Introduction
Hepatopulmonary syndrome (HPS) is defined as the
presence of the triad of an arterial oxygenation defect,
intrapulmonary vasodilation, and the presence of liver disease
(1,2). It usually occurs in patients
diagnosed with cirrhosis and is characterized by a lack of arterial
oxygen (1,2). However, HPS is under-diagnosed in
patients with late-stage liver disease as there is no consensus on
the diagnostic criteria. Liver transplantation is hypothesized to
be a relatively effective treatment for HPS (3–5). The
molecular mechanism underlying HPS is reported to be associated
with numerous factors such as lipopolysaccharide, nitric oxide and
tumor necrosis factor (TNF)-α among others (6–11);
however it remains unclear. Thus, further research is required.
MicroRNAs (miRNAs) are small noncoding RNAs
~22-nucleotides in length, which function as important regulators
in gene expression (12,13). miRNAs inhibit the expression of
target genes by specifically binding and cleaving mRNAs or
inhibiting their translation. There are 2,578 miRNAs that are
involved in various cellular physiological and pathological
processes (12,13). However, to date, there are no
reports on the role of miRNAs in HPS.
To investigate the involvement of miRNAs in the
regulation of pulmonary microvascular endothelial cell (PMVEC)
proliferation, an HPS model was induced and PMVECs were isolated.
This study aimed to identify the molecular mechanism of the effect
of miR-101 on pulmonary microvascular endothelial cells in the HPS
rat model.
Materials and methods
Reagents and antibodies
Rabbit anti-human phospho-janus kinase (Jak)1
polyconal antibody (cat. no. 3331), rabbit anti-human Jak1
polyconal antibody (cat. no. 3332), phospho-Jak2 rabbit mAb (cat.
no. 3376), JAK2 polyconal antibody (cat. no. 3773), phospho-Jak3
rabbit mAb (cat. no. 5031), Jak3 polyconal antibody (cat. no.
3775), phospho-Stat3 rabbit mAb (cat. no. 9154) and Stat3 mouse mAb
(cat. no. 9139) were all purchased from Cell Signaling Technology,
Inc. (Danvers, MA, USA). Each antibody was used at a dilution of
1:1,000. Pim-1 mAb (cat. no. sc-374116) and Mcl-1mAb (cat. no.
sc-377487) were purchased from Santa Cruz Biotechnology Inc. (Santa
Cruz, CA, USA) and the dilution was 1:500. Primary GAPDH mAb and
secondary antibodies conjugated to horseradish peroxidase (1:5,000)
were ordered from Kang-Chen Biotech (Shanghai, China).
HPS rat model
HPS rat models were constructed according to the
protocols of the National Institutes of Health. The procedure of
the study was approved by the committee on Animal Research of First
Affiliated Hospital of Harbin Medical University. A total of 20
male Sprague Dawley rats (weight, 180–220 g; age, 12 weeks) were
used in this study. The HPS rat models were successfully
constructed in our study. At PaO2 <85 mm Hg, the
blood serum of rats with P(A-a)O2 > 18 mm Hg and
pulmonary vasodilatation (PVD) found on pathological sections was
used for this study. Serum was separated from CBDL blood samples
(centrifugation at 2,000 x g/min for 10 min at 4°C) within 20 min
of collection, was carefully handled to avoid hemolysis, and was
stored at −80°C.
PMVEC isolation and culture
PMVECs were isolated from the rat lung as described
previously (11). Briefly, rat
peripheral tissue (2–5 g) was extracted from normal Sprague Dawley
rats. The tissue was washed using phosphate-buffered saline, cut
into sections, digested in 0.3% type II collagenase (Sigma-Aldrich
Canada Co., Oakville, ON, Canada) for 45 min at 37°C, filtered
through 100-µm mesh and centrifuged at 500 x g. After
washing with PBS, the cell pellet was resuspended in binding buffer
and incubated for 20 min at 4°C with magnetic microbeads (Dynal
Inc., Lake Success, NY, USA) coated with anti-CD31 antibody. The
microbeads were used to recruit cells, which were then washed five
times using PBS, resuspended in 10% Endothelial Growth Medium-2
(EGM-2, Cambrex Bio Science Inc., Walkersville, MD, USA) and
incubated for 8–12 h at 37°C in 5% CO2/21%
O2. The growth medium was changed to fresh medium on the
third day of culture in order to remove unattached cells. The
fibroblasts were removed 7–10 days after seeding the cells to make
PMVEC clusters grow. PMVECs were purified again with
anti-CD31-coated magnetic microbeads. PMVECs were seeded in a
25-cm2 flask, passaged at a ratio of 1:3 and used for
experiments at passage 2–3.
Lentiviral vector construction and
transduction
Mature miR-101 antagomir oligonucleotide was
synthesized, amplified and cloned into GV232-Puro Vectors by
Genechem Co., Ltd. (Shanghai, China). The nucleotides of the
insertions were confirmed by DNA sequencing. Lentivirus packaging
was performed in 293T cells using Lipofectamine 2000 (Invitrogen
Life Technologies, Carlsbad, CA, USA) according to the
manufacturer's instructions. When analyzing cellular function,
cells were transduced with the lentivirus and selected with
puromycin. miR-101 expression was verified by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR).
Small interfering (si)RNA
transfection
Transfection was performed according to the
manufacturer's instructions using Lipofectamine 2000 reagent. JAK2
and STAT3 siRNAs were ordered from Santa Cruz Biotechnology Inc.
Briefly, 2×105 PMVECs were seeded in 6-well plates. The
following day, the cells were transfected with JAK2 or STAT3 siRNA
(100 nM) and growth medium was changed after 5 h. JAK2 and STAT3
mRNA and protein levels were examined by RT-qPCR and western
blotting, respectively. The transfected cells were used for various
assays. For reporter assays, the pGL3-basic vector was
co-transfected with siRNAs and luciferase assays were performed 48
h after transfection.
miRNA target prediction dual luciferase
assay
Cells were seeded on 24-well plates and transfected
with plasmids carrying the 3′ untranslated region (UTR) of JAK2 and
miR-101 using Lipofectamine 2000 according to the manufacturer's
instructions. After transfection (6 h), the medium was changed, and
the cells were incubated at 37°C for 2 days. Cells were washed once
with phosphate-buffered saline and luciferase assays were performed
using the Dual Luciferase Assay system (Promega Corporation,
Madison, WI, USA) according to the manufacturer's instructions.
