Introduction
Smith-Magenis Syndrome (SMS) is a rare
neurodevelopmental disorder caused by a microdeletion on chromosome
17p11.2 and has an incidence of ~1 in 25,000 live births (1). Individuals with SMS exhibit wide
phenotypic variability. Certain patients show mild to moderate
mental retardation, while others exhibit atypical facial features,
and cardiac, renal and otolaryngologic abnormalities (1–3).
However, the correlation between genotype and phenotype is not well
understood in SMS (3–5).
For a number of decades, monozygotic (MZ) twin
comparisons have been used to identify the contributions of nature
(heredity) and nurture (environment) (6). Recently, several studies have
suggested that genetic and epigenetic factors exhibit a role in
phenotypic variance (2,3,7).
Phenotypic variability is often observed between unrelated or
related individuals or twins with the same microdeletion syndrome,
such as 22q11.2 (3,8–10).
Observation of the genetic history of a syndrome in MZs often leads
to a greater understanding of the phenotypic variability (11). Recently, a high resolution single
nucleotide polymorphism (SNP)-array allowed for the detection of
copy number variants (CNVs) as well as SNPs. Halder et al
(12) reported a case in which
twins carrying the 22q11.2DS microdeletion had discordant
phenotypes with a different sized genetic deletion. Another
technique, exome sequencing, offers an efficient and affordable
method to investigate the genetic factors involved in human
diseases (13–16). B.D. Solomon performed exome
sequencing on MZs discordant for VACTERL (vertebral anomalies, anal
atresia, cardiac malformations, tracheo-esophageal fistula, renal
anomalies and limb abnormalities) association-type congenital
malformations (17). It was
hypothesized that this method may reveal discordant variants that
are able to explain the cause(s) of disease (17). Hicks et al (11) reported a case of MZs with SMS with
different phenotypes; however, further molecular investigation of
the discordant phenotypes was not conducted. The standard study
design in these cases is to use current molecular techniques, such
as an SNP array and exome sequencing, in order to identify
correlations between SMS and the concomitant phenotypes, in
addition to investigating the factor that may contribute to the
phenotypic variability in MZs with SMS. In the present study,
current molecular techniques, including as an SNP array and exome
sequencing, were used un order to identify correlations between SMS
