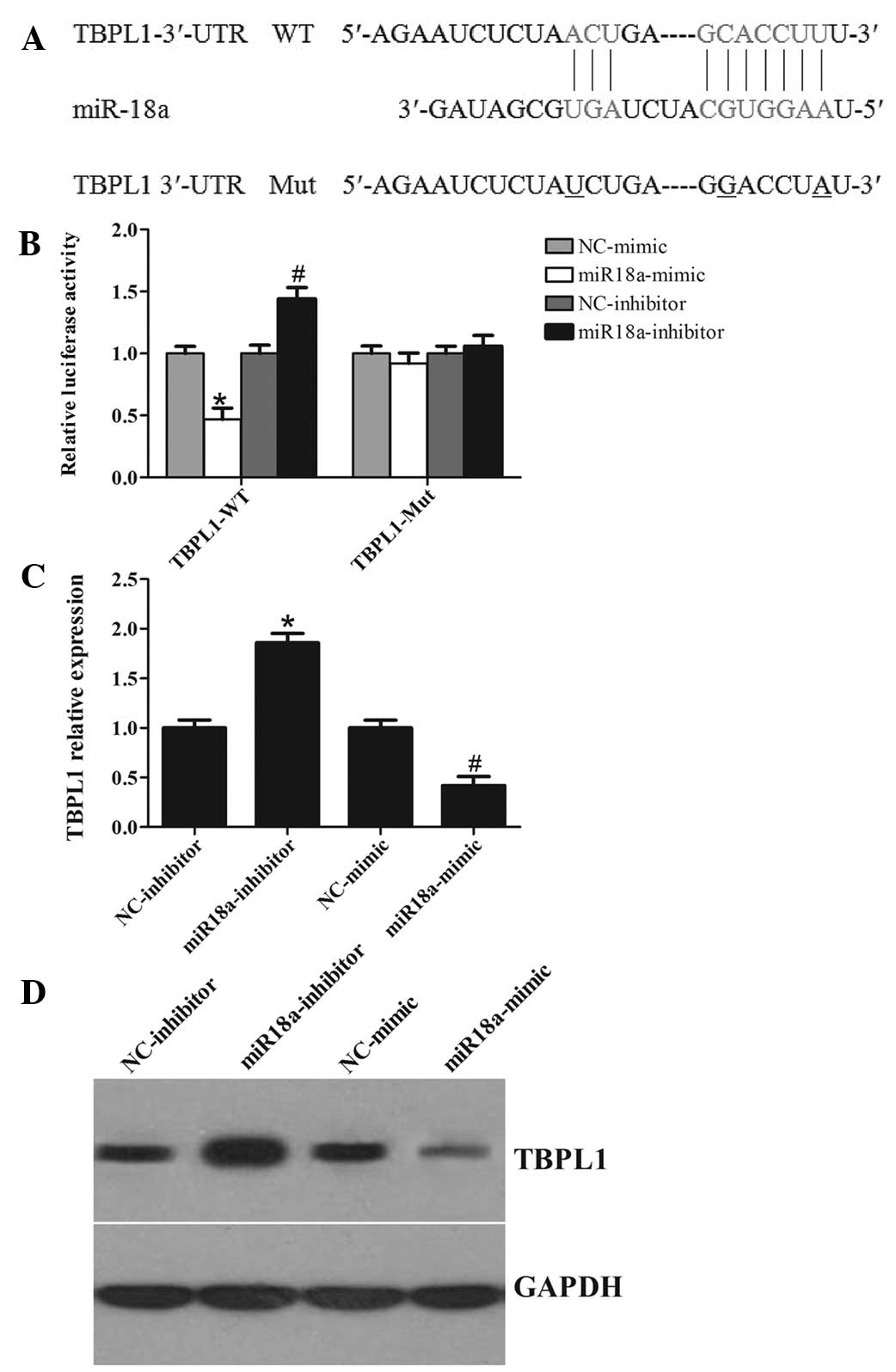
TBPL1 is identified as a direct target of
miR-18a
It is well-known that miRNAs exert their function
through suppressing the expression of their target genes. To better
understand the molecular mechanisms of miR-18a, bioinformatics
analyses was performed using the miRNA target analysis tools
PicTar, TargetScan and microRNA.org in
order to predict putative miRNAs binding to the TBPL1 3′-UTR.