Readings were taken with a Lumat LB 9507 (Berthold Technologies
GmbH, Bad Wildbad, Germany). miR-101 target genes were predicted
based on the online bioinformatics software, including TargetScan
(http://www.targetscan.org/vert_61/)
and miRbase (http://www.mirbase.org).
RT-qPCR
Total RNA was extracted from the PMVECs transfected
with miRNA or siRNA, or treated with interleukin (IL)-6 at
indicated time points using the Tripure Isolation Reagent. Firstly,
total RNA (2 µg) was reverse transcribed into cDNA in the
reaction system with Oligo (dT)15 (Sangon Biotech Co. Ltd.,
Shanghai, China), dNTP (Sangon Biotech Co. Ltd.), and the reaction
buffer supplied with the M-MLV reverse transcriptase (Promega
Corporation). qPCR was performed using Fast SYBR Green master mix
(Life technologies, Grand Island, NY, USA). Primer sequences were
as follows: TNF-α, forward 5′-cgacgtggaactggcagaag-3′ and reverse
5′-ccgagaactgccgtctc-3′; IL-1, forward
5′-gccttgaaggtactgtatctgcaca-3′ and reverse
5′-cgaaggccactccacgtctc-3′; IL-6, forward 5′-gatgctggtgacaaccacg-3′
and reverse 5′-gacttcctgagaccgaaaca-3′; IL-8, forward
5′-taggcatcttcgtccgtccctgt-3′ and reverse 5′-agagggttactttctata-3′;
miR-101, forward 5′-acactccagctgggtacagtactgtgat-3′ and reverse
5′-ctcaactggtgtcgtggagtcggcaattcagttgagtattgact-3′; JAK1, forward
5′-agctgtgcatcagggccgccc-3′ and reverse 5′-cgccgggactacgtgtcga-3′;
JAK2, forward 5′-caccaacattacagaggcataata-3′ and reverse
5′-gaacgacgaagcttctttctgag-3′; JAK3, forward
5′-accgagaccttccgtgtgggg-3′ and reverse
5′-aggcgaaggtcctacaccggcag-3′; STAT3, forward
5′-cacacgctacctggagcagctg-3′ and reverse
5′-actatctcctgtaacctgag-3′; GAPDH, forward
5′-tgtgaaccacggagagggt-3′ and reverse 5′-ggcatggactgtggtcatga-3′;
snRNA, forward 5′-ctcgcttcggcagcaca-3′ and reverse
5′-aacgcttcacgaatttgcgt-3′. TaqMan miRNA assays were purchased from
Life technologies and used to quantify mature miRNAs following the
manufacturer's instructions. U6 snRNA was used as an internal
control. GAPDH and U6 RNA was used the initial control for RNA and
miRNA respectively. Specific primer were ordered from Invitrogen
Life Technologies.
Western blotting
Total protein was extracted from the cells and its
concentration was determined using a Bradford assay (Bio-Rad,
Philadelphia, PA, USA). Protein was separated by SDS-PAGE and
transferred to membranes (Millipore, Bedford, MA, USA) at 80 V for
2 h at 4°C. The membranes were blocked in 5% non-fat dry milk in
PBST then incubated with primary antibodies in PBS, washed with
PBST and then incubated with secondary antibodies conjugated to
horseradish peroxidase in PBST for 1 h at room temperature.
Membranes were washed again in PBST three times and bands were
visualized on X-ray films using an enhanced chemiluminescence
detection system.
MTT assay
Cells (103–104) were
transfected with miR-101, miR-control or STAT3 siRNA, or treated
with IL-6 in 6-cm plates, and added into each well of a 96-well
plate after 24-h transfection. The cells were cultured for 1, 2, 3,
4 and 5 days. MTT solution (20 µl of 5 mg/ml) was added, and
the plate was incubated for 4 h at 37°C. The supernatant was
discarded and 150 µl dimethyl sulfoxide was added to each
well until crystals dissolved completely. The absorbance value of
each well was measured using an ELISA reader with 490 nm as the
test wavelength and 280 nm as the reference wavelength.
Apoptosis assay
Cells (2×105) were seeded in 6-well
plates, cultured and then harvested. The cells were washed with PBS
and then stained with 5 µl Annexin V and 5 µl
propidium iodide (PI) for 15 min at room temperature in the dark
according to the manufacturer's instructions (BD Biosciences, San
Jose, CA, USA). The apoptosis rate (%) of the stained cells was
analyzed using a Beckman Coulter Epics Altra II cytometer (Beckman
Coulter, Brea, CA, USA).
Statistical analysis
Data were analyzed using SPSS 13.0 software (SPSS,
Inc., Chicago, IL, USA) and presented as the mean ± standard error
of the mean ± standard error of the mean of at least three
independent experiments. Two-tailed Student's t-test was used for
comparisons of two independent groups. Gene expression was analyzed
by a Mann-Whitney U test. P<0.05 was considered to indicate a
statistically significant difference.
