Introduction
Patients with severe acute exacerbation of chronic
hepatitis B virus (HBV) infection, including acute-on-chronic
pre-liver failure (pre-ACLF), are at risk for potential progression
to acute-on-chronic liver failure (ACLF) with high rates of
mortality (1,2). Therefore, it is important to
investigate therapies, which can prevent the progression of severe
acute episodes to liver failure in patients with chronic HBV. The
role of antiviral treatments, including nucleoside analogs in
preventing the progression of severe HBV to liver failure remains
to be elucidated. However, glucocorticoids are effective for the
treatment of pre-ACLF, severe and potentially life-threatening
exacerbation of chronic HBV and acute liver failure (3,4).
Studies have indicated that the mechanisms by which glucocorticoids
prevent the progression of liver disease include: (i) Prevention of
HBV-induced primary liver injuries by inhibiting excessive immune
responses; (ii) prevention of endotoxin-induced secondary liver
injuries by inhibiting the production of oxygen-free radicals and
cytokines (5); (iii) prevention of
cytolysis of ballooned hepatocytes by stabilizing the lysosomal
membrane, and inhibiting the production of lysosomal proteases and
circulating toxic substances (6);
and (iv) improvement in the functional activity of the residual
hepatocytes (7). However, the
mechanisms by which gluco-corticoid treatment alleviates various
liver conditions remain to be fully elucidated. Similar to hepatic
necrosis, apoptosis of hepatocytes is also a key feature of almost
all acute and chronic cases of HBV, including acute liver failure
(8). Therefore, investigating the
role of glucocorticoids and their underlying mechanism in
preventing the apoptosis of hepatocytes is necessary for improved
treatment of severe chronic HBV and acute liver failure associated
with HBV infection. Dexamethasone (DEX) is a synthetic
glucocorticoid, which inhibits apoptosis in certain cells,
including human neutrophils, hair cells, human fibroblasts and
primary rat hepatocytes; and induces apoptosis in other cells,
including thymocytes and lymphocytes (9,10).
DEX also enhances trichosanthin-induced apoptosis in the HepG2
hepatoma cell line (11). However,
the precise mechanism by which DEX inhibits or induces apoptosis
remains to be elucidated.
Multidrug resistant (MDR) P-glycoprotein (P-gp),
encoded by the ABCB1 gene, was first discovered in MDR tumor cells,
where it reduces the cellular accumulation of chemotherapeutic
agents (12,13). In addition to being expressed in
cancer cells, P-gps are also expressed in normal tissues, where
they have an important protective role in limiting the absorption
and/or facilitating the excretion of a wide range of substrates, by
actively transporting substrates from the inner to the outer
leaflets of the cell membranes (14). In addition to their ability to
discharge toxins, P-gps can also inhibit apoptosis, which is
induced by a wide array of cell death stimuli that rely on the
activation of intracellular caspases for complete functionality
(15–17). Therefore, the present study
hypothesized that the expression of P-gps in hepatocytes also have
an important hepatoprotective role. A number of studies have
reported that glucocorticoids, particularly DEX, are able to
improve the blood-brain barrier and placental barrier functions by
increasing the expression and function of P-gps (18,19).
DEX increases the expression of pregnane X receptor (PXR) at the
transcriptional level, and PXR mediates the spironolac-tone-induced
expression of P-gp in HepG2 cells (20).
Based on evidence that DEX and P-gps have the
ability to prevent the apoptosis induced by a wide array of cell
death stimuli, and that DEX alone induces the expression and
function of P-gps in several organs and cells; the present study
hypothesized that DEX prevents apoptosis in human hepatocytes by
upregulating the expression of P-gp. At present, few studies
detailing the role of P-gps in the inhibition of hepatocyte
apoptosis by DEX have been performed. The present study was
performed to determine the anti-apoptotic effects of DEX on the
L-02 normal human liver cell line, and to ascertain whether the
anti-apoptotic effects are associated with an upregulation in the
expression of P-gp. The results from these investigation aim to
establish whether DEX protects L-02 normal human liver cells from
tumor necrosis factor-related apoptosis-inducing ligand
(TRAIL)-induced apoptosis, by upregulating the expression of
P-gps.
Materials and methods
Materials
The L-02 normal human liver cell line was purchased
from the Shanghai Institute of Biochemistry and Cell Biology,
Chinese Academy of Sciences (Shanghai, China). RPMI 1640 medium was
purchased from Gibco Life Technologies (Carlsbad, CA, USA) and
newborn bovine serum was purchased from Lanzhou Minhai
Bioengineering Co., Ltd. (Lanzhou, China). Trypsin (0.25%) was
purchased from GE Healthcare Life Sciences (Logan, UT, USA) and DEX
was purchased from Chongqing Xi'nan Pharmaceutical Group Co., Ltd.
(Chongqing, China). Primary mouse anti-human monoclonal P-gp
antibody (cat. no. ab3366) was obtained from Abcam (Cambridge, UK).
An avidin-free, polymer detection kit [PV-9000; containing a
polymer helper (reagent 1) and polyperoxidase anti-mouse/rabbit IgG
(reagent 2)] for immunocytochemical staining and hematoxylin were
purchased from Zhongshan Golden Bridge Biotechnology Co., Ltd.,
(Beijing, China). Hydrogen peroxide was purchased from Chongqing
Chuandong Chemical Group Co., Ltd. (Chongqing, China) IC fixation
buffer (cat. no. 00-8222) and permeabilization buffer (cat. no.
00-8333) were purchased from eBioscience, Inc. (San Diego, CA,
USA). Fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse
secondary antibody was purchased from Zhongshang Golden Bridge
Biotechnology Co., Ltd. (cat. no. ZF-0312).
Radioimmunoprecipitation assay (RIPA) cell lysis buffer, the
bicinchoninic acid (BCA) protein assay kit and horseradish
peroxidase-labeled goat anti-mouse secondary antibody (cat. no.
A0216) were purchased from Beyotime Institute of Biotechnology
(Jiangshu, China). Polyvinylidene fluoride (PVDF) membranes were
purchased from EMD Millipore (Billerica, MA,USA). SDS was purchased
from Sigma-Aldrich (St. Louis, MO, USA), and acrylamide was
purchased from Bio-Rad Laboratories Inc. (Hercules, CA, USA). The
UNIQ-10 TRIzol total RNA extraction kit and paraformaldehyde were
obtained from Sangon Biotech Co., Ltd. (Shanghai, China). The
QuantScript reverse transcription kit was obtained from Tiangen
Biotech Co., Ltd. (Beijing, China). The M×3000P Real-Time QPCR
System was obtained from Agilent Technologies, Inc. (Santa Clara,
CA, USA). The FastStart Universal SYBR Green Master kit was
obtained from Roche Diagnostics (Basel, Switzerland). The primers
for the ABCB1 gene, encoding P-gp, were synthesized according to
the method reported by Jigore et al (21). The primers for the β-actin gene
were synthesized by Sangon Biotech. All the primers used for the
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR) analysis were purchased from Sangon Biotech Co., Ltd. The
sequences of the primer pairs were as follows: ABCB1, forward,
5′-GCCAAAGCCAAAATATCAGC-3′ and reverse, 5′-TTCCAATGTGTTCGGCATTA-3′;
and β-actin, forward 5′-GACGACATGGAGAAAA-3′ and reverse
5′-AAGGCTGGAAGAGTGC-3′. Recombinant Human sTRAIL/Apo2 L (TRAIL) was
obtained from PeproTech, Inc. (Rocky Hill, NJ, USA). The in
situ Cell Death Detection kit, POD, was purchased from Roche
Diagnostics and tariquidar (TQD) was purchased from Selleck
Chemicals (Shanghai, China).