According to the analysis, all three programs predicted that a
binding sequence in the 3′-UTR of TBPL1 was a good match for the
miR-18a seed (Fig. 4A). The
bioinformatics analysis thus indicated a potential functional link
between TBPL1 and miR-18a.
Furthermore, a luciferase reporter assay was
performed to further confirm whether miR-18a can directly target
the 3′-UTR region of TBPL1 in CRC cells. The TBPL1 wild-type 3′-UTR
containing the miR-18a-binding site and a mutated TBPL1 3′-UTR
sequence were cloned into modified pGL-3 luciferase reporter
vectors, which were co-transfected into SW620 cells together with
NC, the miR-18a mimic, or the miR-18a inhibitor. As shown in
Fig. 4B, miR-18a overexpression
significantly reduced the luciferase reporter activity by the TBPL1
3′-UTR in a consistent manner, and inhibition of miR-18a had the
opposite effect. However, TBPL1 3′-UTR luciferase reporter activity
was unaffected by point mutations in the miR-18a-binding seed
region. Collectively, these data suggest that miR-18a may inhibit
post-transcriptional TBPL1 expression by targeting its 3′-UTR. As
predicted, western blot analysis showed that, at 48 h after
transfection, the enhanced miR-18a in SW620 cells significantly
repressed TBPL1 protein expression compared with cells transfected
with a scrambled control. By comparison, downregulation of miR-18a
by inhibitors in SW620 cells led to a moderate increase in the
TBPL1 mRNA level (Fig. 4C). In
addition, apparent alterations in TBPL1 protein expression were
also observed (Fig. 4D). Together,
these data provide strong evidence that TBPL1 is a specific target
of miR-18a in CRC cells.
Discussion
CRC is a highly malignant tumor that leads to
>600,000 fatalities worldwide annually (20). Therefore, it is important to
investigate and validate novel biomarkers that are involved in CRC
development. Deregulation of miRNAs has been observed in various
types of human cancer (21,22);
however, the molecular mechanisms by which miRNAs modulate
carcinogenesis and cancer progression remain unclear. Metastasis is
the major reason for a poor prognosis in CRC, and increasing
evidence supports the contribution of miRNAs to cancer progression.
miRNAs have been found to be involved in the regulation of multiple
pathological processes that contribute to tumorigenesis. A number
of miRNAs have been identified that regulate tumor metastasis
(23,24). Recent studies have increasingly
linked microRNAs to CRC. Although aberrant expression of several
miRNAs occurs in CRC metastasis, such as miR-7 (25), miR-29a (26), miR-132 (16), miR-103 and miR-107 (17), and miR-143 (27), novel candidates with a definite
molecular mechanism that are involved in promoting CRC metastasis
require further investigation. The pathological role and regulatory
mechanism of miR-18a in the development and progression of
colorectal cancer still remain unclear.
In a previous study, overexpression of miR-18a in
the plasma of patients with pancreatic cancer as compared with
healthy individuals was revealed, suggesting that miR-18a may be
involved in pancreatic tumorigenesis (19). Conversely, it was reported that
miR-18a* can suppress K-Ras to inhibit tumor growth
(28). Recent findings have shown
high level expression of miR-18a in breast (29), bladder (18) and pancreatic (19) cancer. In the present study, miR-18a
expression was markedly downregulated in CRC cells and clinical
tissues samples compared with normal colonic cells and adjacent
noncancerous tissues from the same patient, respectively. Until
now, no functional evidence of such a role has been documented. The
present study aimed to identify the role of miR-18a in CRC by
investigating its effects on cell proliferation, migration and
invasion. It was demonstrated that overexpression of miR-18a could
effectively reduce CRC cell growth, migration and invasion in
vitro, whereas knockdown of miR-18a expression promoted CRC
cell proliferation, migration and invasion. Although the precise
mechanisms are not fully understood, these findings nevertheless
suggest that miR-18a may be important in the proliferation of
CRC.