Results
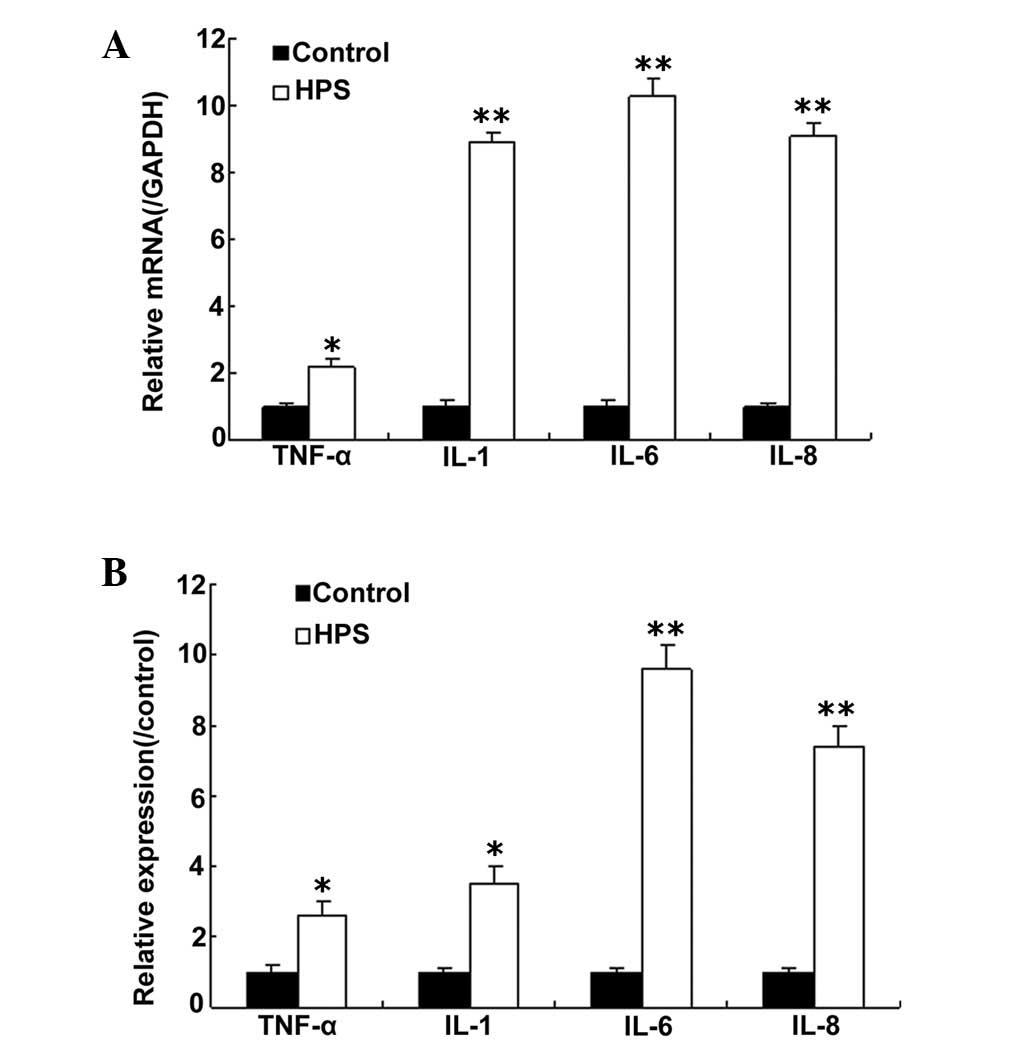
IL-6 expression in rat models of HPS
To investigate whether cytokine production is
induced in HPS, rat models were established. The cytokine profile
was assayed by a micro-array analysis. The data showed that TNF-α,
IL-8, IL-1 and IL-6 mRNA were increased compared with the controls.
This result was confirmed by RT-qPCR (Fig. 1A). Notably, IL-6 mRNA levels were
significantly increased compared with TNF-α, IL-8 and IL-1. Next,
IL-6 protein from the cultured lung tissue was analyzed ELISA
(Fig. 1B). The result indicated
that IL-6 protein was also increased in the rat models.
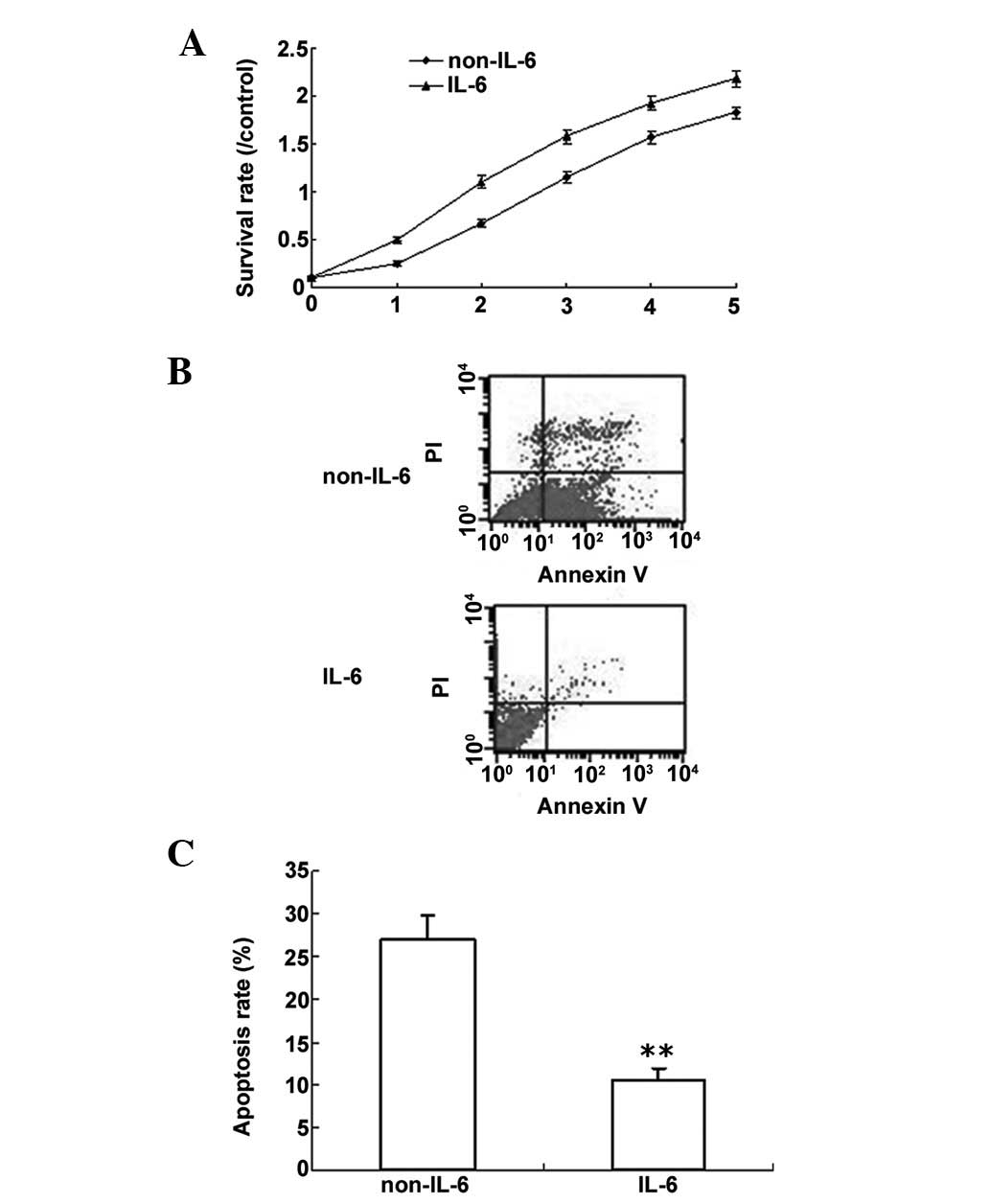
IL-6 promotes proliferation and inhibits
LPS-induced apoptosis in PMVECs
Based on the above data, PMVECs were used to
investigate the role of IL-6. PMVEC proliferation was analyzed by
the MTT method. The data showed that IL-6 promoted cell
proliferation (Fig. 2A). LPS can
induce cell apoptosis; thus, in order to determine whether IL-6
protects cells from apoptosis, PWVECs were treated with LPS, with
or without IL-6, and cell apoptosis was analyzed by flow cytometry.