and the concomitant phenotypes, in addition to investigating the
factors, which may contribute to the phenotypic variability in MZs
with SMS
Patients and methods
Clinical description
A 24-year-old woman, gravida 3, para 1, with a
monochorionic diamniotic twin pregnancy was referred to the
prenatal department of the Department of Obstetrics and
Gynaecology, The First Affiliated Hospital of of Sun Yat-Sen
University (Guangzhou, China) at 28 weeks and 4 days gestation. The
patient presented with the presence of a ventricular septal defect
and stenosis of the main pulmonary artery in one twin; the other
twin was normal. Her two previous pregnancies were uncomplicated,
and the third pregnancy was conceived spontaneously with a
25-year-old man. Ultrasound examination (GE Voluson 730 Expert; GE
Healthcare Life Sciences, Vienna, Austria) at the cessation of
menstruation, at 9 weeks, and at 16 weeks revealed that the twins
were developmentally delayed by 2 weeks. These ultrasound findings
and her delayed menstrual cycle postponed the expected delivery
date by 2 weeks. A thin dividing membrane between the fetuses was
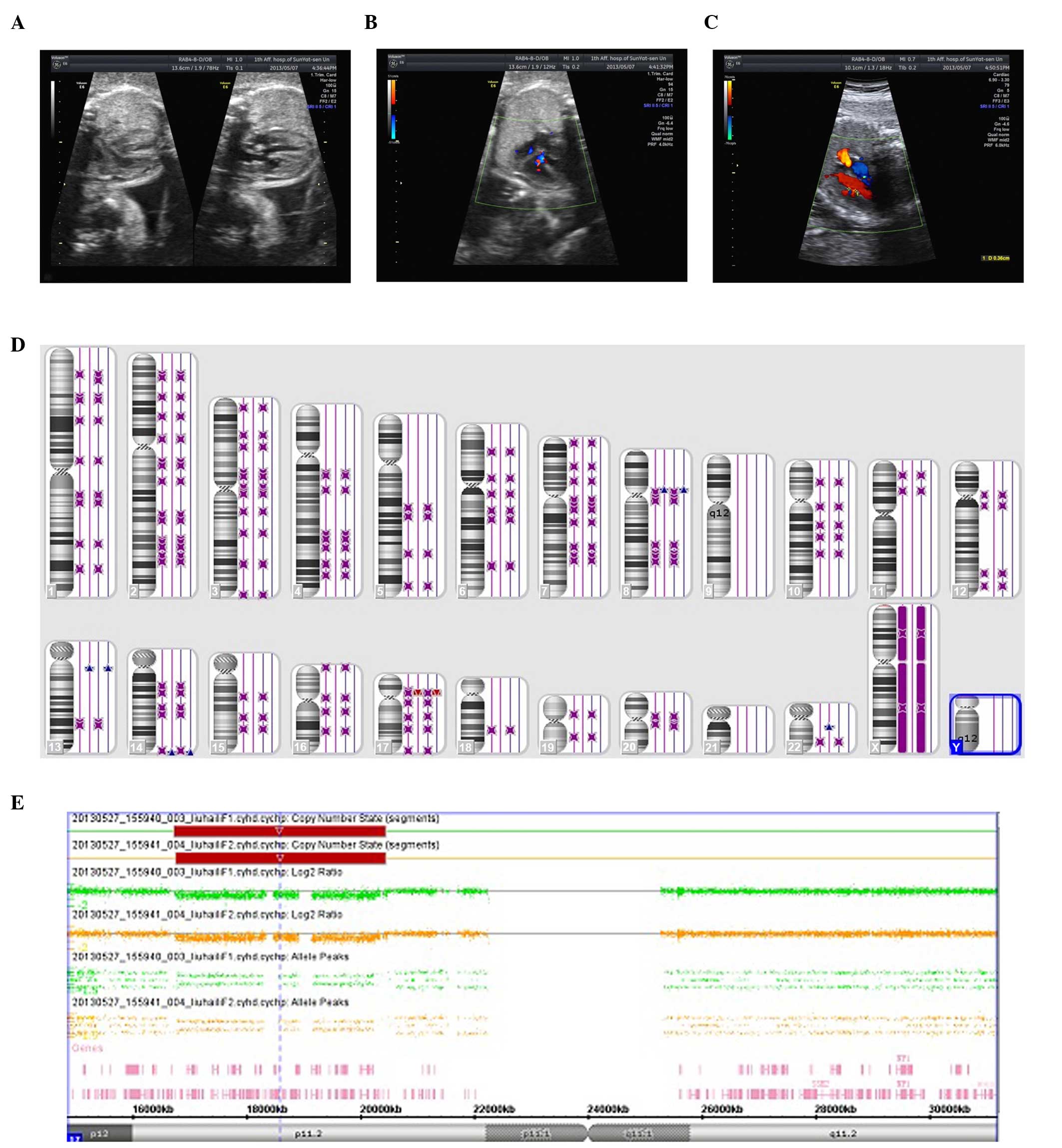
visualized on ultrasound. The twins (Fig. 1A) were diagnosed with fetal growth
retardation (FGR) at 28 weeks and 4 days gestation due to the
2-week developmental delay. A sonographic examination suggested
measurable growth discordance. In addition, one twin (twin 1) was
observed to have a ventricular septal defect (3 mm) and stenosis of
the main pulmonary artery (Fig.
1C), while the other twin (twin 2), with the exception of FGR
showed no abnormalities (Fig. 1B).
The amniotic fluid volume (AFV) of twin 1 and twin 2 was 48
mm3 and 31 mm3, respectively. Following
genetic counseling, an ultrasound-guided amniocentesis of the
fetuses was performed for a cytogenetic analysis and chromosomal
microarray analysis (CMA) at 29 weeks and 4 days gestation. The
ultrasound examination of the AFV of twin 1 and 2 prior to
cordocentesis were 13 and 21 mm3, respectively.
One week following surgery, the AFV of the two
fetuses recovered to normal with volumes of 37 and 28
mm3, in twins 1 and 2, respectively, but the ventricular
septal defect was enlarged to 3.5 mm in twin 1. Additionally, twin
1 developed a more severe form of FGR and absent end-diastolic flow
of the umbilical artery. The ultrasound testing performed 2 weeks
after amniocentesis revealed oligohydramnion of both fetuses, with
AFVs of 9.7 mm3 (twin 1) and 12 mm3 (twin 2).