Generally, miRNAs function as posttranscriptional
repressors that exert their biological function by suppressing
their target genes. Each miRNA can potentially downregulate a
number of target genes through binding their 3′-UTRs. In the
present study, predicted by bioinformatics analysis and validated
by luciferase reporter gene assay, TBPL1 was identified as the
major regulatory target of miR-18a. Thus, it was deduced that
miR-18a may function by interacting with the TBPL1 gene. Consistent
with this hypothesis, RT-qPCR and western blot analysis
demonstrated that miR-18a could decrease the expression of TBPL1 at
the mRNA and protein levels. In addition, inhibiting miR-18a
expression could increase TBPL1 expression. A luciferase assay also
showed that miR-18a could interact with the 3′-UTR of TBPL1. These
observations indicate that TBPL1 is a bona fide miR-18a-targeted
gene. Further investigations are required to identify and
characterize other miR-18a targets in order to fully elucidate the
functional role of miR-18a in CRC development, progression and
metastasis.
In conclusion, the results showed that miR-18a, acts
as an oncogenic miRNA, significantly reducing TBPL1 expression,
which subsequently inhibits CRC cell proliferation, migration and
invasion by its functional target TBPL1. The results of this study
indicate that miR-18a may exhibit a suppressive role in the
development and progression of CRC. To the best of our knowledge,
this study is the first to reveal a novel gene, TBPL1, as a target
for miR-18a in CRC. This may be used as a potential biomarker or
target for CRC diagnosis and chemotherapy. Further exploration of
the molecular link between miR-18a and TBPL1 may advance our
understanding of the intricate molecular regulation of CRC cells
and may provide novel targets for anticancer treatment.
References
|
1
|
Edwards BK, Ward E, Kohler BA, Eheman C,
Zauber AG, Anderson RN, Jemal A, Schymura MJ, Lansdorp-Vogelaar I,
Seeff LC, et al: Annual report to the nation on the status of
cancer, 1975–2006, featuring colorectal cancer trends and impact of
interventions (risk factors, screening and treatment) to reduce
future rates. Cancer. 116:544–573. 2010. View Article : Google Scholar
|
|
2
|
Chaffer CL and Weinberg RA: A perspective
on cancer cell metastasis. Science. 331:1559–1564. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wood LD, Parsons DW, Jones S, Lin J,
Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al: The
genomic landscapes of human breast and colorectal cancers. Science.
318:1108–1113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Croce CM: Oncogenes and cancer. New Engl J
Med. 358:502–511. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Deng S, Calin GA, Croce CM, Coukos G and
Zhang L: Mechanisms of microRNA deregulation in human cancer. Cell
Cycle. 7:2643–2646. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nicoloso MS, Spizzo R, Shimizu M, Rossi S
and Calin GA: MicroRNAs-the micro steering wheel of tumour
metastases. Nat Rev Cancer. 9:293–302. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
He L and Hannon GJ: MicroRNAs: Small RNAs
with a big role in gene regulation. Nat Rev Genet. 5:522–531. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bueno MJ, Pérez de Castro I and Malumbres
M: Control of cell proliferation pathways by microRNAs. Cell Cycle.
7:3143–3148. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qin J and Luo M: MicroRNA-221 promotes
colorectal cancer cell invasion and metastasis by targeting RECK.
FEBS Lett. 588:99–104. 2014. View Article : Google Scholar
|
|
12
|
Zhang Y, Zheng L, Huang J, Gao F, Lin X,
He L, Li D, Li Z, Ding Y and Chen L: MiR-124 radiosensitizes human
colorectal cancer cells by targeting PRRX1. PloS One. 9:e939172014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang N, Li X, Wu CW, Dong Y, Cai M, Mok
MT, Wang H, Chen J, Ng SS, Chen M, et al: microRNA-7 is a novel
inhibitor of YY1 contributing to colorectal tumorigenesis.
