Introduction
Toll-like receptors (TLRs) were first characterized
as being able to recognize various pathogen-associated molecular
patterns and forming part of the first line of defense against
pathogens (1). Furthermore, TLRs
were shown to have a broad range of biological activities, as they
are involved in immune regulation, tissue repair and have
anti-viral and anti-neoplastic effects. TLR ligands have been
considered as potential targets for the development of therapeutic
drugs for infectious diseases, autoimmune diseases and cancer
(2,3). 1-[2-methylpropyl]-1H
-imidazo[4,5-c]quinolin-4-amine) (imiquimod) is a synthetic agonist
of TLR7 that has been used as topical therapeutic for certain skin
neoplasms such as basal cell carcinoma (4). Accumulating evidence indicated that
the mechanism of action of imiquimod is not restricted to the
elicitation of the innate and adaptive immune responses, but also
directly induces autophagy or apoptosis in certain types of tumor
cell (5–7).
Autophagy is a physiological process during which
the cell's cytosol and organelles are sequestered within
double-membrane vesicles termed autophagosomes, which are then
fused with lysosomes for degradation and recycling of cytoplasmic
constituents (8). Autophagy
provides a survival advantage for the organism under multiple
stresses, including starvation, oxygen deprivation and intoxication
(9). However, if autophagy is
overactivated and allowed to escalate, it eventually leads to cell
death due to depletion of proteins important for the functioning of
complex signaling pathways (10).
Dysfunctional autophagy has been observed in numerous types of
human cancers and increasing evidence suggested that the
cytotoxicity of certain anti-tumor agents is based on the induction
of autophagy or promotion of a non-apoptotic, autophagic cell death
(11).
For the past few decades, gastric cancer mortality
has decreased in most areas of the world due to the availability of
screening for early detection and improved treatments (12); however, resistance to chemotherapy
and tumor recurrence still make this disease the second leading
cause of cancer-associated mortality worldwide (13). Therefore, novel and effective
therapies for gastric cancer are in demand. Based on the previously
demonstrated efficacy of imiquimod on colon cancer cells (5), the present study hypothesized that
imiquimod may have suppressive effects against other
gastrointestinal cancers, including gastric cancer.
To the best of our knowledge, no previous study has
investigated whether imiquimod can decrease the viability of
gastric cancer cells, and its mechanisms of its action have also
remained elusive. Therefore, the present study assessed the
anti-proliferative and cytotoxic effects of the TLR7 agonist
imiquimod on a gastric cancer cell line and investigated the
probable molecular mechanisms of its action.
Materials and methods
Cell culture and reagents
The human gastric cancer cell line SGC-7901 was
obtained from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China). Cells were maintained in RPMI-1640 medium
containing 10% fetal bovine serum (both from Invitrogen; Thermo
Fisher Scientific, Waltham, MA, USA) with 100 U/ml penicillin and
100 µg/ml streptomycin (both from Gibco; Thermo Fisher
Scientific) at 37°C in a humidified incubator containing 5%
CO2. Prior to all incubations with imiquimod (Enzo Life
Sciences, Farmingdale, NY, USA), cells were seeded into dishes and
allowed to attach for 24 h to reach the logarithmic growth
phase.
Cell viability assay
The effects of imiquimod on cell viability was
evaluated in vitro using the
3-(4,5-dimeth-ylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma-Aldrich, St. Louis, MO, USA) assay. Cells were seeded in
96-well plates at 5×103 cells/well and then incubated
with various doses of imiquimod (25, 50, 100 or 200 µg/ml)
for various time periods (12, 24, 48 and 72 h). Subsequently, 20
µl MTT solution (5 mg/ml) was added to each well and the
plates were re-incubated at 37°C for 4 h. The supernatant was
carefully removed, formazan products were dissolved in 150
µl dimethyl sulfoxide (Sigma-Aldrich) with agitation for 5
min, and the absorbance was measured at 490 nm using a PowerWave XS
microtiter plate reader (Bio-Tek, Winooski, VT, USA). The effects
of autophagy regulators on cell viability were detected following
1-h pre-incubation with 5 mM 3-methyladenine (3-MA) followed by
treatment with imiquimod for 24 h.