It was determined that IL-6 decreased cell apoptosis induced by LPS
(Fig. 2B and C).
IL-6 activates the JAK/STAT3 signaling
pathway in pulmonary microvascular endothelial cells
IL-6 is induced during HPS. To examine the
possibility that secretory factors from PMVECs could activate the
JAK/STAT3 signaling pathway, the cells were treated with IL-6 for
12 h, and phosphorylation of JAKs was then monitored by western
blot analysis. As shown in Fig.
3A, phosphorylation of JAK2 was increased significantly, when
the cells were treated with IL-6 for 12 h, but not JAK1 and JAK3
phosphorylation. mRNA levels of JAK1, JAK2 and JAK3 were not
changed, but STAT3 mRNA levels increased greatly in the cells
following IL-6 treatment. (Fig.
3B).
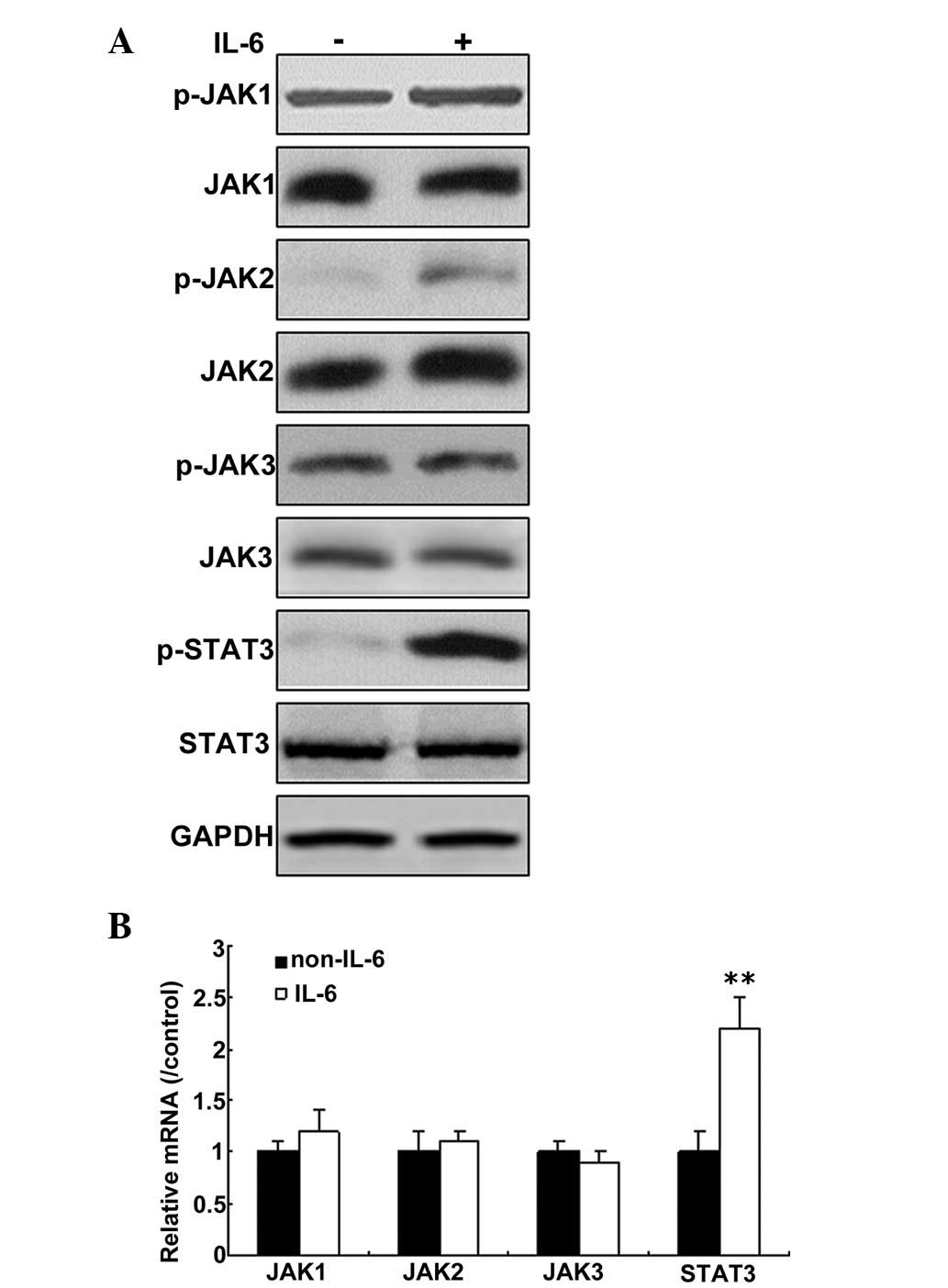 | Figure 3IL-6 activates the JAK/STAT3 signaling
pathway in PMVECs. (A) IL-6 upregulated JAK and STAT3 expression in
PMVECs. Total protein was isolated from the cells, and p-JAK, JAK1,
2 and 3, p-STAT3, STAT3, and GAPDH protein were detected by western
blotting. (B) IL-6 was not observed to effect the levels of JAK and
STAT3 mRNA in PMVECs. **P<0.01 vs. non-IL-6. Total
RNA was isolated from the cells, and JAK and STAT3 mRNA was