Doppler investigations (GE Voluson 730 Expert; GE Healthcare Life
Sciences) of the umbilical arteries and ductus venosus revealed no
abnormal findings. Analysis of the amniocentesis sample by CMA
revealed a 17p11.2 deletion, which was characteristic of SMS. The
patient was called back for additional genetic counseling to
discuss the results and prognosis of both fetuses. The patient
decided to continue the pregnancy after multiple discussions.
Ultrasound examination at 34 weeks and 3 days
gestation revealed the intrauterine fetal death of both twins and
the absence of amniotic fluid. The two fetuses were delivered by
stillbirth at the Obstetric Department of The First Affiliated
Hospital of Sun Yat-Sen University). Both fetuses were visually
nearly normal. However, organ malformations could not be determined
as the parents did not consent to a fetal autopsy.
Ethical approval was obtained for this study from
the Ethics Committee of the First Affiliated Hospital of Sun
Yat-Sen University. All data were collected with the informed
consent of the patient.
Cytogenetic analysis
Routine cytogenetic analysis using G-banding
techniques at a resolution of 550 bands was performed. Briefly, a
10 ml sample of amniotic fluid was collected and subjected to
amniocyte culture according to the standard cytogenetic protocol
(18). A 5 ml sample of parental
blood was collected from each parent and subjected to lymphocyte
culture according to the standard blood cytogenetic protocol
(18).
Whole-genome high-resolution SNP
array
CMA on the uncultured amniotic fluid was performed
using an Affymetrix cyto HD Array (Affymetrix, High Wycombe, UK).
DNA was amplified, labeled, and hybridized to a CytoScan HD array
platform, according to the manufacturer's protocol. The array was
designed specifically for cytogenetics research and offers more
than two million markers across the genome, including SNP probes
and probes to detect copy number variations (Cyto-arrays). CEL
files, obtained by scanning the CytoScan arrays, were analyzed with
Chromosome Analysis Suite software (Affymetrix) using the
annotations of genome version GRCH37 (hg19). Only those achieving
the manufacturer's quality cut-off measures (MAPD≤0.25; SNP
QC≥15.0; waviness standard deviation ≤0.12) were included in the
analysis. Gains and losses that affected a minimum of 50 markers
over a 100-kbp length were initially considered. Changes in copy
number were compared with the CNVs catalogued in the Database of
Genomic Variants (DGV; http://dgv.tcag.ca/dgv/app/home) and the University of
Santa Clara in California (UCSC; http://genome.ucsc.edu/) genome browser. The gene
content of the CNVs of interest was determined using the UCSC
browser, based on the Genome Reference Consortium Human Genome
(GRCH; build 37; http://genome.ucsc.edu/).
Exome sequencing
The zygosity of the twins was confirmed using small
tandem repeat markers. Polymorphic DNA marker analysis was
performed on the parental and fetal DNAs using an ABI Prism 3500
(Applied Biosystems, Foster City, CA, USA). Solution hybridization
exome capture was performed using the SureSelect Human All Exon v5
systems (Agilent Technologies, Santa Clara, CA, USA) using
biotinylated RNA baits to hybridize sequences that corresponded
with exons (19). The
manufacturer's software version 1.5, which is compatible with
Illumina paired-end sequencing software (Illumina, San Diego, CA,
USA), was used. The manufacturer's specifications for the v5 kit
report that the capture regions total ~50 Mb, which corresponded
with the Consensus Conserved Domain Sequences database (http://www.sanger.ac.uk/resources/databases/encode/)
that contains >1,000 non-coding RNAs as well as Gencode Project
defined exons. Targeted regions included the exons of 18,113 genes
of the Consensus Conserved Domain Sequences database, exons of
additional genes, miRNAs, and non-coding RNA genes, totaling 30,241
genomic features and 51,646,629 targeted bases. Flowcell
preparation and sequencing were performed according to the protocol
for the HiSeq 2000 sequencer (Illumina) using 100-bp paired-end
reads to generate sufficient data such that ≥85% of the targeted
bases were accurately genotyped, with an average coverage of 150×.