Human L-02 cellculture and induction with
DEX
The L-02 cells were seeded in 6-well plates or
75-cm2 culture flasks (Nunc, Roskilde, Denmark) at a
density of 5×105 cells/ml, and cultured in RPMI 1640
medium supplemented with 10% (v/v) newborn bovine serum at 37°C and
5% CO2. Following treatment with varying concentrations
(1 and 10 µM) of DEX at 37°C for 24–72 h, the cells were
harvested for detection of the expression of P-gp. Untreated cells
were used as a control group.
Immunocytochemistry
Following fixation with 4% paraformaldehyde for 10
min, the cells were processed for immunocytochemistry by staining
using a PV-9000 avidin-free polymer detection system. The
procedures included the following steps: i) Incubation with 3%
hydrogen peroxide for 10 min to inhibit the activity of endogenous
peroxidase; ii) incubation with the primary mouse anti-human
monoclonal P-gp antibody (cat. no. ab3366; 1:50 dilution; Abcam) at
4°C overnight, followed by incubation with the polymer helper
(reagent 1) at 37°C for 20 min; iii) incubation with polyperoxidase
anti-mouse/rabbit IgG (reagent 2) at 37°C for 20 min; iv)
development of the peroxidase reaction with 3,3′-Diaminobenzidine
(DAB) tetrahydrochloride following three washes with
phosphate-buffered saline (PBS); and v) counterstaining of the
cells with hematoxylin. In the negative control group, the primary
antibody (cat. no. ab3366) was substituted with PBS. The cells were
examined and images were captured under an Olympus IX70 microscope
(Olympus Corporation, Beijing, China). Image-Pro Plus image
analysis software v6.0 (Media Cybernetics, Inc., Rockville, MD,
USA) was used for quantitatively assessing the optical
densities.
Flow cytometric analysis
Flow cytometry was performed on a BD FACSCalibur
flow cytometer (BD Biosciences, San Jose, CA, USA). Briefly,
106 cells were trypsinized in each well of a 6-well
plate, washed with PBS and resuspended in PBS. The cells were fixed
and permeabilized using the IC fixation and permeabilization
buffers, respectively, according to the manufacturer's instructions
(eBioscience, Inc.). The cells were incubated with primary mouse
anti-human monoclonal P-gp antibody (ab3366; 1:50 dilution; Abcam)
overnight at 4°C, followed by incubation with the FITC-conjugated
goat anti-mouse secondary antibody (1:50 dilution; cat. no.
ZF-0312; Zhongshan Golden Bridge Biotechnology Co., Ltd.) for 20
min in the dark. Following three washes, the samples were analyzed
using the flow cytometer and FlowJo software (v 6.4.7; Tree Star,
Inc. Ashland, OR, USA).
Protein extraction and western blot
analysis
Total protein was extracted from the cells using
RIPA cell lysis buffer, containing 20 mM Tris (pH 7.5), 150 mM
NaCl, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM EDTA, 1%
Na3VO4 and 0.5 mg/ml leupeptin. A BCA protein
assay kit was used for measuring the protein concentrations in the
cell extracts. Subsequently, the proteins (20 µg) were
separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis on 6% gels, and were then transferred onto PVDF
membranes by electroelution. The membranes were incubated overnight
with the primary mouse anti-human monoclonal P-gp antibody (ab3366;
1:50 dilution; Abcam), followed by incubation with the horseradish
peroxidase-labeled goat anti-mouse secondary antibody (cat. no.
A0216; 1:1,000 dilution; Beyotime Institute of Biotechnology) for 2
h at room temperature. The bands were visualized using enhanced
chemiluminescence (Pierce Biotechnology, Inc., Rockford, IL, USA)
and images were captured using a ChemiDoc XRS+ system (Bio-Rad
Laboratories, Inc.). The relative intensities in each band were
quantified densitometrically using Quantity One Software (v.
4.4.0.36, Bio-Rad Laboratories, Inc.) and calculated by comparing
the band densities with those of the reference protein,
β-actin.
Total RNA purification and RT-Qpcr
Total RNA was extracted from the cells using a
UNIQ-10 TRIzol total RNA extraction kit, according to the
manufacturer's instructions. The extracted RNA samples were reverse
transcribed into cDNA using a QuantScript reverse transcription
kit. qPCR was performed on an M×3000P Real-Time QPCR System using
the FastStart Universal SYBR Green master kit. qPCR was performed
in 96-well plates, with each well containing 1X FastStart universal
SYBR Green master mix, 2X each of the forward and reverse primers,
ddH2O and template cDNA, in a 20 µl total reaction volume.
The DNA template (1 µl) was pre-incubated at 95°C for 2 min
prior to the amplification cycle, followed by 40 cycles of
denaturation at 95°C for 15 sec, and annealing and extension at
60°C for 40 sec. β-actin was used as the reference gene. The
relative mRNA expression levels were calculated using the
2−ΔΔCt method (22).
All the experiments were performed in triplicate.
Effects of DEX and TQD on apoptosis
induced by TRAIL/Apo2 L
It has been reported that P-gps can protect cells
from apoptosis induced by a wide range of drugs (23,24).
To examine the effects of DEX on apoptosis induced by TRAIL in the
present study, the L-02 cells were treated with 20 ng/ml TRAIL for
24 h to induce apoptosis, followed by treatment with either 1 or 10
µM DEX for 24 or 48 h. Untreated cells were used as a
control group. A TUNEL assay was performed to analyze the induction
of apoptosis by TRAIL in the cells.
For investigating the effects of TQD on apoptosis,
the L-02 cells were divided into six groups: i) control group
comprising untreated L-02 cells; ii) L-02 cells pretreated with 10
µM DEX for 24 h, followed by incubation with TRAIL for 24 h;
iii) L-02 cells pretreated with 10 µM DEX for 24 h, followed
by a 24 h incubation with TRAIL and TQD (25 nM); iv) L-02 cells
pretreated with 10 µM DEX for 24 h, followed by a 24 h
incubation with TRAIL and TQD (50 nM); v) L-02 cells pretreated
with 10 µM DEX for 24 h, followed by a 24 h incubation with
TRAIL and TQD (100 nM); vi) L-02 cells incubated with TRAIL for 24
h. All the groups were incubated at 37°C, and with the exception of
the control, were treated with TRAIL to induce apoptosis. Following
the various treatments, all groups reached the same final cell
density of ~90% confluence. Apoptosis was evaluated using the TUNEL
assay, and the number of apoptotic cells in each group were
counted.
The apoptosis status of the L-02 cells was measured
using the TUNEL assay, using a POD In Situ Cell Death
Detection kit, according to the manufacturer's instructions.
Briefly, following treatment with 4% paraformaldehyde for 1 h at
room temperature and 3% hydrogen peroxide for 10 min at room
temperature, the cells (~90% confluence) were washed twice in PBS
for 5 min. Following incubation with 0.1% Triton X-100 on ice for 5
min and two washes with PBS for 5 min, the labeling reactions were
performed using 50 µl TUNEL reagent for 1 h at 37°C for each
group. Following washing with PBS, the cells were incubated with a
converter reagent for 30 min at 37°C, developed using DAB for 10
min, and then counter-stained with hematoxylin. Following
dehydration in ethanol and clearing in xylene, the cells were
mounted on slides and examined under a light microscope (Olympus
IX70; Olympus Corporation). Apoptotic cells were quantified by
counting the number of TUNEL-positive cells in ten random
microscopic fields (magnification, ×200).