detected by reverse transcription-quantitative polymerase chain
reaction. The results are representative of three independent
experiments with similar results. IL, interleukin; JAK, janus
kinase; STAT, signal transducer and activator of transcription; p-,
phosphorylated; PMVECs, pulmonary microvascular endothelial
cells. |
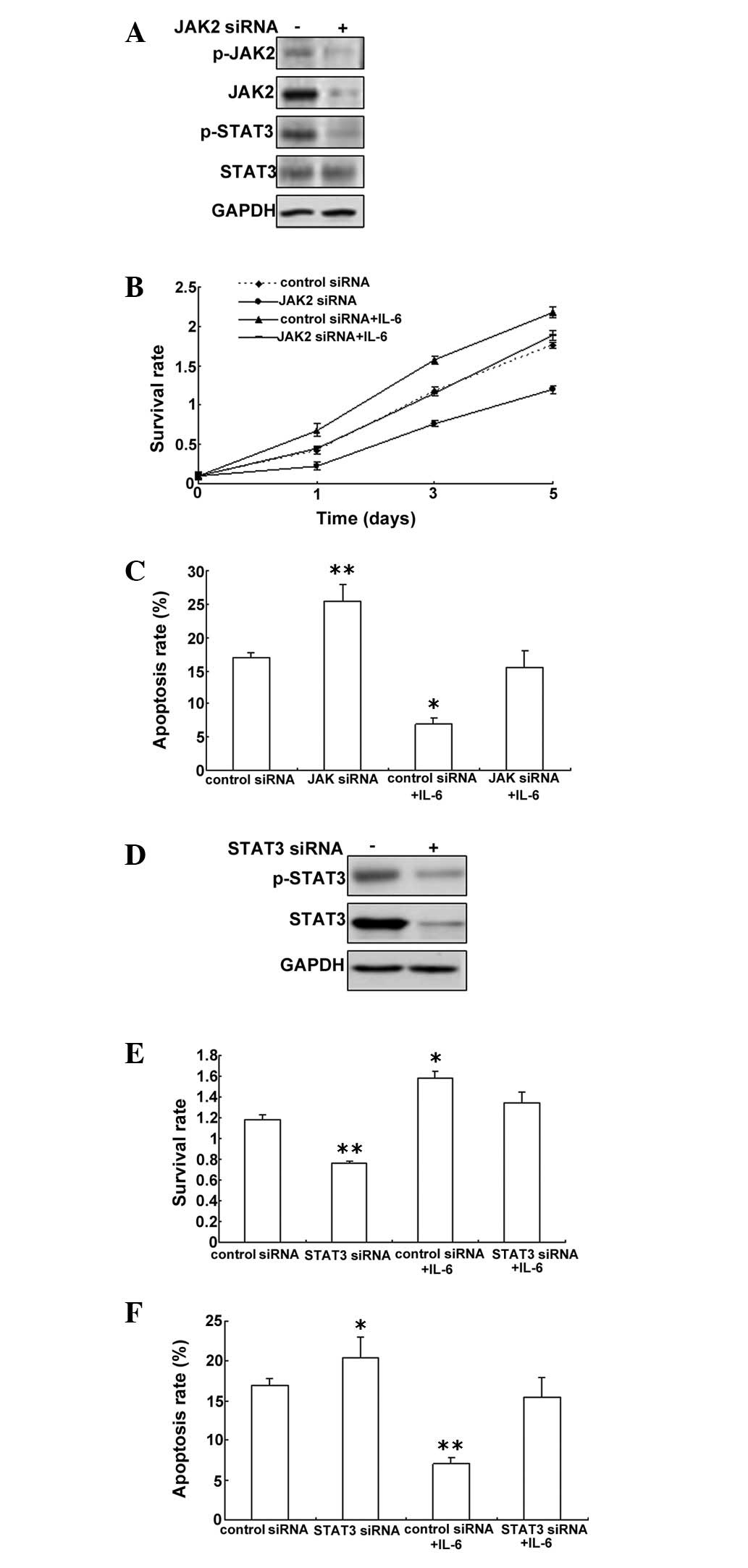
IL-6-induced downregulation of JAK
expression inhibits proliferation and induces apoptosis in
PMVECs
To investigate the role of JAK2/STAT3 in
IL-6-induced cell proliferation, an MTT assay was performed on
PMVECs with JAK2 down-regulation to analyze cell proliferation. The
data showed that JAK2 was knocked down effectively by RNAi and
STAT3 was inactive (Fig. 4A).
Downregulation of JAK2 could inhibit cell proliferation of PWVECs
exposed to IL-6 (Fig. 4B). After
JAK2 downregulation, the cell apoptosis rate increased, but when
the cells were treated with IL-6, the apoptosis rate began to
decrease (Fig. 4C). To further
investigate whether STAT3 has a role in PMVECs, STAT3 was knocked
down and not active in the cells with STAT3 siRNA (Fig. 4D). STAT3 was observed to promote
PMVEC cell proliferation and inhibit apoptosis (Fig. 4E and F).