Image analysis and base calling were performed on all data lanes
using the Illumina HiSeq Control Software (HCS) v2.0.5 and RTA
software v1.17.20 (Illumina) with default parameters.
Results
Chromosome analysis
The chromosome analysis revealed a normal male
karyotype in both fetuses. The parents also had a normal karyotype.
However, the CMA indicated that the fetuses had a deletion at
chromosome 17 as arr 17p11.2 (16, 761, 814-20, 433, 502)x1 in one
fetus, and, arr 17p11.2 (16, 761, 814-20, 433, 502)x1 in the other
fetus. Both harbored the RAI1 gene, which is a critical gene
involved in SMS (Fig. 1E). The
parents had a normal CMA result, indicating a de novo
deletion in both fetuses.
CNV identification
With the exception of the 17p11.2 deletion, the CMA
identified a total of 9 CNVs. The identified CNVs were classified
as being absent in the DG) or present in the DGV (Table I). The sizes of these CNVs vary
from 114 to 656 kb. In the fetuses, the duplication at 13q12.31 was
inherited from the father, and the other CNV at 8p11.22 was a de
novo event. Exome sequencing revealed the same disorder in the
twins. A polymorphic DNA marker showed that the twins were a
perfect match monozygosity (data not provided) and confirmed the
twin pairs by high-density SNP microarray analysis (Fig. 1D). In brief, neither microarray
analysis nor exome sequencing revealed an obvious discordant
genetic anomaly (CNV or exome mutation) that would readily explain
the presence of the congenital anomalies in one of the twins.
 | Table IIdentity of copy number variants in
the family members. |
Table I
Identity of copy number variants in
the family members.
| Family member | CNVs | Size (kb) | Gene | Type | Inherited |
|---|
| Father | 13q12.13 (26, 803,
491-26, 918, 775)x3 | 115 | CDK8 | Novel | |
| 14q32.33 (106, 072,
250-106, 728, 149)x3 | 656 | KIAA0125, ADAM6 | PD | |
| 16p11.2 (33, 596,
761-33, 774, 726)x1 | 178 | | PD | |
| 19p12 (20, 588,
836-20, 716, 153)x1 | 127 | ZNF826P | PD | |
| 22q11.22 (23, 063,
020-23, 258, 369)x3 | 195 | MIR650, IGLL5 | PD | |
| Mother | 1p11.2 (121, 225,
582-121, 339, 317)x3 | 114 | EMBP1 | PD | |
| 12p13.31 (8, 006,
510-8, 124, 048)x3 | 118 | SLC2A14, SLC2A3 | PD | |
| 19p12 (20, 588,
836-20, 720, 705)x1 | 132 | ZNF826P | PD | |
| 14q32.33 (106, 251,
147-106, 728, 149)x3 | 477 | KIAA0125, ADAM6 | PD | |
| Twin 1 | 13q12.13 (26, 803,
491-26, 918, 933)x3 | 115 | CDK8 | Novel | pat |
| 8p11.22 (39, 247,
097-39, 386, 952)x3 | 140 | ADAM5P, ADAM3A | PD | dn |
| 14q32.33 (106, 072,
264-106, 728, 149)x3 | 656 | KIAA0125, ADAM6 | PD | pat |
| Twin 2 | 13q12.13 (26, 803,
491-26, 927, 389)x3 | 124 | CDK8 | Novel | pat |
| 8p11.22 (39, 254,
032-39, 384, 337)x3 | 130 | ADAM5P, ADAM3A | PD | dn |
| 14q32.33 (106, 072,
264-106, 728, 149)x3 | 656 | KIAA0125, ADAM6 | PD | pat |
| 22q11.22 (23, 063,
020-23, 258, 369)x3 | 195 | MIR650, IGLL5 | PD | pat |
Discussion
The present study reports a pair of MZ twins, with a
3.7 Mb micro-deletion at 17p11.2. This type of deletion is 'common'
as 70% of patients with SMS have an ~3.7 Mb microdeletion in the
17p11.2 region (https://decipher.sanger.ac.uk/syndrome/8#overview).
However, the twin pair presented with discordant phenotypes: One
with a ventricular septal defect and stenosis of the main pulmonary
artery, and the other as nearly normal.