Transmission electron microscopy
For electron microscopy, the SGC-7901 cells were
treated with 100 µg/ml imiquimod for 12 h, fixed in ice with
cold glutaraldehyde (Wolsen, Xi'an, China; 2.5% solution in
phosphate buffer) at 4°C for a minimum of 4 h and then fixed in 1%
osmium tetroxide (Wolsen). Following dehydration in a graded
ethanol series, samples were embedded in Agar 100 resin (Agar
Scientific, Stansted, UK). Ultrathin sections were stained with
uranyl acetate and lead citrate and analyzed by transmission
electron microscopy (H-7650; Hitachi, Tokyo, Japan).
Monodansylcadaverine (MDC) staining
MDC, a specific marker of autophagic vacuoles, was
employed to stain autophagosomes. 1×105 cells were
seeded into a six-well plate and allowed to attach for 24 h. The
cells were treated with 100 µg/ml imiquimod for 12 h and
then stained with 0.05 mM MDC (Sigma-Aldrich) in fresh RPMI-1640
for 30 min at 37°C in the dark. After three washes with
phosphate-buffered saline (PBS), the cells were harvested and
rinsed with PBS. Intracellular MDC intensity was measured using a
flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) at an
excitation wavelength of 360 nm.
Western blot analysis
Cells were seeded in six-well plates at a density of
1×105 cells/well and treated with imiquimod (0, 25, 50
or 100 µg/ml for 0, 6, 8, 12, 24 or 36 h) with or without
3-MA (5 mM; Sigma-Aldrich). Following two washes with cold PBS,
cells were lysed in radioimmunoprecipitation assay lysis buffer on
ice for 1 h. The lysates were centrifuged at 20,627 × g at 4°C for
20 min and the supernatant was collected. The protein concentration
was measured using a bicinchoninic acid protein assay kit (Pierce™
BCA Protein Assay Reagent A; cat no. 23222; Thermo Fisher
Scientific). Equal amounts of protein (20–30 µg) were
applied to the SDS-PAGE gel (12% for Bcl-2, Bax, LC3 and cleaved
caspase 3 and 10% for beclin-1, cleaved caspase 9). After
electrophoresis, the resolved proteins were transferred to a
polyvinylidene difluoride membrane (Wolsen). The membrane was
blocked in Tris-buffered saline supplemented with 5% skimmed milk
for 2 h and subsequently incubated with the following primary
antibodies at 4°C overnight: Rabbit anti-beclin 1 monoclonal
antibody (mAb; cat. no. 2026-1; 1:1,000 dilution; Epitomics,
Burlingame, CA, USA), rabbit anti-light chain (LC)3 polyclonal
antibody (pAb) (cat no. L7543; 1:500 dilution; Sigma-Aldrich),
rabbit anti-B-cell lymphoma 2 (Bcl-2) pAb (cat. no. BS3711; 1:500
dilution; Bioworld Technology, Nanjing, China), rabbit
anti-Bcl-2-associated X protein (Bax) pAb (cat. no. BS2538; 1:500
dilution; Bioworld Technology), rabbit anti-cleaved caspase-3 pAb
(cat. no. 9661; 1:1,000 dilution; Cell Signaling Technology, Inc.,
Danvers, MA, USA), rabbit anti-cleaved caspase-9 pAb (cat no. 9505;
1:1,000 dilution; Cell Signaling Technology, Inc.) or mouse
anti-GAPDH mAb (cat. no. Sc-32233; 1:200 dilution; Santa Cruz
Biotechnology, Inc.). Subsequently, membranes were incubated with
the secondary anti-mouse and anti-rabbit antibodies (1:5,000
dilution; Bioworld Technology) for 2 h at 37°C. GAPDH was used as
the loading control. The specific protein expression levels were
detected using an enhanced chemiluminescence kit (Western Blotting
Luminol Reagent; cat no. sc-2048; Santa Cruz Biotechnology, Dallas,
TX, USA) in accordance with the manufacturer's instructions. Images
of blots were captured using a Chemiluminescence Imaging System
(CoreBio, Seoul, Republic of Korea).