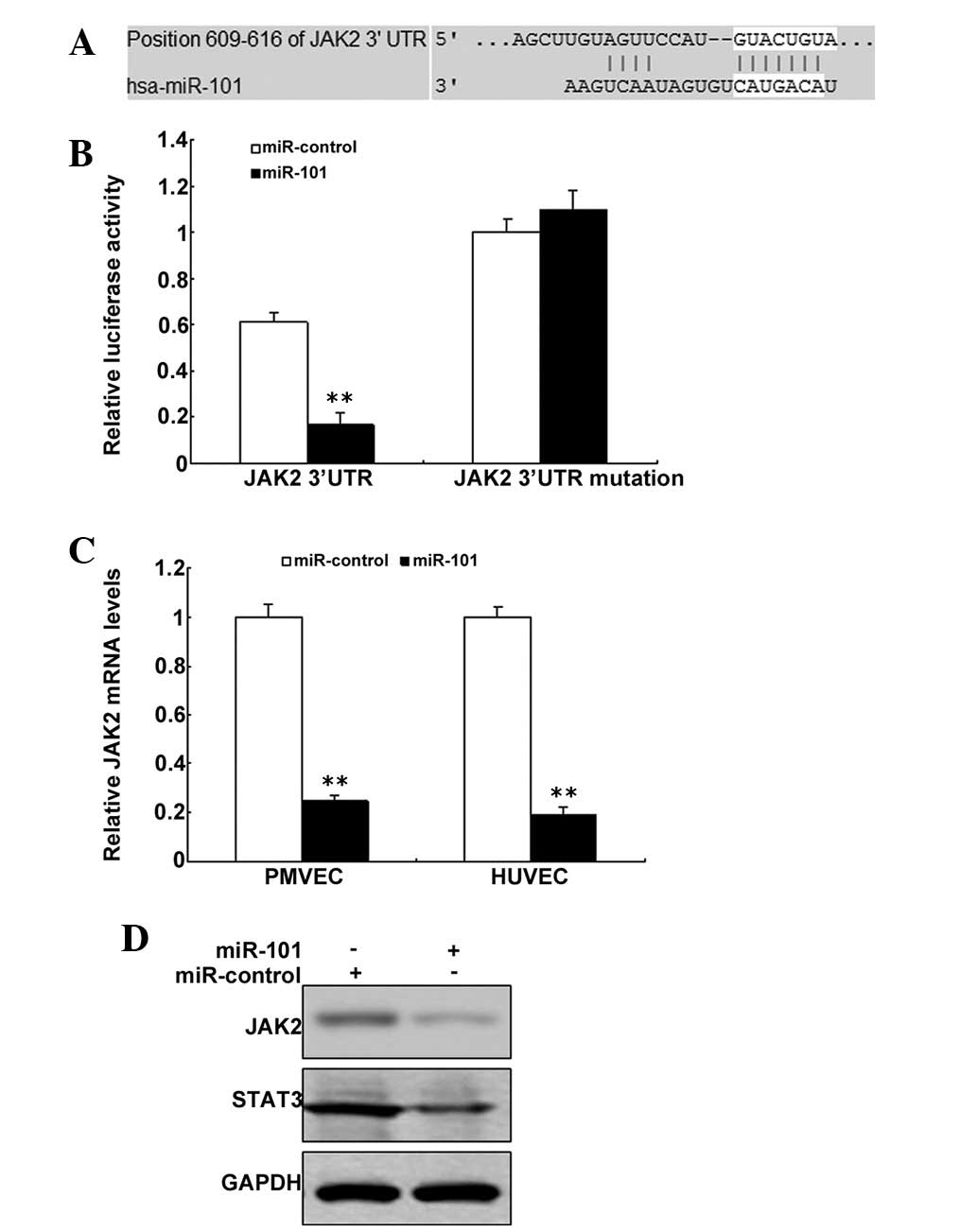
JAK2 is a target gene of miR-101
Bioinformatic prediction indicated that JAK2 may be
a target gene of miR-101 (Fig.
5A). The pGL3 plasmid was modified by adding the human 3′-UTR
or the 3′-UTR with mutations in regions complementary to miR-101
seed regions downstream of the firefly luciferase gene. PMVECs were
transiently co-transfected with negative control (mock) or miR-101
together with the indicated luciferase constructs, and luciferase
activity was analyzed after 48 h. When the PMVECs were
co-transfected with miR-101 or luciferase vectors and pGL3-JAK2-Mut
or pGL3-JAK2-WT, the luciferase activity was markedly lower in
cells with pGL3-JAK2-WT transfection and was rescued in the cells
with pGL3-JAK2-Mut transfection (Fig.
5B). JAK2 mRNA was decreased in the PMVECs with miR-101
treatment than the control (Fig.
5C). JAK2 protein was also decreased in PMVECs with miR-101
treatment compared with the control (Fig. 5D).
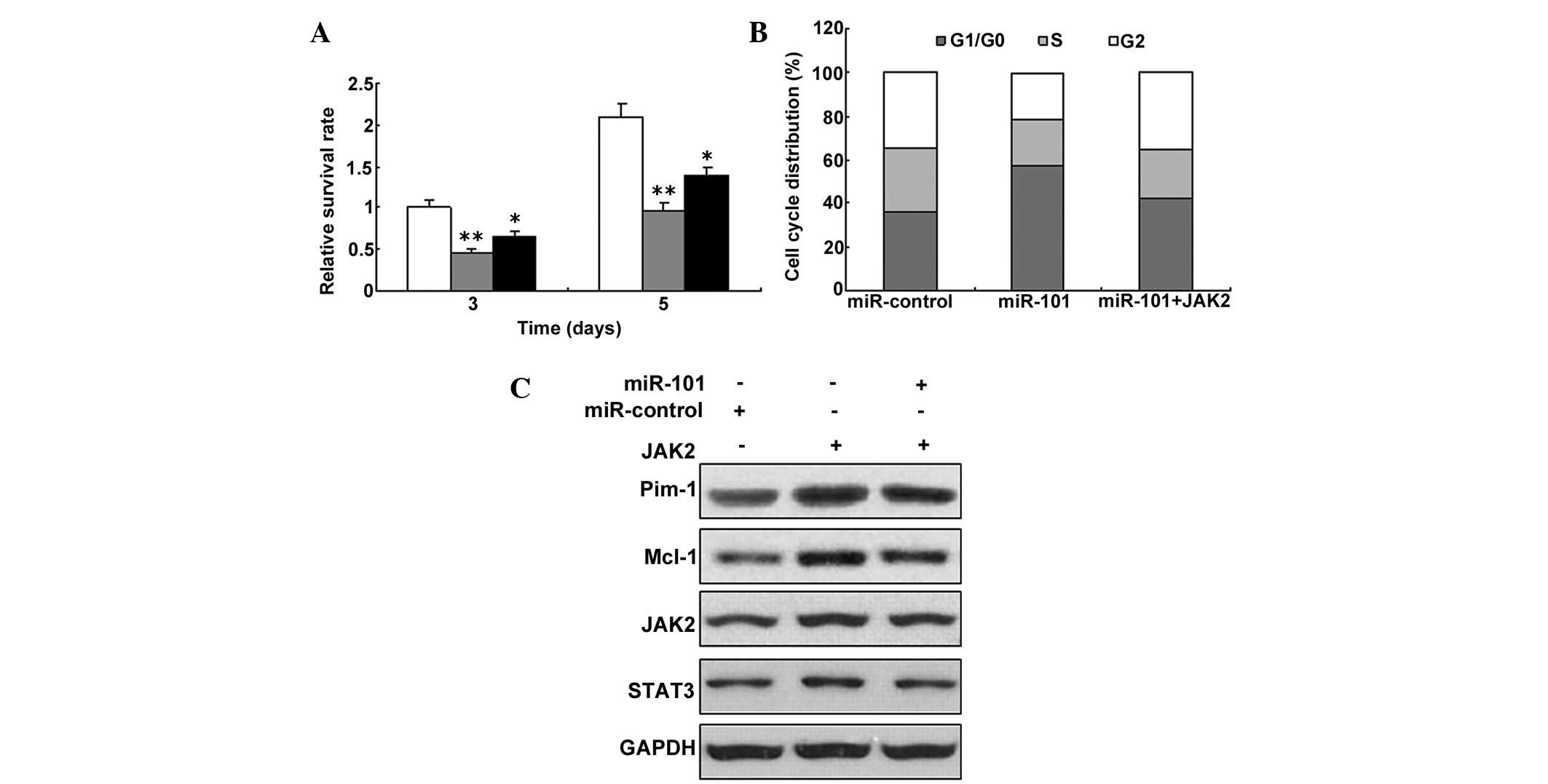
miR-101 inhibits pPMVEC proliferation by
targeting the IL-6/JAK2/STAT3 pathway
Given the fact that JAK2 was the target gene of
miR-101, the present study aimed to determine whether miR-101
regulates the proliferation of PMVECs. It was tested whether
miR-101 inhibits the proliferation of the cells with overexpression
of JAK2. The result showed that miR-101 in PMVECs inhibited cell
proliferation with JAK2 overexpression (Fig. 6A). Since the JAK/STAT3 signaling
pathway is an important pathway involved in PMVECs, the levels of
several proteins and downstream proteins in the JAK/STAT3 signaling
pathway were analyzed. To further investigate cell proliferation,
the cell cycle was analyzed by flow cytometry, and the results
showed that miR-101 could induce G1 phase arrest (Fig. 6B). A downstream protein of the
JAK/STAT3 singling pathway, Pim-1, was decreased by miR-101
over-expression. A similar result was obtained for Mcl-1, an
important downstream protein of this pathway (Fig. 6C).
Discussion
In the present study, the mechanism of proliferation
of PMVECs in HPS was investigated. It was determined that IL-6 was
enhanced in rat models of HPS and promoted cell proliferation and
inhibited apoptosis. In addition, cell proliferation was inhibited
and apoptosis was induced in the cells with JAK2 or STAT3
downregulation. Thus a novel role of miR-101 in regulating cell
proliferation by suppressing JAK2 expression in PMVECs was
determined.
IL-6 is a pro-inflammatory cytokine characterized as
a potent activator of STAT3. They function cooperatively to promote
cellular proliferation and inhibit apoptosis (14). As shown in Fig. 1, a significant difference in the
expression of IL-6 was observed between HPS and normal rats. This
study focused on the IL-6/JAK/STAT3 signaling pathway and
investigated its function in PMVECs. As expected, the presence of
high IL-6 and p-STAT3 expression was observed in PMVECs, and a
significant correlation was observed between the two. This