Statistical analysis
The data are presented as the mean ± standard
deviation. One-way analysis of variance was used for determining
significant differences. All analyses were performed using the SPSS
13.0 statistical software (SPSS, Inc., Chicago, IL, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
DEX increases the mRNA and protein
expression levels of P-gp in the L-02 cell line
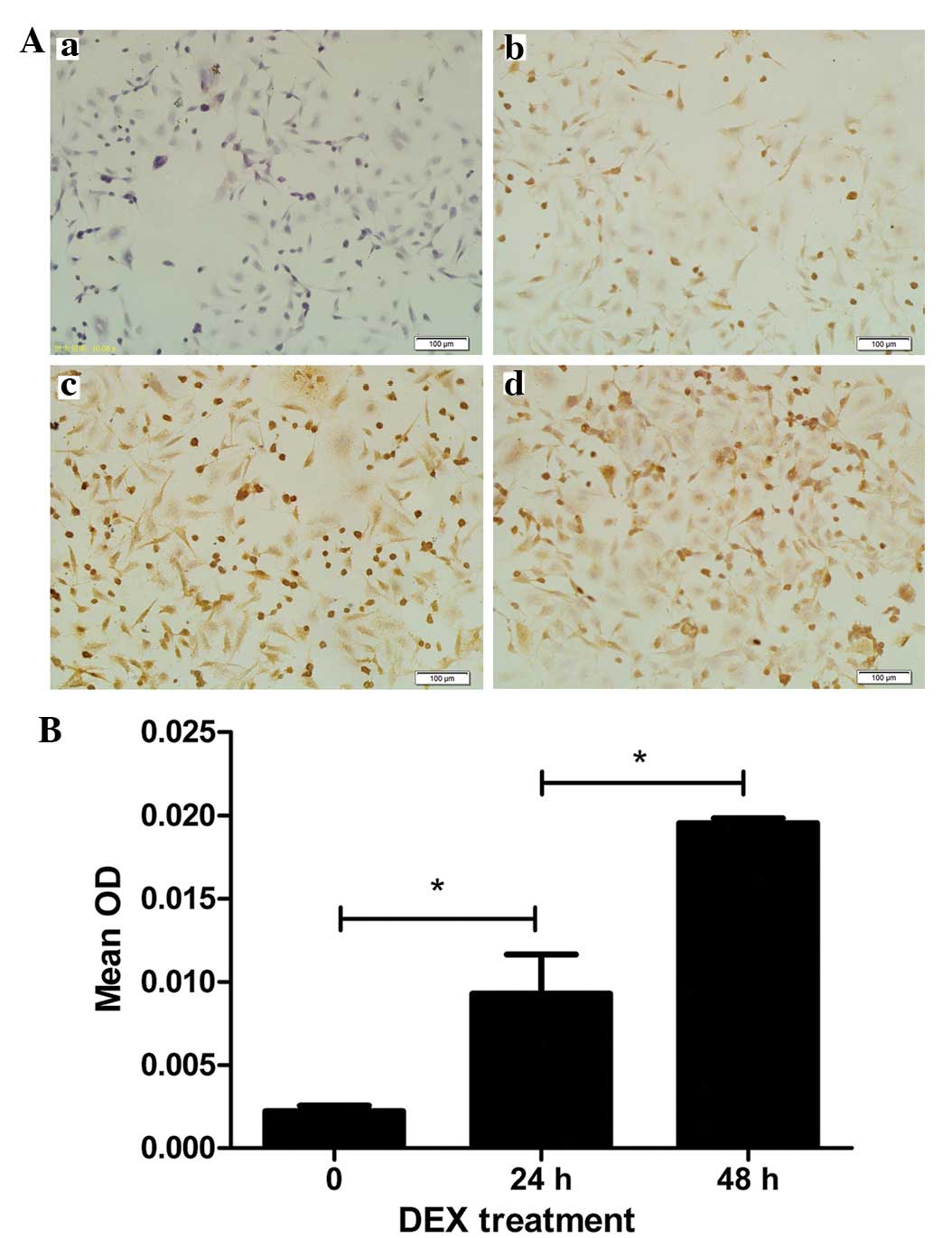
The results from the immunocyto-chemistry assays
demonstrated definitive expression of P-gp in the L-02 normal human
liver cell lines, compared with the negative control (Fig. 1Aa and b). Treatment of the L-02
cells with 10 µM DEX for 24 and 48 h resulted in marked P-gp
staining in the membrane and in the cytoplasm of the majority of
the cells (Fig. 1Ac and d). The
protein expression levels of P-gp, presented as mean densitometric
units, in the L-02 cells following treatment with 10 µM DEX
for 24 and 48 h were 9.284×10−3±0.578×10−2
and 19.530×10−3±0.719×10−3, respectively,
which were significantly higher than in the cells without DEX
treatment (2.228×10−3±0.770×10−3; Fig. 1B). The cells, which were incubated
for 48 h with DEX had higher levels of P-gps, compared with those
incubated for 24 h (P<0.01).
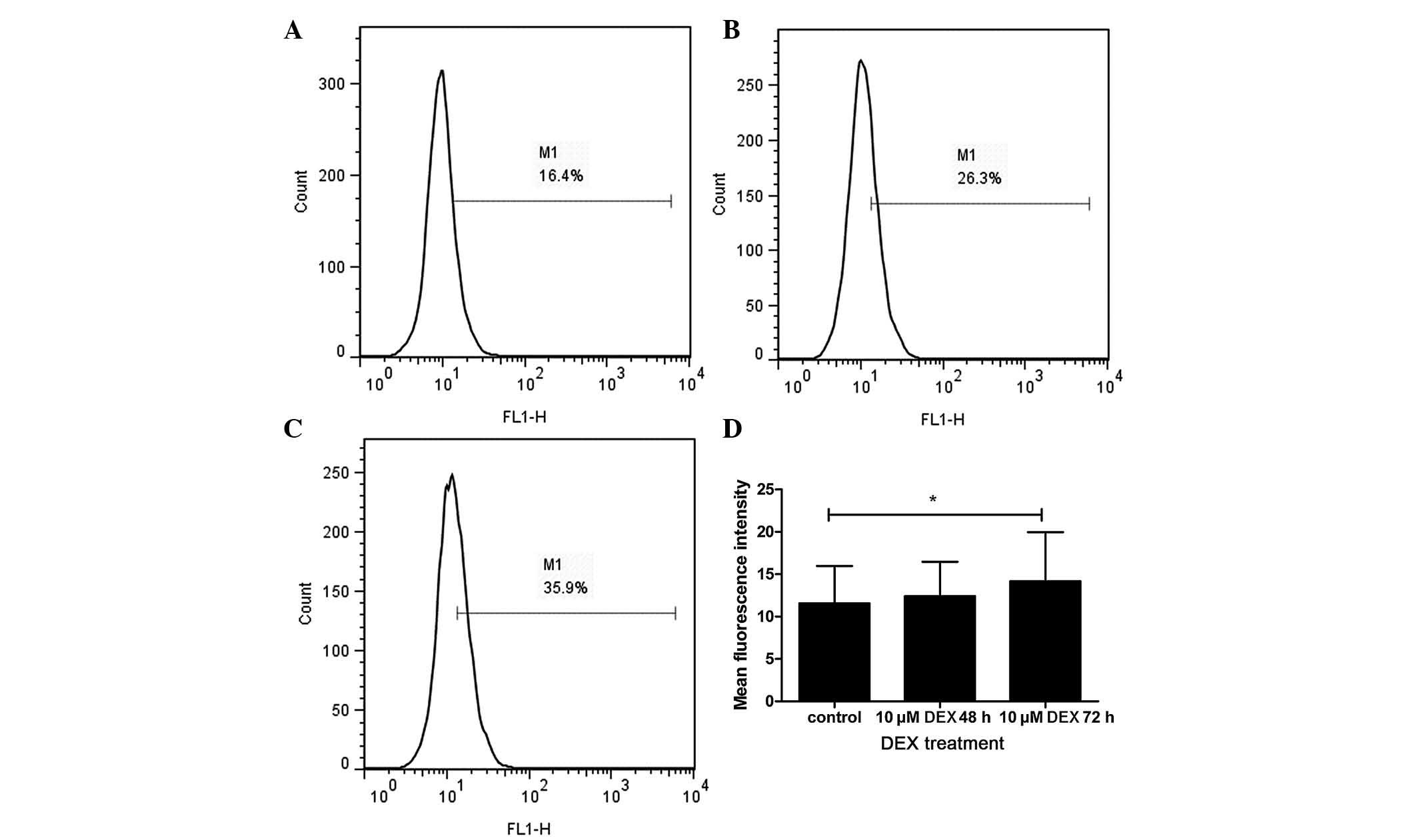
The results of the flow cytometric analysis
(Fig. 2) demonstrated that the
levels of P-gp were significantly higher in the L-02 cells treated
with 10 µM DEX for 48 h, compared with levels in the
untreated control group (P<0.01). Additionally, the levels of
P-gp were higher in the cells, which were treated with DEX for 48
h, compared with those treated for 24 h (P=0.014). The L-02 cells,
which were treated with DEX for 24 h exhibited a degree of
upregulation in the expression of P-gp, compared with the untreated
cells; however, no statistical difference was observed between
these two groups (P=0.054).