The majority of SMS features are due to an RAI1
haploinsufficiency (20), while
the variability and severity of the disorder are modified by other
genes in the 17p11.2 region. The functional role of RAI1 is not
completely understood, but based on homology and preliminary
studies, it is likely to be involved in transcription (21,22).
A phenotypic comparison between patients with deletions and
patients with RAI1 mutations shows that 21 out of 30 SMS features
are the result of an RAI1 haploinsufficiency, whereas cardiac
anomalies, speech and motor delay, hypotonia, short stature and
hearing loss are associated with 17p11.2 deletions rather than RAI1
mutations (7). In addition to
RAI1, the 3.7 Mb region deleted in the present case contains at
least 50 genes, including mitogen-activated protein kinase 7
(MAPK7) activated by an upstream cascade of kinases in response to
a wide variety of extracellular stimuli. Specific PRKM kinases
(MAPKKs or PRKMKs) have been shown to phosphorylate and activate
specific PRKMs in a given signaling pathway. Hayashi et al
(23) concluded that the MAPK7
pathway is critical for endothelial function and the maintenance of
blood vessel integrity. In addition, a common cause of intrauterine
growth restriction in humans is uteroplacental vascular
insufficiency, which increases the incidence of perinatal asphyxia
and neurodevelopmental disorders (24). A recent study showed that the
cardiac dimensions are spared and may be used for gestational age
estimation in growth-restricted fetuses resulting from
uteroplacental insufficiency (25). A case report of MZ twins with SMS
also shows both fetuses presenting with FGR (11), which is concordant with the present
study. However, whether a fetus with SMS is more prone to FGR and
whether haploinsufficiency of the PRKM7 gene is connected with
uteroplacental insufficiency requires further investigation.
The twins also had a duplication at 13q12.31 which
was inherited from the father. It was absent in the DGV and there
was no reported CNV in this region. The CNV only included one
protein-encoding gene CDK8 (cyclin-dependent kinases 8,*603184),
which encodes a member of the mediator complex, located at
13q12.13, a region of recurrent copy number gain in a substantial
fraction of colon cancers (26).
Spore investigation confirmed the importance of Cdk8 at multiple
stages of Dictyostelium development, although the severity of the
defect in spore production depends on the genetic background
(27). Although, it is important
to consider the possibility of incomplete penetrance, the family
did not know of any history of cancer. CDK8 is also a
haploinsufficiency gene in DECIPHER (28). While, deletions in the region have
clinical relevance, duplication of the same interval may be benign
(29). The present study also used
phenotype databases, such as OMIM or gene/mutation-specific
databases included in the specific CNV, and, considering the normal
phenotype of the father, the duplication at 13q12.31 appears to be
a benign polymorphism in the family and did not affect the twins
SMS-associated features.
The zygosity of the twins, first confirmed by a
polymorphic DNA marker and genomic DNA analysis on a
high-resolution SNP array, showed the twins were a perfect match,
except for the size of the CNV. However, except for experimental
error and its proximal endpoint located in a low copy number,
repeat or segmental duplication region, as evidenced on the UCSC
genome browser (hg19), no CNV or exonic mutations were identified
or affected known disease-associated loci that would explain the
congenital anomalies for the possibility of incomplete penetrance.
In this study, a high-resolution SNP array and exome sequencing of
the MZ but phenotypically discordant twins did not explain why only
one member of the twin pair was affected with features associated
with SMS. There are multiple possible explanations: The presence of
a regulatory factor that affects gene expression or a coding-region
CNV, the presence of mutations only in the affected individual, or
the mutations occurred in a region that may not be revealed by
current methods of CNV analysis. Eventually, developments in genome
testing may be able to evaluate these hypotheses, and there may be
a discordant mutation not detected by the applied CNV. Microarray
studies were conducted on DNA extracted from amniotic fluid, and
similar testing based on other tissue types may yield greater
success, as has recently been demonstrated in Proteus syndrome
(30). The occurrence of a twin
pregnancy may act on susceptible alleles to result in congenital
malformations. Although parental studies were performed in this
twin pair, molecular studies indicated that there were variants
present in the parents but not the 17p11.2 deletion. Additionally,
other testing modalities, such as methylation analysis, in
conjunction with CNV, may shed more light on disease pathogenesis.