Flow cytometry
SGC-7901 cells were seeded in six-well plates at
1×105 cells/well and following 1 h of pre-incubation
with 5 mM 3-MA, cells were treated with imiquimod (100
µg/ml) for 24 h. Cells were harvested, washed twice with
pre-cooled PBS and centrifuged at 20,627 × g for 10 min. Cells were
then re-suspended in 500 µl binding buffer, and incubated in
5 µl annexin V-fluorescein isothiocyanate (FITC) and 10
µl propidium iodide (PI) included in a Annexin V-FITC
Apoptosis 81 kit (cat no. K101-100; BioVision, Milpitas, CA, USA)
at room temperature in the dark for 15 min. Staining was then
quantified using flow cytometry (Beckman Coulter, Brea, CA, USA)
within 1 h.
Statistical analysis
All experiments were repeated at least three times,
and value are expressed as the mean ± standard deviation. Data were
analyzed using Student's two-tailed t-test or analysis of variance
using SPSS 16.0 statistical software (SPSS, Inc., Chicago, IL, USA)
to determine the significance of differences between groups.
P<0.01 was considered to indicate a statistically significant
difference between values.
Results
Anti-proliferative effects of
imiquimod
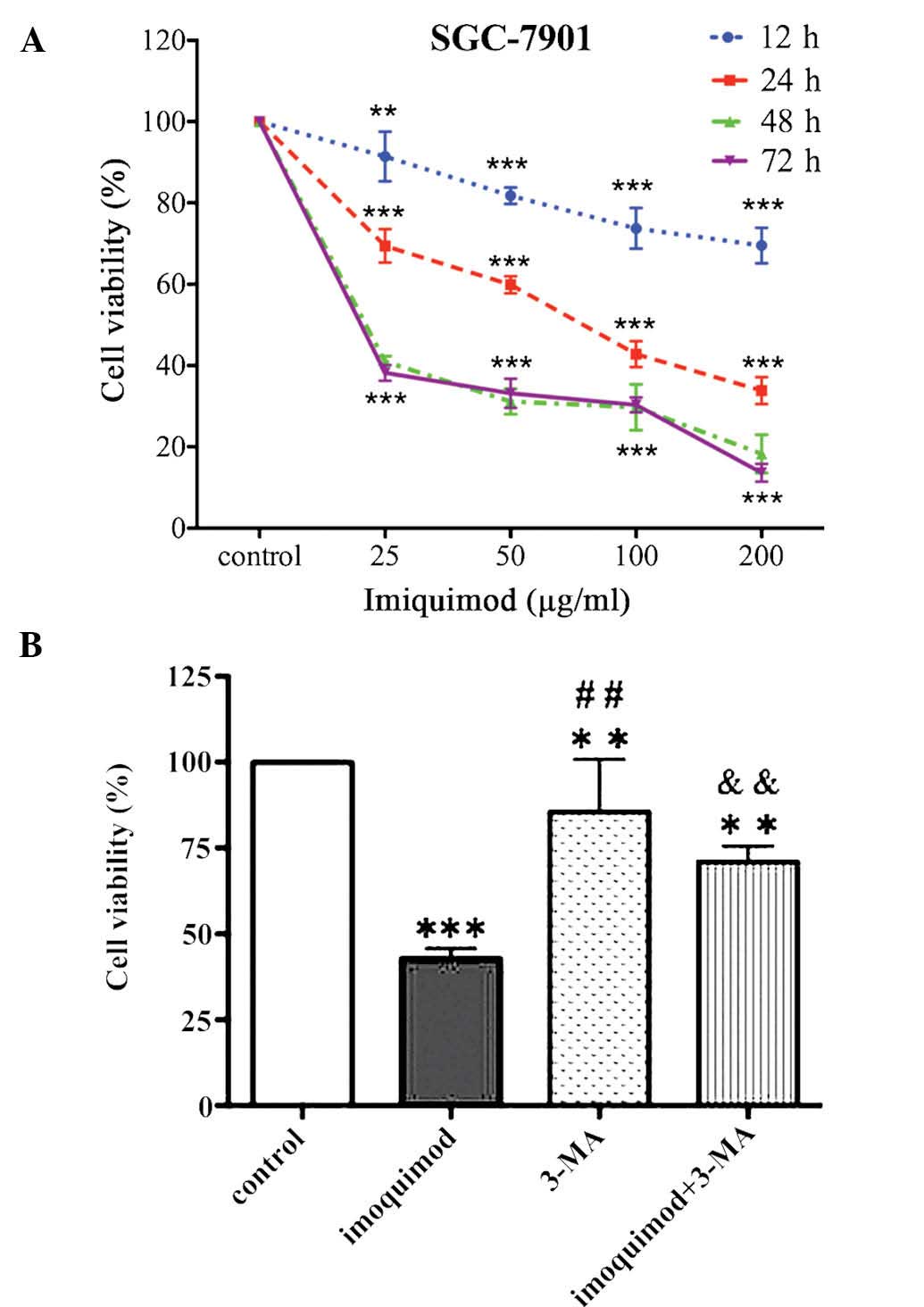
As shown in Fig.
1A, imiquimod inhibited the proliferation of SGC-7901 cells in
a dose- and time-dependent manner. The IC50 value of
imiquimod at 24 h was 71.13±7.81 µg/ml. While effects of low
doses of imiquimod (25 and 50 Mg/ml) on the cell viability were
obvious at as early as 12 h of treatment, high doses of imiquimod
(100 and 200 µg/ml) led to a >25% reduction in cell
viability at 48 and 72 h of treatment. Following treatment of
SGC-7901 cells with the selective autophagy inhibitor 3-MA (5 mM),
the viability of SGC-7901 cells was reduced compared with that of
control cells; furthermore, pre-treatment with 5 µM of 3-MA
significantly attenuated the growth-inhibitory effects of imiquimod
(100 µg/ml) (Fig. 1B).
Imiquimod increases the occurrence of
autophagy in gastric cancer cells
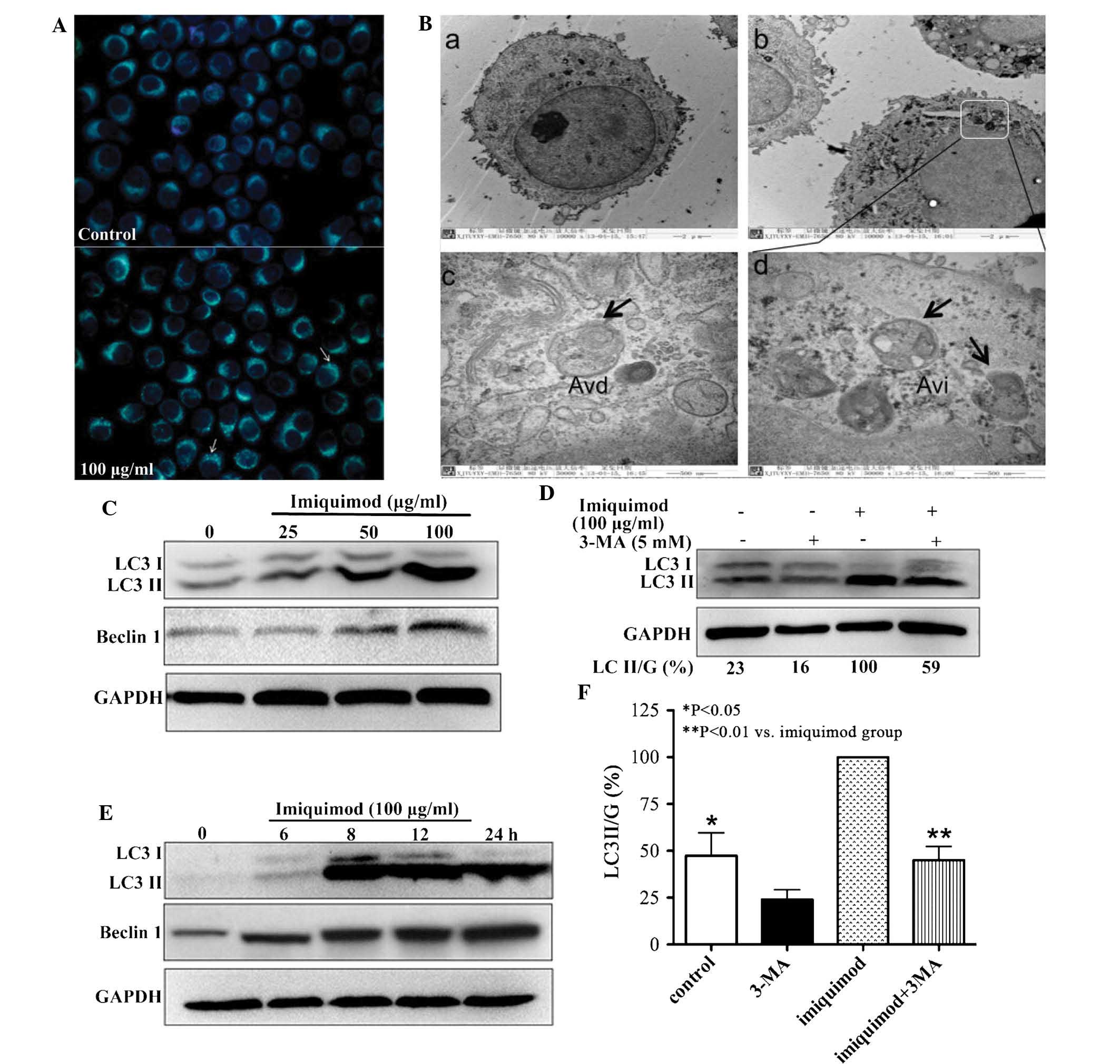
To test the autophagic activity induced by
imiquimod, MDC staining was performed fist. As shown in Fig. 2A, the control cells showed faint
fluorescence, while the SGC-7901 cells treated with 100
µg/ml imiquimod accumulated MDC into granular structures of
high fluorescence intensity. Furthermore, transmission electron
microscopic analysis was performed to examine the formation of
autophagic vesicles. Compared with the untreated cells, an
abundance of autophagosome-like vacuoles containing double-membrane
structures were identified in the cytoplasm of 100 µg/ml
imiquimod-treated SGC-7901 cells following 12 h of incubation
(Fig. 2B).
Imiquimod enhances the expression of
autophagy-associated proteins
The levels of LC3II, which is considered the
hallmark of mammalian autophagy (14), as well as the expression of
autophagy-associated protein Beclin1 were examined by western blot
analysis. After treatment of SGC-7901 cells with various
concentrations of imiquimod for 24 h, a marked and
concentration-dependent upregulation of LC3II expression and an
increase in the LC3II/LC3I ratio were observed compared with the
untreated control cells. Furthermore, imiquimod increased Beclin1
levels in a time-dependent manner (Fig. 2C). The induction of LC3II and
Beclin1 expression was observed at as early as 6 h of treatment
with imiquimod (Fig. 2D).
Furthermore, the increase of LC3II expression was blocked in the
presence of the autophagy inhibitor 3-MA (Fig. 2E).