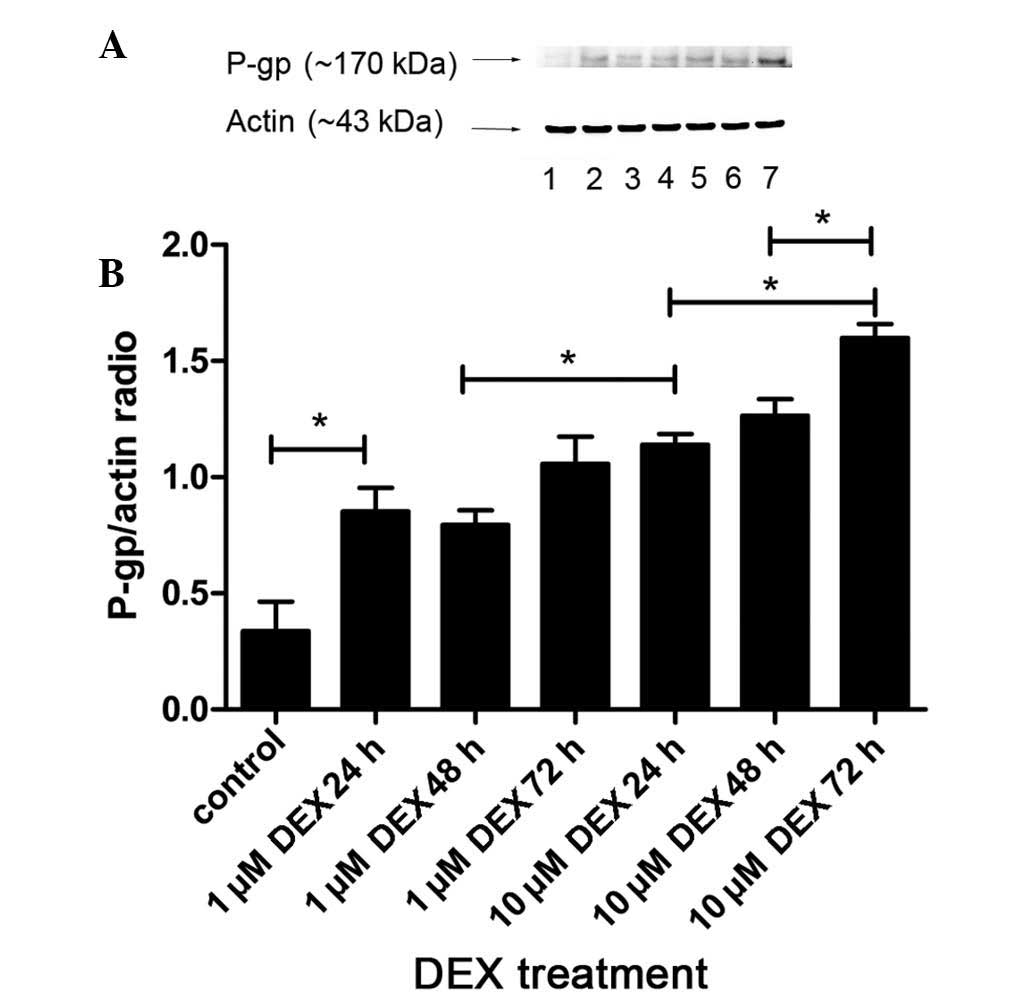
The protein expression levels of P-gp in the L-02
cells were also determined by western blot analysis, using a
monoclonal antibody against P-gp (JSB-1). As shown in Fig. 3, the results revealed a band at 170
kDa, which corresponded with P-gp. The results were analyzed
quantitatively, according to the grayscale values. The expression
of P-gp in the group treated with 1 µM DEX for 24 h were
significantly increased, compared with the control group (P=0.001).
Treatment with 10 µM DEX for 24 h resulted in higher
expression levels of P-gp than that following 1 µM DEX
treatment for 48 h (P=0.006). In addition, the protein expression
levels in the cells treated with 10 µM DEX for 72 h were
markedly upregulated, compared with those in the groups treated
with 10 µM DEX for 24 and 48 h (P<0.05). Therefore, it
was concluded that the protein expression levels of P-gp increased
in a dose-dependent manner, between 1 and 10 µM DEX, and a
time-dependent manner, between 24 and 72 h DEX stimulation.