Finally, it is possible that the causes of these congenital
malformations may not be directly gene-related and may involve a
primary, currently unidentified, environmental factor. Finally, on
a level more specific to SMS, it is likely that multiple
interacting genetic and environmental factors are involved in
determining phenotype.
Further studies are ongoing related to several
genetic variants of high interest (not located in the genes
previously shown to be associated with human disease) that were
found in the two twins and that may act in concert as
susceptibility factors.
Acknowledgments
The authors would like to thank the staff of the
Prenatal Center (Department of Obstetrics and Gynaecology, The
First Affiliated Hospital of Sun Yat-Sen University) for providing
patient data.
References
|
1
|
Smith ACM, Boyd KE, Elsea SH, et al:
Smith-Magenis syndrome. Genereviews. Pagon RA, Adam MP, Bird TD,
Dolan CR, Fong CT and Stephens K: Seattle (WA): 1993
|
|
2
|
Madduri N, Peters SU, Voigt RG, Llorente
AM, Lupski JR and Potocki L: Cognitive and adaptive behavior
profiles in Smith-Magenis syndrome. J Dev Behav Pediatr.
27:188–192. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Andrieux J, Villenet C, Quief S, Lignon S,
Geffroy S, Roumier C, de Leersnyder H, de Blois MC, Manouvrier S,
Delobel B, et al: Genotype phenotype correlation of 30 patients
with Smith-Magenis syndrome (SMS) using comparative genome
hybridisation array: Cleft palate in SMS is associated with larger
deletions. J Med Genet. 44:537–540. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gamba BF, Vieira GH, Souza DH, Monteiro
FF, Lorenzini JJ, Carvalho DR and Morreti-Ferreira D: Smith-Magenis
syndrome: Clinical evaluation in seven Brazilian patients. Genet
Mol Res. 10:2664–2670. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
De Leersnyder H: Smith-Magenis syndrome.
Handb Clin Neurol. 111:295–296. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hall JG: Twinning. Lancet. 362:735–743.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Girirajan S, Vlangos CN, Szomju BB,
Edelman E, Trevors CD, Dupuis L, Nezarati M, Bunyan DJ and Elsea
SH: Genotype-phenotype correlation in Smith-Magenis syndrome:
Evidence that multiple genes in 17p11.2 contribute to the clinical
spectrum. Genet Med. 8:417–427. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Singh SM, Murphy B and O'Reilly R:
Monozygotic twins with chromosome 22q11 deletion and discordant
phenotypes: Updates with an epigenetic hypothesis. J Med Genet.
39:e712002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Desmaze C, Scambler P, Prieur M, Halford
S, Sidi D, Le Deist F and Aurias A: Routine diagnosis of DiGeorge
syndrome by fluorescent in situ hybridization. Hum Genet.
90:663–665. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Van Hemel JO, Schaap C, Van Opstal D,
Mulder MP, Niermeijer MF and Meijers JH: Recurrence of DiGeorge
syndrome: Prenatal detection by FISH of a molecular 22q11 deletion.
J Med Genet. 32:657–658. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hicks M, Ferguson S, Bernier F and Lemay
JF: A case report of monozygotic twins with Smith-Magenis syndrome.