Apoptosis is induced by imiquimod
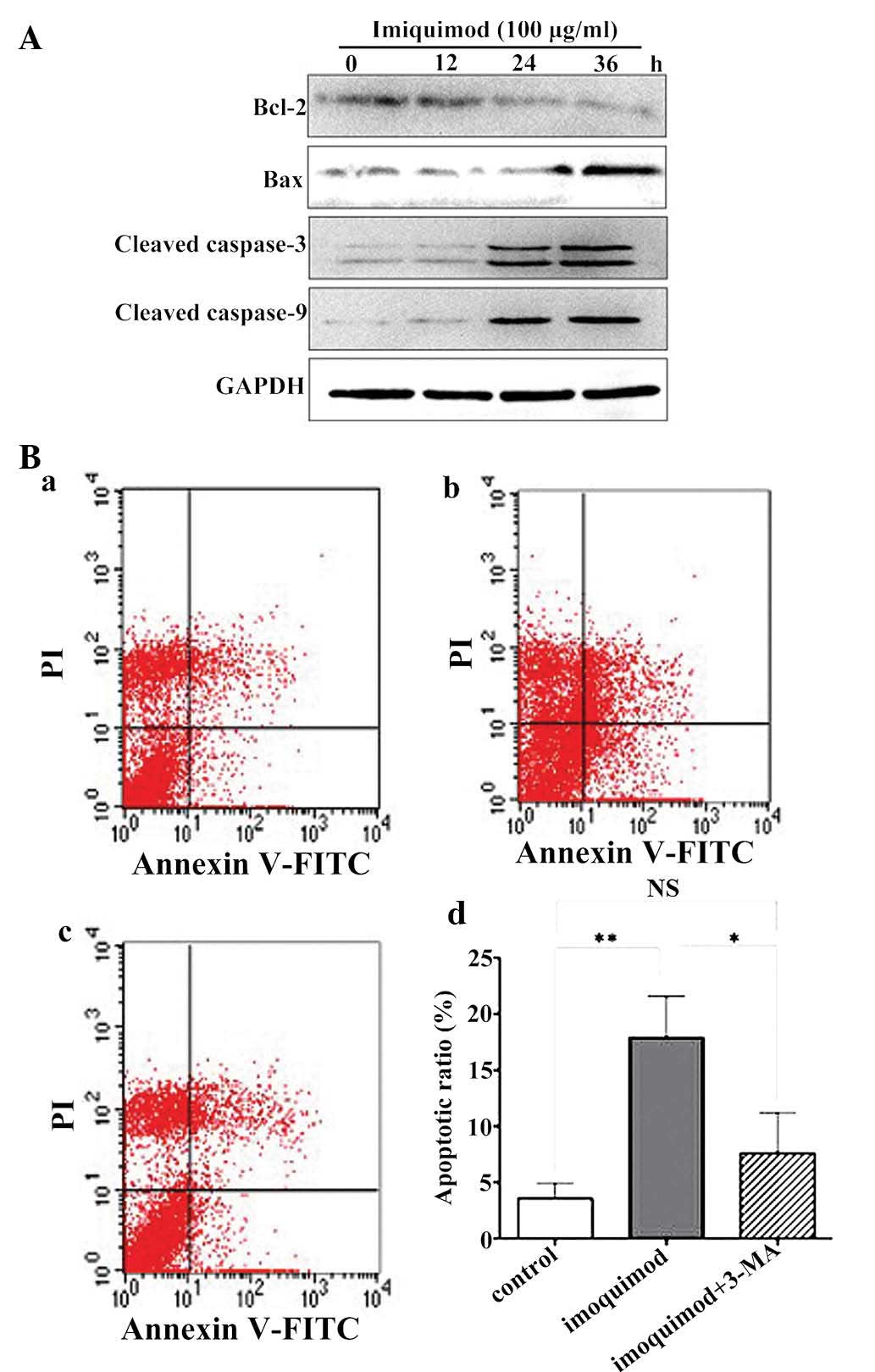
To confirm that imiquimod was able to induce
apoptosis in SGC-7901 cells, the present study assessed the
expression of apoptotic signaling proteins by western blot
analysis. After treatment with 100 µg/ml imiquimod, a
substantial decrease in the expression of Bcl-2 and increased
cleavage of caspase-3, caspase-9 and Bax was observed at 24 and 36
h of treatment with imiquimod (Fig.
3A). Next, the present study assessed whether imiquimod-induced
autophagy has a protective or an apoptosis-promoting effect on
SGC-7901 cells. For this purpose, the apoptotic rate was assessed
by flow cytometry following Annexin V-FITC/PI staining. As shown in
Fig. 3B, the number of apoptotic
cells was enhanced following 24 h of treatment with 100
µg/ml imiquimod, which was, however, significantly decreased
by pre-treatment with 5 mM 3-MA. This result indicated that
imiquimod-induced apoptosis of SGC-7901 cells proceeded, at least
in part, via an autophagic process, and that autophagy had a
pro-apoptotic function.