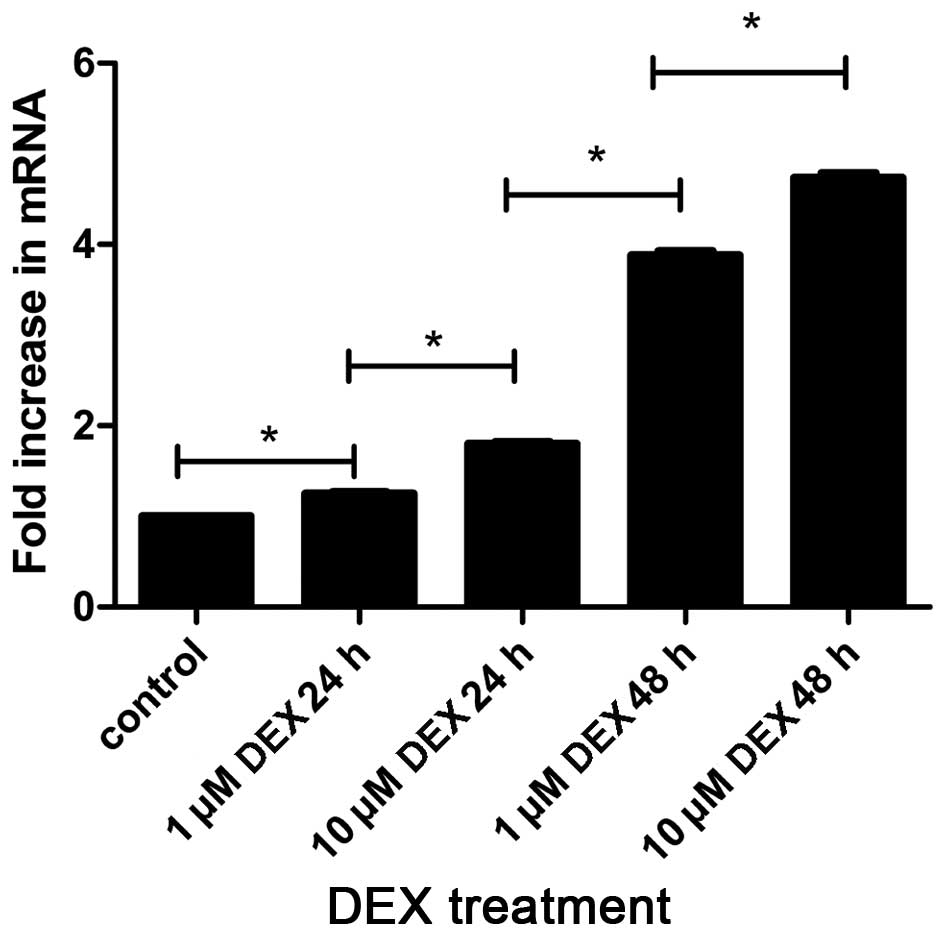
RT-qPCR was used for detecting the mRNA expression
levels of the ABCB1 gene, encoding P-gp, in the L-02 cells. As
shown in Fig. 4, the present study
detected for the first time, to the best of our knowledge,
transcription of the ABCB1 gene in the L-02 cell lines. In
addition, the effects of DEX on the transcription of ABCB1 mRNA
were also demonstrated in the L-02 cell line. The results
demonstrated that the mRNA levels of the ABCB1 gene increased in
response to treatment with 1 µM DEX for 24 h, compared with
the control (P=0.004), similar to the results of the protein
levels. During the same time interval, treatment with 1 µM
and 10 µM DEX exhibited differences in mRNA levels, which
were significantly higher in the 10 µM DEX group, compared
with the 1 µM group (P<0.05). Additionally, treatment
with DEX for 48 h lead to increased mRNA levels, compared with
treatment with DEX for 24 h (P<0.05). These results suggested
that treatment with DEX increased the expression of P-gp at the
mRNA and protein levels, in a dose- and time-dependent manner.
Effects of DEX on apoptosis induced by
TRAIL
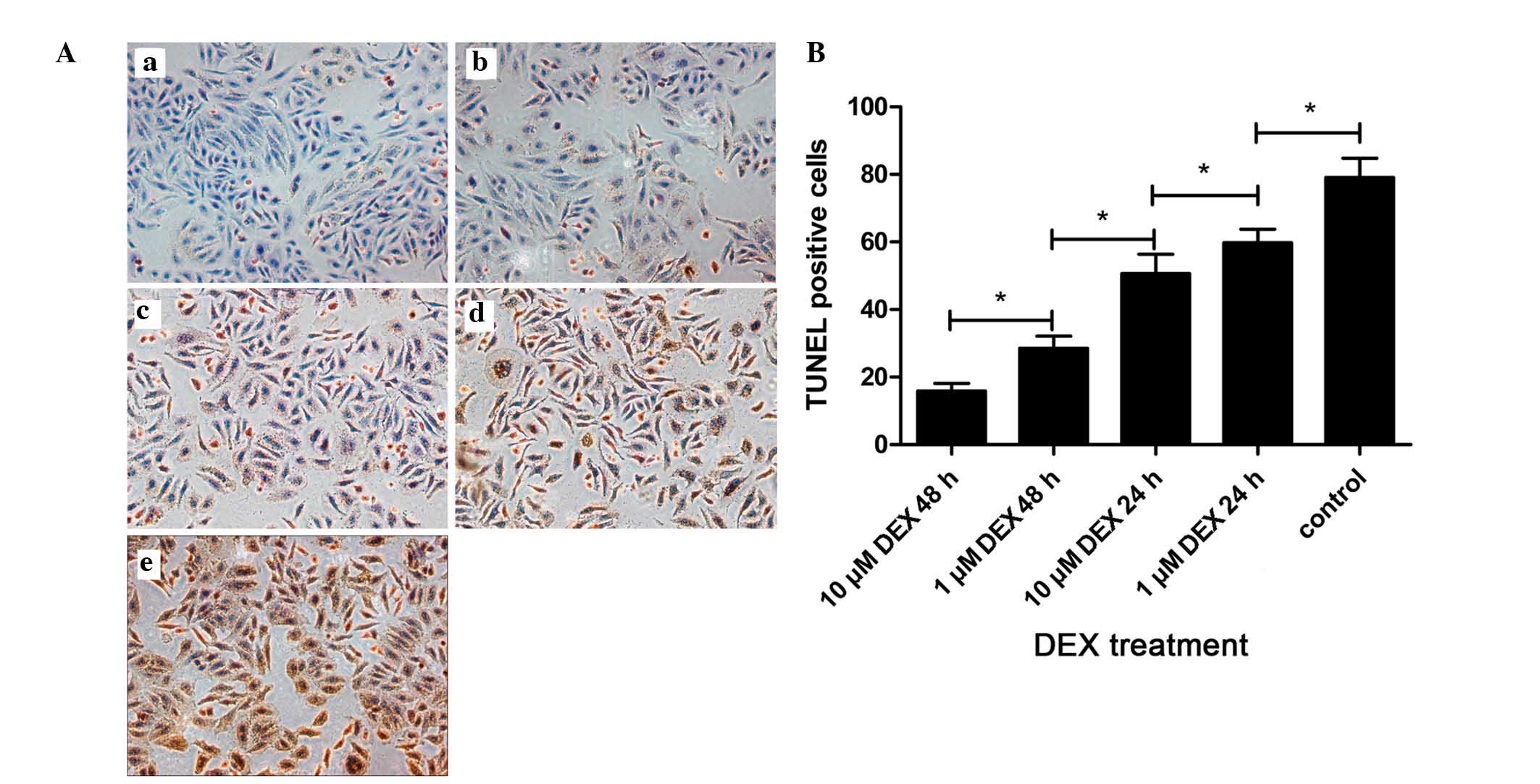
As shown in Fig. 5,
all the nuclei were stained blue with hematoxylin, whereas the
brown color indicated the presence of apoptotic nuclei, which were
visualized following staining with DAB reagent. The highest degree
of apoptosis was observed in the control group, which was incubated
with TRAIL in the absence of DEX. By contrast, treatment with 10
µM DEX for 48 h resulted in the lowest observed levels of
apoptosis in the L-02 cells. In addition, the results demonstrated
that the degree of DNA fragmentation decreased with prolonged
exposure to DEX, between 24–48 h, and increased concentrations of
DEX between 1 and 10 µM (P<0.05).