indicated the activation of JAK/STAT3 signaling in HPS. Similar
results were obtained by Jiang et al (15), where it was reported that IL-6
promoted STAT3 activation significantly at the posttranslational
level in vitro and indicated that IL-6/STAT3 signaling was
involved in human biliary epithelial cell migration and wound
healing.
Binding of IL-6 to the IL-6 receptor can
phosphorylate the tyrosine of gp130, and further activate JAK
family members including JAK1, JAK2, and tyrosine kinase2 (Tyk2).
STAT3 is phosphorylated by activated JAK and translocates into the
nucleus to control the expression of substrates (14). STAT3 mediates signal transduction
and transcription via various cytokines and growth factors. p-STAT3
accelerates the cell cycle, promotes cellular differentiation and
inhibits apoptosis through regulating the expression of Cyclin D1,
C-myc, Bcl-xl and vascular endothelial growth factor (VEGF), which
is an essential factor in cell proliferation (16). The present results are consistent
with previous results and propose that the IL-6/JAK/STAT3 signaling
pathway is active in PMVECs and may represent a novel therapeutic
target. In recent years, inhibitors of the IL-6/JAK/STAT3 signaling
pathway have emerged as a promising treatment option for certain
diseases. For instance, siRNA-STAT3 therapy was demonstrated to be
effective in ovarian cancer, and a suppressor of cytokine signaling
treatment was also demonstrated to be active in blocking the STAT3
signaling in liver disease such as cirrhosis (17–21).
This study has provided insight into a potential strategy for the
treatment of tumors and other proliferative diseases, such as
HPS.
There are certain studies that demonstrated that
miR-101 is important in cancer cell proliferation by targeting
ZEB1, ZEB2, Rac1, RanBP9, Cox-2, EZH2, CPEB1 and Stathmin1
(22–28). However, in HPS, there has not
previously been a study to determine the role of miRNA-101. The
present study identified that JAK2 was regulated by miR-101 in
PMVECs, and that miR-101 inhibited cell proliferation.
In conclusion, the present study demonstrated that
miR-101 inhibited the cell proliferation of PMVECs by
downregulation of JAK2. Low miR-101 expression may be an
unfavorable prognostic factor in patients with HPS. The data in the
present study indicates that miR-101 may be a therapeutic target in
HPS. In addition, the present findings expand the knowledge of the
pathogenic mechanisms underlying HPS.
References
|
1
|
Eshraghian A, Kamyab AA and Yoon SK:
Pharmacological treatment for hepatopulmonary syndrome. Biomed Res
Int. 2013:6701392013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Polavarapu N and Tripathi D: Liver in
cardiopulmonary disease. Best Pract Res Clin Gastroenterol.
27:497–512. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grace JA and Angus PW: Hepatopulmonary
syndrome: Update on recent advances in pathophysiology,
investigation and treatment. J Gastroenterol Hepatol. 28:213–219.