Discussion
Imiquimod, the most commonly used TLR7 agonist, has
been approved by the United States Food and Drug Administration as
the first-line topical treatment for basal cell carcinoma (15). In addition, an increasing number of
studies have clearly demonstrated that imiquimod shows
anti-proliferative effects in a variety of benign and malignant
carcinomas, including colorectal (5), prostate (16), bladder (17), renal (18) and oral cancer (19). However, whether imiquimod is able
to decrease the viability of gastric cancer cells as well as its
mechanisms of action have remained elusive.
In the present study, an MTT assay showed that
imiquimod markedly decreased the viability and proliferation of
SGC-7901 cells in a dose- and time-dependent manner. Treatment with
100 µg/ml imiquimod started to inhibit cell viability at an
early time-point (12 h), while cell viability was reduced to
>25% at 36 h. A previous study by our group (20) indicated that incubation with 100
µg/ml imiquimod for 24 h resulted in SGC-7901-cell apoptosis
as demonstrated by an increased ratio of early apoptotic cells and
marked ultrastructural changes in the cells, while this phenomenon
was not observed at 12 h. In order to prove that imiquimod induces
apoptosis in gastric cancer cells, the present study assessed the
expression of a number of apoptotic proteins. Activation of cleaved
caspase-3 and -9 indicated that imiquimod-induced apoptosis may
proceed via the caspase-dependent pathway. A significant change in
the levels of apoptosis-associated proteins was observed at 24 and
36 h of treatment with imiquimod, while autophagy-associated
proteins were enhanced at as early as 8 h. Therefore, present study
aimed to further investigate the mechanism by which imiquimod
induced cell death in gastric cancer cells; in particular, the
early effects of imiquimod treatment leading to cell death and the
implication of autophagy were investigated.
Autophagy, classified as type II programmed cell
death, is a mechanism which can be clearly distinguished from
apoptosis. Autophagy was originally described as a mechanism to
maintain homeostasis through the depredation of long-lived proteins
and damaged organelles (21). It
is usually induced as a mechanism of stress tolerance when cells
are exposed to starvation, hypoxia, reactive oxygen species or
anti-tumor treatment. It has been suggested that dysregulation of
autophagy has a vital role in gastric tumorigenesis (22). Previous studies also indicated that
imiquimod exerts a direct autophagic effect through the activation
of autophagy-associated molecules to promote cell death in certain
tumor types (5,7). The present study demonstrated that
the rate of autophagy was significantly higher in imiquimod-treated
cells compared with that in the untreated controls, as evidenced by
the obvious increase in autophagic markers, including autophagosome
formation and positivity for MDC staining. Treatment with imiquimod
also dose-dependently induced the upregulation of autophagy makers
LC3II and Beclin1. 3-MA, an inhibitor of autophagy, was observed to
decrease the toxicity of imiquimod, while it did not completely
prevent cell death. The apoptotic rate was increased following
treatment with 100 µg/ml imiquimod for 24 h, while it was
significantly decreased by pre-treatment with 3-MA. These results
indicated that in SGC-7901 cells, autophagy is a cell death
mechanism activated by imiquimod, and that blocking of autophagy
with 3-MA inhibited the activation of apoptosis.
In conclusion, to the best of our knowledge, the
present study was the first to demonstrate that imiquimod exhibited
efficacy against gastric cancer cells in vitro. The results
indicated that imiquimod induced autophagy as well as apoptosis,
which led to the death of SGC-7901 cells; furthermore, early
induction of autophagy by imiquimod may trigger apoptosis. Based on
these findings, it is concluded that imiquimod may be a potential
anti-tumor agent for the treatment of gastric cancer.
Acknowledgments
The present study was supported by the Fundamental
Research Funds for the Central Universities (National twelfth
five-year plan of science and technology; grant no.