Effects of TQD on apoptosis induced by
TRAIL
As shown in Fig. 6,
the lowest degree of apoptosis was observed in the control group,
which comprised L-02 cells cultured in media alone with no
additional treatments. These results indicated that the levels of
apoptosis in the L-02 cells were intrinsically low. Following
exposure to TRAIL, the levels of apoptosis in the L-02 cells
increased markedly. By contrast, pretreatment of the cells with DEX
inhibited this apoptosis. Notably, the addition of TQD, which is a
P-gp inhibitor, increased the levels of apoptosis. When the
concentrations of TQD were increased to 50 and 100 nM, statistical
significance in the levels of apoptosis were observed, compared
with the group treated with DEX and TRAIL (P<0.05). No
significant difference was observed between the TRAIL-treated and
TQD-treated (100 nM) groups.
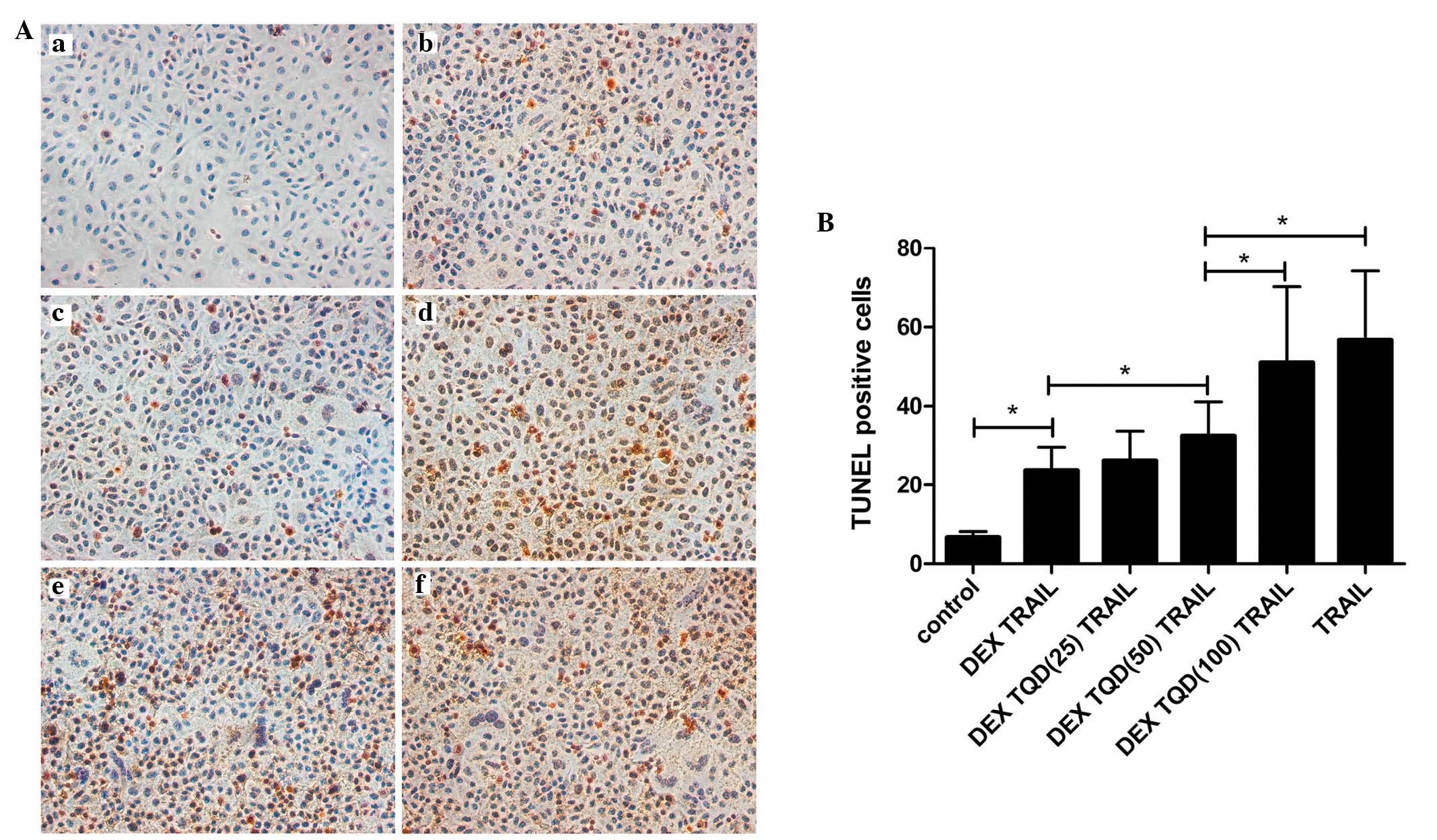 | Figure 6DEX protects L-02 cells from
TRAIL-induced apoptosis by upregulating P-gp. Apoptosis was
evaluated using a TUNEL assay, and the number of TUNEL-positive
cells in each group were counted. (A) Representative
photomicrographs of the TUNEL assay. L-02 cells were divided into
six groups based on their treatment regimens, (a) control untreated
group, (b) pretreated with 10 µM DEX for 24 h, followed by
incubation with TRAIL for 24 h, (c) pretreatment with 10 µM
DEX for 24 h, followed by incubation with TRAIL and 25 nM TQD for
24 h, (d) pretreated with 10 µM DEX for 24 h, followed by
incubation with TRAIL and 50 nM TQD for 24 h, (e) pretreated with
10 µM DEX for 24 h, followed by incubation with TRAIL and
100 nM TQD for 24 h, and (f) incubated with TRAIL for 24 h. All the
groups were treated with TRAIL for the induction of apoptosis, with
the exception of the control group. Magnification, ×200. (B)
Quantitative analysis of the levels of apoptosis. Data are
presented as the mean ± standard deviation. *P<0.05.
DEX, dexamethasone; TRAIL, tumor necrosis factor-related
apoptosis-inducing ligand; TQD, tariquidar; TUNEL, terminal
deoxynucleotidyl transferase dUTP nick end labeling. |
Discussion
Studies have reported that DEX can induce the
expression and function of P-gp in the blood-brain barrier,
placenta and other tissues (18,19).
Therefore, the present study examined whether DEX increases the
levels of P-gps in normal human liver cells. The results of the
immunocytochemical and flow cytometric analyses revealed that
pretreatment of L-02 cells with DEX for 24–72 h significantly
upregulated the levels of P-gps (Figs.
1B and 2B). The results of the
western blot analysis demonstrated that pretreatment of the cells
with DEX increased the expression P-gp in a time- and
dose-dependent manner (Fig. 3). In
addition, DEX also enhanced the mRNA levels of the ABCB1 gene,
similar to the results obtained for the protein levels. These
results suggested that pretreatment with DEX induced the expression
of P-gp at the mRNA and protein levels in the L-02 cell line.
The results from previous studies reporting the
effects of glucocorticoids on the expression of P-gps are
inconsistent. In several studies, glucocorticoids, including DEX
have been reported to upregulate the expression of P-gps, in
agreement with the results of the present study. Salje et al
reported the ability of DEX to upregulate the expression of P-gps
in the liver (25), and the
expression of P-gp also increased in the blood-brain barriers of
DEX-treated animals in a report by Chan et al (19). These results are consistent with
those reported by Petropoulos et al, which demonstrated the
induction of placental P-gp, mediated by DEX (26). By contrast, other studies have
demonstrated either no change or decreased expression levels of
P-gps upon treatment of cells or animals with DEX. For example,
Nishimura et al reported that DEX had a tendency to decrease
the mRNA levels of the ABCB1 gene in monkey hepatocytes (27). Mark et al demonstrated that
pre-treatment with glucocorticoids decreases the expression levels
of P-gp in the placenta (28). Of
note, DEX exerts varied effects on P-gps in different tissues and
cell types. Previous studies have demonstrated that DEX
pretreatment increases the levels of P-gp in the intestine, but not
in the liver (29). Another study
reported that DEX rapidly increases the expression levels of P-gps
in the liver and lungs, but decreases their expression in kidneys
(30). Glucocorticoid treatment
exerts differing effects, even in the case of cell lines derived
from the same tissue; the expression of P-gp is induced in LS180
cells, but downregulated in Caco-2 cells upon treatment (31). There is no definitive explanation
for these contrasting results; however, it is possible that it is
due to tissue specificity, as well as the complex regulation of
this efflux transporter by glucocorticoids in certain organs.