2013. View Article : Google Scholar
|
|
4
|
Zhang J and Fallon MB: Hepatopulmonary
syndrome: Update on pathogenesis and clinical features. Nat Rev
Gastroenterol Hepatol. 9:539–549. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sussman NL, Kochar R and Fallon MB:
Pulmonary complications in cirrhosis. Curr Opin Organ Transplant.
16:281–288. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang ZJ and Yang CQ: Progress in
investigating the pathogenesis of hepatopulmonary syndrome.
Hepatobiliary Pancreat Dis Int. 9:355–360. 2010.PubMed/NCBI
|
|
7
|
Valenti A and Caimi G: Physiopathological,
clinical and therapeutic aspects of hepatopulmonary syndrome. Clin
Ter. 161:e123–e128. 2010.PubMed/NCBI
|
|
8
|
Macêdo LG and Lopes EP: Hepatopulmonary
syndrome: An update. Sao Paulo Med J. 127:223–230. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ho V: Current concepts in the management
of hepatopulmonary syndrome. Vasc Health Risk Manag. 4:1035–1041.
2008.
|
|
10
|
Rodríguez-Roisin R and Krowka MJ:
Hepatopulmonary syndrome-a liver-induced lung vascular disorder. N
Engl J Med. 358:2378–2387. 2008. View Article : Google Scholar
|
|
11
|
Varghese J, Ilias-basha H, Dhanasekaran R,
Singh S and Venkataraman J: Hepatopulmonary syndrome-past to
present. Ann Hepatol. 6:135–142. 2007.PubMed/NCBI
|
|
12
|
Kartha RV and Subramanian S: Competing
endogenous RNAs (ceRNAs): New entrants to the intricacies of gene
regulation. Front Genet. 5:82014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Banno K, Iida M, Yanokura M, Kisu I, Iwata
T, Tominaga E, Tanaka K and Aoki D: MicroRNA in cervical cancer:
OncomiRs and tumor suppressor miRs in diagnosis and treatment.
Scientific World Journal. 2014:1780752014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chang Q, Bournzou E, Sansone P, Berishaj
M, Gao SP, Daly L, Wels J, Theilen T, Granitto S, Zhang X, et al:
The IL-6/JAK/STAT3 feed-forward loop drives tumorigenesis and
metastasis. Neoplasia. 15:848–862. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang GX, Zhong XY, Cui YF, et al:
IL-6/STAT3/TFF3 signaling regulates human biliary epithelial cell
migration and wound healing in vitro. Mol Biol Rep. 37:3813–3818.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang G, Qian P, Jackson FR, Qian G and Wu
G: Sequential activation of JAKs, STATs and xanthine
dehydrogenase/oxidase by hypoxia in lung microvascular endothelial
cells. Int J Biochem Cell Biol. 40:461–470. 2008. View Article : Google Scholar
|
|
17
|
Xiong H, Zhang ZG, Tian XQ, Sun DF, Liang
QC, Zhang YJ, Lu R, Chen YX and Fang JY: Inhibition of JAK1,
2/STAT3 signaling induces apoptosis, cell cycle arrest and reduces
tumor cell invasion in colorectal cancer cells. Neoplasia.
10:287–297. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Januma N, Shima H, Nakamura K and Kikuchi
K: Protein tyrosine phosphatase epsilonC selectively inhibits
interleukin-6- and IL-10- induced JAK-STAT signaling. Blood.
98:3030–3034. 2001. View Article : Google Scholar
|
|
19
|
Takeda K and Akim S: Stat family of
transcription factors in cytokine-mediated biological responses.
Cytokine Growth Factor Rev. 11:199–207. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kuczkowski J, Sakowicz-Burkiewica M,
Izycka-Swieszewska E, Mikaszewski B and Pawełczyk T: Expression of
tumor necrosis factor-α, interleukin-1α, interleukin-6 and
interleukin-10 in chronic otitis media with bone osteolysis. ORL J
Otorhinolaryngol Relat Spec. 73:93–99. 2011. View Article : Google Scholar
|
|
21
|
Nason R, Jung JY and Chole RA:
Lipopolysaccharide-induced osteoclastogenesis from mononuclear
precursors: A mechanism for osteolysis in chronic otitis. J Assoc
Res Otolaryngol. 10:151–160. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guo F, Cogdell D, Hu L, Yang D, Sood AK,
Xue F and Zhang W: miR-101 suppresses the epithelial-to-mesenchymal
transition by targeting ZEB1 and ZEB2 in ovarian carcinoma. Oncol
Rep. 31:2021–2028. 2014.PubMed/NCBI
|
|
23
|
Lin X, Guan H, Li H, Liu L, Liu J, Wei G
and Zhang W: miR-101 inhibits cell proliferation by targeting Rac1
in papillary thyroid carcinoma. Biomed Rep. 2:122–126.
2014.PubMed/NCBI
|
|
24
|
Barbato C, Pezzola S, Caggiano C,
Antonelli M, Frisone P, Ciotti MT and Ruberti F: A lentiviral
sponge for miR-101 regulates RanBP9 expression and amyloid
precursor protein metabolism in hippocampal neurons. Front Cell
Neurosci. 8:372014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lin C, Huang F, Zhang YJ, Tuokan T and
Kuerban G: Roles of MiR-101 and its target gene Cox-2 in early
diagnosis of cervical cancer in Uygur women. Asian Pac J Cancer
Prev. 15:45–48. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Y, Xiang W, Wang M, Huang T, Xiao X,
Wang L, Tao D, Dong L, Zeng F and Jiang G: Methyl jasmonate
sensitizes human bladder cancer cells to gambogic acid-induced
apoptosis through downregulation of EZH2 expression by miR-101. Br
J Pharmacol. 171:618–635. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xiaoping L, Zhibin Y, Wenjuan L, Zeyou W,
Gang X, Zhaohui L, Ying Z, Minghua W and Guiyuan L: CPEB1, a
histone-modified hypomethylated gene, is regulated by miR-101 and
involved in cell senescence in glioma. Cell Death Dis. 4:e6752013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang R, Wang HB, Hao CJ, Cui Y, Han XC, Hu
Y, Li FF, Xia HF and Ma X: MiR-101 is involved in human breast
carcinogenesis by targeting Stathmin1. PLoS One. 7:e461732012.
View Article : Google Scholar : PubMed/NCBI
|