2012BAJ18B03-03).
References
|
1
|
Akira S and Takeda K: Toll-like receptor
signalling. Nat Rev Immunol. 4:499–511. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Suzuki N, Suzuki S and Yeh WC: IRAK-4 as
the central TIR signaling mediator in innate immunity. Trends
Immunol. 23:503–506. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tse K and Horner AA: Update on toll-like
receptor-directed therapies for human disease. Ann Rheum. 66(Suppl
3): iii77–iii80. 2007. View Article : Google Scholar
|
|
4
|
Miller RL, Gerster JF, Owens ML, Slade HB
and Tomai MA: Imiquimod applied topically: A novel immune response
modifier and new class of drug. Int J Immunopharmacol. 21:1–14.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yi JY, Jung YJ, Choi SS, Hwang J and Chung
E: Autophagy-mediated anti-tumoral activity of imiquimod in Caco-2
cells. Biochem Biophys Res Commun. 386:455–458. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schön MP, Wienrich BG, Drewniok C, Bong
AB, Eberle J, Geilen CC, Gollnick H and Schön M: Death
receptor-independent apoptosis in malignant melanoma induced by the
small-molecule immune response modifier imiquimod. J Invest
Dermatol. 122:1266–1276. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang SW, Liu KT, Chang CC, Chen YJ, Wu
CY, Tsai JJ, Lu WC, Wang YT, Liu CM and Shieh JJ: Imiquimod
simultaneously induces autophagy and apoptosis in human basal cell
carcinoma cells. Br J Dermatol. 163:310–320. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rubinsztein DC, Gestwicki JE, Murphy LO
and Klionsky DJ: Potential therapeutic applications of autophagy.
Nat Rev Drug Discov. 6:304–312. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schönthal AH: Endoplasmic reticulum stress
and autophagy as targets for cancer therapy. Cancer Lett.
275:163–169. 2009. View Article : Google Scholar
|
|
10
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Crew KD and Neugut AI: Epidemiology of
gastric cancer. World J Gastroenterol. 12:354–362. 2006.PubMed/NCBI
|
|
13
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. IntJ Cancer. 127:2893–2917. 2010. View Article : Google Scholar
|
|
14
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tyring S, Conant M, Marini M, Van Der
Meijden W and Washenik K: Imiquimod; an international update on
therapeutic uses in dermatology. Int J Dermatol. 41:810–816. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Han JH, Lee J, Jeon SJ, Choi ES, Cho SD,
Kim BY, Kim DJ and Park JH and Park JH: In vitro and in vivo growth
inhibition of prostate cancer by the small molecule imiquimod. Int
J Oncol. 42:2087–2093. 2013.PubMed/NCBI
|
|
17
|
Smith EB, Schwartz M, Kawamoto H, You X,
Hwang D, Liu H and Scherr DS: Antitumor effects of
imidazoquinolines in urothelial cell carcinoma of the bladder. J
Urol. 177:2347–2351. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schwartz MJ, Liu H, Hwang DH, Kawamoto H
and Scherr DS: Antitumor effects of an imidazoquinoline in renal
cell carcinoma. Urology. 73:1156–1162. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ahn MY, Kwon SM, Cheong HH, Park JH, Lee
J, Min SK, Ahn SG and Yoon JH: Toll-like receptor 7 agonist,
imiquimod, inhibits oral squamous carcinoma cells through apoptosis
and necrosis. J Oral Pathol Med. 41:540–546. 2012.PubMed/NCBI
|
|
20
|
Jiang J, Dong L, Qin B, Guo X, Li H, Shi H
and Liu Y: Expression of Toll-like receptor 7 (TLR7) in gastric
cancer cell lines and effects of TLR7 agonist on proliferation and
apoptosis of SGC-7901 cells in vitro. J South Med Univ.
34:1606–1610. 2014.In Chinese.
|
|
21
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu D and Gao M: A hypothesis. Med Sci
Monit. 19:794–796. 2013. View Article : Google Scholar : PubMed/NCBI
|