Notably, incubation with DEX in the present study
led to the transcriptional levels of the ABCB1 gene mRNA increasing
in parallel with that of P-gp protein levels. This correlation
between the ABCB1 gene mRNA and the P-gp protein has been reported
in previous studies, which are in good agreement with our studies.
Narang et al demonstrated that DEX increases the expression
of P-gp in primary rat brain microvascular endothelial cells, and
an increase in the mRNA expression of the ABCB1 gene results in a
change in the protein expression of P-gp (18). According to Salje et al, the
mRNA and protein levels of P-gp are significantly induced by DEX in
the fetal brain and liver (25).
However, there are results contradicting those described above.
Katayama et al demonstrated that the expression of P-gp can
be increased without affecting its mRNA levels (32). Similarly, Micuda et al
demonstrated that pretreatment with DEX results in an increase in
the protein expression of P-gp, but a decrease in the mRNA
expression of the ABCB1 gene in the liver (33). The present study hypothesized that
the these differences may be due to the complex transcriptional
regulation of MDR1, which involves several signaling pathways
(34).
Several studies have provided evidence suggesting
that DEX can protect several cells from apoptosis induced by drugs.
Nieuwenhuis et al reported that DEX protects human
fibroblasts from apoptosis (9).
According to a study by Haake et al, DEX treatment also
protects hair cells against apoptosis (10). Similarly, other studies have
demonstrated that P-gps can exert anti-apoptotic effects. Ruefli
et al reported that P-gps can specifically inhibit
Fas-induced caspase activation and apoptosis (15), whereas Tainton et al
demonstrated that P-gps inhibit apoptotic stimuli in lymphoma cells
(16). In acute myeloid leukemia,
resistance to apoptosis is associated with the expression of P-gp,
as reported by Pallis et al (17). In view of these observations, the
present study investigated whether DEX had similar anti-apoptotic
effects on L-02 cells. The results revealed that pretreatment with
DEX for 24–48 h effectively protected the L-02 cells from
TRAIL-induced apoptotic cell death. A gradual increase in the
protective effect was observed when the duration of treatment was
increased between 24 and 48 h, and when the concentration of DEX
was increased between 1 and 10 µM. Together, these findings
indicated that DEX pretreatment suppressed TRAIL-induced apoptosis
in the L-02 cell line.
Previous studies have demonstrated that, in addition
to their role in MDR, P-gps may also protect cells at two levels,
by decreasing the accumulation of toxins in the cells and by
inhibiting the apoptotic pathways induced by toxins and other
stressors. Ruefli et al reported that P-gp can specifically
inhibit caspase-8 activation and apoptosis (15). Similarly, Pallis et al
demonstrated that the expression and activity of P-gps are
associated with resistance to apoptosis in acute myeloid leukemia
cells (17).
Therefore, the present study subsequently examined
the anti-apoptotic effects of DEX on the cells, in which the
functions of P-gps were inhibited by treatment with TQD, a P-gp
inhibitor. The results revealed that, in addition to inducing the
expression of P-gp, DEX alleviated cell apoptosis at increased
concentrations and incubation periods. By contrast, treatment with
TQD aggravated apoptosis in the L-02 cells, compared with the
untreated cells. Additionally, the numbers of apoptotic cells were
elevated with increasing concentrations of TQD. Based on these
results, it is possible that DEX exerted anti-apoptotic and
cell-protective effects through the induction of P-gps in the L-02
cell line.
In conclusion, the present study provided evidence
that P-gps exist in L-02 cells, a normal human liver cell line, and
the effect of DEX on the expression of P-gps in this cell line were
investigated. It was demonstrated that pretreatment with DEX
suppressed TRAIL-induced apoptosis in the L-02 cells. In addition,
the results indicated that the induction of P-gp is likely a
molecular mechanism underlying the anti-apoptotic effects of DEX.
The results also suggest that the L-02 cell line is a suitable cell
model for investigating the effects of glucocor-ticoids on
transport proteins, including P-gp. In addition, this model may
also assist in understanding the benefits of DEX for the treatment
of hepatic failure and other liver diseases. Additional
investigations are required for understanding the underlying
molecular mechanisms by which DEX regulates the expression of P-gp
in the L-02 cell line.
Abbreviations:
|
DEX
|
dexamethasone
|
|
P-gp
|
P-glycoprotein
|
|
TRAIL
|
tumor necrosis factor-related
apoptosis-inducing ligand
|
|
TUNEL
|
terminal deoxynucleotidyl transferase
dUTP nick end labeling
|
|
pre-ACLF
|
acute-on-chronic pre-liver failure
|
|
ACLF
|
acute-on-chronic liver failure
|
|
NA
|
nucleoside analog
|
|
MDR
|
multidrug resistance
|
|
PXR
|
pregnane X receptor
|
|
TQD
|
tariquidar
|
Acknowledgments
This study was supported by a grant from the Chinese
National Natural Science Foundation project (grant. no.
81270525).
References
|
1
|
Levy P, Marcellin P, Martinot-Peignoux M,
Degott C, Nataf J and Benhamou JP: Clinical course of spontaneous
reactivation of hepatitis B virus infection in patients with
chronic hepatitis B. Hepatology. 12:570–574. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sarin SK, Kumar A, Almeida JA, Chawla YK,
Fan ST, Garg H, de Silva HJ, Hamid SS, Jalan R, Komolmit P, et al:
Acute-on-chronic liver failure: Consensus recommendations of the
Asian Pacific Association for the study of the liver (APASL).
Hepatol Int. 3:269–282. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang XQ, Jiang L, You JP, Liu YY, Peng J,
Zhang HY, Xu BY and Mao Q: Efficacy of short-term dexamethasone
therapy in acute-on-chronic pre-liver failure. Hepatol Res.
41:46–53. 2011. View Article : Google Scholar
|
|
4
|
Fujiwara K, Yasui S, Yonemitsu Y, Fukai K,
Arai M, Imazeki F, Suzuki A, Suzuki H, Sadahiro T, Oda S and
Yokosuka O: Efficacy of combination therapy of antiviral and
immunosuppressive drugs for the treatment of severe acute
exacerbation of chronic hepatitis B. J Gastroenterol. 43:711–719.