J Dev Behav Pediatr. 29:42–46. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Halder A, Jain M, Chaudhary I and Varma B:
Chromosome 22q11.2 microdeletion in monozygotic twins with
discordant phenotype and deletion size. Mol Cytogenet. 5:132012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fahiminiya S, Almuriekhi M, Nawaz Z,
Staffa A, Lepage P, Ali R, Hashim L, Schwartzentruber J, Abu
Khadija K, Zaineddin S, et al: Whole exome sequencing unravels
disease-causing genes in consanguineous families in Qatar. Clin
Genet. 86:134–141. 2014. View Article : Google Scholar
|
|
14
|
Kono M, Sugiura K, Suganuma M, Hayashi M,
Takama H, Suzuki T, Matsunaga K, Tomita Y and Akiyama M:
Whole-exome sequencing identifies ADAM10 mutations as a cause of
reticulate acropigmentation of Kitamura, a clinical entity distinct
from Dowling-Degos disease. Hum Mol Genet. 22:3524–3533. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nuytemans K, Bademci G, Inchausti V,
Dressen A, Kinnamon DD, Mehta A, Wang L, Züchner S, Beecham GW,
Martin ER, et al: Whole exome sequencing of rare variants in EIF4G1
and VPS35 in Parkinson disease. Neurology. 80:982–989. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ravenscroft G, Thompson EM, Todd EJ, Yau
KS, Kresoje N, Sivadorai P, Friend K, Riley K, Manton ND, Blumbergs
P, et al: Whole exome sequencing in foetal akinesia expands the
genotype-phenotype spectrum of GBE1 glycogen storage disease
mutations. Neuromuscul Disord. 23:165–169. 2013. View Article : Google Scholar
|
|
17
|
Solomon BD, Hadley DW, Pineda-Alvarez DE,
Kamat A, Teer JK, Cherukuri PF, Hansen NF, Cruz P, Young AC,
Berkman BE, et al NISC Comparative Sequencing Program: Incidental
medical information in whole-exome sequencing. Pediatrics.
129:e1605–e1611. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Verma R and Babu A: Human Chromosomes:
Principles & Techniques. 2nd edition. McGraw-Hill; New York,
NY: pp. 4191995, book review.
Mol Reprod Devel. 43:1341996.
|
|
19
|
Gnirke A, Melnikov A, Maguire J, Rogov P,
LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C,
et al: Solution hybrid selection with ultra-long oligonucleotides
for massively parallel targeted sequencing. Nat Biotechnol.
27:182–189. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Carmona-Mora P, Canales CP, Cao L, Perez
IC, Srivastava AK, Young JI and Walz K: RAI1 transcription factor
activity is impaired in mutants associated with Smith-Magenis
Syndrome. PLoS One. 7:e451552012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lacaria M, Gu W and Lupski JR: Circadian
abnormalities in mouse models of Smith-Magenis syndrome: Evidence
for involvement of RAI1. Am J Med Genet A. 161A:1561–1568. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Girirajan S, Elsas LJ II, Devriendt K and
Elsea SH: RAI1 variations in Smith-Magenis syndrome patients
without 17p11.2 deletions. J Med Genet. 42:820–828. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hayashi M, Kim SW, Imanaka-Yoshida K,
Yoshida T, Abel ED, Eliceiri B, Yang Y, Ulevitch RJ and Lee JD:
Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular
integrity and leads to endothelial failure. J Clin Invest.
113:1138–1148. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Burke C, Sinclair K, Cowin G, Rose S, Pat
B, Gobe G and Colditz P: Intrauterine growth restriction due to
uteroplacental vascular insufficiency leads to increased
hypoxia-induced cerebral apoptosis in newborn piglets. Brain Res.
1098:19–25. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Uerpairojkit B, Manotaya S,
Tanawattanacharoen S, Wuttikon sammakit P and Charoenvidhya D: Are
the cardiac dimensions spared in growth-restricted fetuses
resulting from uteroplacental insufficiency? J Obstet Gynaecol Res.
38:390–395. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Firestein R, Bass AJ, Kim SY, Dunn IF,
Silver SJ, Guney I, Freed E, Ligon AH, Vena N, Ogino S, et al: CDK8
is a colorectal cancer oncogene that regulates beta-catenin
activity. Nature. 455:547–551. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Greene DM, Bloomfield G, Skelton J, Ivens
A and Pears CJ: Targets downstream of Cdk8 in Dictyostelium
development. BMC Dev Biol. 11:22011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang N, Lee I, Marcotte EM and Hurles ME:
Characterising and predicting haploinsufficiency in the human
genome. PLoS Genet. 6:e10011542010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Grayton HM, Fernandes C, Rujescu D and
Collier DA: Copy number variations in neurodevelopmental disorders.
Prog Neurobiol. 99:81–91. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lindhurst MJ, Sapp JC, Teer JK, Johnston
JJ, Finn EM, Peters K, Turner J, Cannons JL, Bick D, Blakemore L,
et al: A mosaic activating mutation in AKT1 associated with the
Proteus syndrome. N Engl J Med. 365:611–619. 2011. View Article : Google Scholar : PubMed/NCBI
|