Oncogene. 32:5078–5088. 2013. View Article : Google Scholar
|
|
14
|
He X, Dong Y, Wu CW, Zhao Z, Ng SS, Chan
FK, Sung JJ and Yu J: MicroRNA-218 inhibits cell cycle progression
and promotes apoptosis in colon cancer by downregulating BMI1
polycomb ring finger oncogene. Mol Med. 18:1491–1498. 2013.
|
|
15
|
Wang H, An H, Wang B, Liao Q, Li W, Jin X,
Cui S, Zhang Y, Ding Y and Zhao L: miR-133a represses tumour growth
and metastasis in colorectal cancer by targeting LIM and SH3
protein 1 and inhibiting the MAPK pathway. Eur J Cancer.
49:3924–3935. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zheng YB, Luo HP, Shi Q, Hao ZN, Ding Y,
Wang QS, Li SB, Xiao GC and Tong SL: miR-132 inhibits colorectal
cancer invasion and metastasis via directly targeting ZEB2. World J
Gastroenterol. 20:6515–6522. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen HY, Lin YM, Chung HC, Lang YD, Lin
CJ, Huang J, Wang WC, Lin FM, Chen Z, Huang HD, et al: miR-103/107
promote metastasis of colorectal cancer by targeting the metastasis
suppressors DAPK and KLF4. Cancer Res. 72:3631–3641. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tao J, Wu D, Li P, Xu B, Lu Q and Zhang W:
microRNA-18a, a member of the oncogenic miR-17-92 cluster, targets
Dicer and suppresses cell proliferation in bladder cancer T24
cells. Mol Med Rep. 5:167–172. 2012.
|
|
19
|
Morimura R, Komatsu S, Ichikawa D,
Takeshita H, Tsujiura M, Nagata H, Konishi H, Shiozaki A, Ikoma H,
Okamoto K, et al: Novel diagnostic value of circulating miR-18a in
plasma of patients with pancreatic cancer. Br J Cancer.
105:1733–1740. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lu J, Getz G, Miska EA, Alvarez-Saavedra
E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA,
et al: MicroRNA expression profiles classify human cancers. Nature.
435:834–838. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Volinia S, Calin GA, Liu CG, Ambs S,
Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et
al: A microRNA expression signature of human solid tumors defines
cancer gene targets. Proc Natl Acad Sci USA. 103:2257–2261. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aigner A: MicroRNAs (miRNAs) in cancer
invasion and metastasis: Therapeutic approaches based on
metastasis-related miRNAs. J Mol Med (Berl). 89:445–457. 2011.
View Article : Google Scholar
|
|
24
|
White NM, Fatoohi E, Metias M, Jung K,
Stephan C and Yousef GM: Metastamirs: A stepping stone towards
improved cancer management. Nat Rev Clin Oncol. 8:75–84. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu K, Chen Z, Qin C and Song X: miR-7
inhibits colorectal cancer cell proliferation and induces apoptosis
by targeting XRCC2. OncoTargets Ther. 7:325–332. 2014. View Article : Google Scholar
|
|
26
|
Tang W, Zhu Y, Gao J, Fu J, Liu C, Liu Y,
Song C, Zhu S, Leng Y, Wang G, et al: MicroRNA-29a promotes
colorectal cancer metastasis by regulating matrix metalloproteinase
2 and E-cadherin via KLF4. Br J Cancer. 110:450–458. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang Y, Wang Z, Chen M, Peng L, Wang X,
Ma Q, Ma F and Jiang B: MicroRNA-143 targets MACC1 to inhibit cell
invasion and migration in colorectal cancer. Mol Cancer. 11:232012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tsang WP and Kwok TT: The
miR-18a* microRNA functions as a potential tumor
suppressor by targeting on K-Ras. Carcinogenesis. 30:953–959. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Song BJ, Matsunaga T, Hardwick JP, Park
SS, Veech RL, Yang CS, Gelboin HV and Gonzalez FJ: Stabilization of
cytochrome P450j messenger ribonucleic acid in the diabetic rat.
Mol Endocrinol. 1:542–547. 1987. View Article : Google Scholar : PubMed/NCBI
|