2008. View Article : Google Scholar
|
|
5
|
Naveau S, Balian A, Capron F, Raynard B,
Fallik D, Agostini H, Grangeot-Keros L, Portier A, Galanaud P,
Chaput JC and Emilie D: Balance between pro and anti-inflammatory
cytokines in patients with acute alcoholic hepatitis. Gastroenterol
Clin Biol. 29:269–274. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang M, Shen F, Shi LH, Xi T, Li XF, Chen
X and Wu MC: Protective effect of prednisolone on ischemia-induced
liver injury in rats. World J Gastroenterol. 14:4332–4337. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thoresen GH, Gjone IH, Gladhaug IP,
Refsnes M, Ostby E and Christoffersen T: Studies of glucocorticoid
enhancement of the capacity of hepatocytes to accumulate cyclic
AMP. Pharmacol Toxicol. 65:175–180. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jaeschke H, Gujral JS and Bajt ML:
Apoptosis and necrosis in liver disease. Liver Int. 24:85–89. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nieuwenhuis B, Lüth A and Kleuser B:
Dexamethasone protects human fibroblasts from apoptosis via an
S1P3-receptor subtype dependent activation of PKB/Akt and Bcl XL.
Pharmacol Res. 61:449–459. 2010. View Article : Google Scholar
|
|
10
|
Haake SM, Dinh CT, Chen S, Eshraghi AA and
Van De Water TR: Dexamethasone protects auditory hair cells against
TNFalpha-initiated apoptosis via activation of PI3K/Akt and
NFkappaB signaling. Hear Res. 255:22–32. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li M, Chen F, Liu CP, Li DM, Li X, Wang C
and Li JC: Dexamethasone enhances trichosanthin-induced apoptosis
in the HepG2 hepatoma cell line. Life Sci. 86:10–16. 2010.
View Article : Google Scholar
|
|
12
|
Gottesman MM and Pastan I: Biochemistry of
multidrug resistance mediated by the multidrug transporter. Annu
Rev Biochem. 62:385–427. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ambudkar SV, Dey S, Hrycyna CA,
Ramachandra M, Pastan I and Gottesman MM: Biochemical, cellular and
pharmacological aspects of the multidrug transporter. Annu Rev
Pharmacol Toxicol. 39:361–398. 1999. View Article : Google Scholar
|
|
14
|
Ruefli AA and Johnstone RW: A role for
P-glycoprotein in regulating cell growth and survival. Clin Appl
Immunol Rev. 4:31–47. 2003. View Article : Google Scholar
|
|
15
|
Ruefli AA, Tainton KM, Darcy PK, Smyth MJ
and Johnstone RW: P-glycoprotein inhibits caspase-8 activation but
not formation of the death inducing signal complex (disc) following
fas ligation. Cell Death Differ. 9:1266–1272. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tainton KM, Smyth MJ, Jackson JT, Tanner
JE, Cerruti L, Jane SM, Darcy PK and Johnstone RW: Mutational
analysis of P-glycoprotein: Suppression of caspase activation in
the absence of ATP-dependent drug efflux. Cell Death Differ.
11:1028–1037. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pallis M, Turzanski J, Grundy M, Seedhouse
C and Russell N: Resistance to spontaneous apoptosis in acute
myeloid leukaemia blasts is associated with p-glycoprotein
expression and function, but not with the presence of FLT3 internal
tandem duplications. Br J Haematol. 120:1009–1016. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Narang VS, Fraga C, Kumar N, Shen J, Throm
S, Stewart CF and Waters CM: Dexamethasone increases expression and
activity of multidrug resistance transporters at the rat
blood-brain barrier. Am J Physiol Cell Physiol. 295:C440–C450.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chan GN, Saldivia V, Yang Y, Pang H, de
Lannoy I and Bendayan R: In vivo induction of P-glycoprotein
expression at the mouse blood-brain barrier: An intracerebral
microdialysis study. J Neurochem. 127:342–352. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rigalli JP, Perdomo VG, Luquita MG,
Villanueva SS, Arias A, Theile D, Weiss J, Mottino AD, Ruiz ML and
Catania VA: Regulation of biotransformation systems and ABC
transporters by benznidazole in HepG2 cells: Involvement of
pregnane X-receptor. PLoS Negl Trop Dis. 6:e19512012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jigorel E, Le Vee M, Boursier-Neyret C,
Parmentier Y and Fardel O: Differential regulation of sinusoidal
and canalicular hepatic drug transporter expression by xenobiotics
activating drug-sensing receptors in primary human hepatocytes.
Drug Metab Dispos. 34:1756–1763. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
23
|
Takeba Y, Sekine S, Kumai T, Matsumoto N,
Nakaya S, Tsuzuki Y, Yanagida Y, Nakano H, Asakura T, Ohtsubo T and
Kobayashi S: Irinotecan-induced apoptosis is inhibited by increased
P-glycoprotein expression and decreased p53 in human hepatocellular
carcinoma cells. Biol Pharm Bull. 30:1400–1406. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sakaeda T, Nakamura T, Hirai M, Kimura T,
Wada A, Yagami T, Kobayashi H, Nagata S, Okamura N and Yoshikawa T:
MDR1 up-regulated by apoptotic stimuli suppresses apoptotic
signaling. Pharm Res. 19:1323–1329. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Salje K, Lederer K, Oswald S, Dazert E,
Warzok R and Siegmund W: Effects of rifampicin, dexamethasone, St.
John's Wort and thyroxine on maternal and foetal expression of
Abcb1 and organ distribution of talinolol in pregnant rats. Basic
Clin Pharmacol Toxicol. 111:99–105. 2012.PubMed/NCBI
|
|
26
|
Petropoulos S, Gibb W and Matthews SG:
Effect of glucocorticoids on regulation of placental multidrug
resistance phosphoglycoprotein (P-gp) in the mouse. Placenta.
31:803–810. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nishimura M, Koeda A, Morikawa H, Satoh T,
Narimatsu S and Naito S: Comparison of inducibility of multidrug
resistance (MDR)1, multidrug resistance-associated protein (MRP)1
and MRP2 mRNAs by prototypical microsomal enzyme inducers in
primary cultures of human and cynomolgus monkey hepatocytes. Biol
Pharm Bull. 31:2068–2072. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mark PJ, Augustus S, Lewis JL, Hewitt DP
and Waddell BJ: Changes in the placental glucocorticoid barrier
during rat pregnancy: Impact on placental corticosterone levels and
regulation by progesterone. Biol Reprod. 80:1209–1215. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yumoto R, Murakami T, Sanemasa M, Nasu R,
Nagai J and Takano M: Pharmacokinetic interaction of cytochrome
P450 3A-related compounds with rhodamine 123, a P-glycoprotein
substrate, in rats pretreated with dexamethasone. Drug Metab
Dispos. 29:145–151. 2001.PubMed/NCBI
|
|
30
|
Demeule M, Jodoin J, Beaulieu E, Brossard
M and Béliveau R: Dexamethasone modulation of multidrug
transporters in normal tissues. FEBS Lett. 442:208–214. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maier A, Zimmermann C, Beglinger C, Drewe
J and Gutmann H: Effects of budesonide on P-glycoprotein expression
in intestinal cell lines. Br J Pharmacol. 150:361–368. 2007.
View Article : Google Scholar
|
|
32
|
Katayama K, Noguchi K and Sugimoto Y:
FBXO15 regulates P-glycoprotein/ABCB1 expression through the
ubiquitin-proteasome pathway in cancer cells. Cancer Sci.
104:694–702. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Micuda S, Fuksa L, Mundlova L,
Osterreicher J, Mokry J, Cermanova J, Brcakova E, Staud F, Pokorna
P and Martinkova J: Morphological and functional changes in
p-glycoprotein during dexamethasone-induced hepatomegaly. Clin Exp
Pharmacol Physiol. 34:296–303. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mizutani T and Hattori A: New horizon of
MDR1 (P-glycoprotein) study. Drug Metab Rev. 37:489–510. 2005.
View Article : Google Scholar : PubMed/NCBI
|