Introduction
Liver fibrosis is a reversible wound-healing
response that is characterized by increased and altered deposition
of extracellular matrix (ECM) in the liver (1). This process is rigorously controlled
by several growth factors and cytokines (2–5). Of
these, transforming growth factor (TGF)-β is the most potent factor
in stimulating type I collagen gene transcription, which is the
predominant ECM component of fibrotic tissue (6).
N-methyl-4-isoleucine cyclosporine (NIM811) is a
derivative of cyclosporine, but does not exhibit an
immunosuppressive effect. NIM811 inhibits the expression of the
mitochondrial permeability transition (mPT) pore protein in
hepatocytes, which results in hepatocyte cytoprotection (7–10).
It also suppresses hepatic stellate cell (HSC) proliferation and
collagen production, as well as stimulating collagenase activity
in vitro (11). These
findings indicate that NIM811 is a potential candidate for
antifibrosis therapy. Indeed, it was found previously that NIM811
attenuates liver fibrosis and inflammation in rats with
CCl4-induced liver fibrosis (12). Therefore, in the present study, the
TGF-β signaling pathway was analyzed with the aim of elucidating
the molecular mechanism underlying the effects of NIM811 in rats
with CCl4-induced liver fibrosis and cultured HSC-T6
cells.
Materials and methods
Animal study
Liver samples were obtained from control rats and
rats with CCl4-induced liver fibrosis, with or without
NIM811 treatment. These liver samples were collected by Dr Hui
Wang. NIM811 was a gift from Novartis (Basel, Switzerland). Male
Wistar rats weighing 230–260 g were obtained from Beijing Vital
River Laboratory Animal Technology Co., Ltd. (Beijing, China).
Liver fibrosis was induced by intraperitoneal injection of 0.2
ml/100 g body weight of 40% CCl4/corn oil twice weekly.
Rats were also treated with different doses of NIM811 (low dose, 10
mg/kg/day; and high dose, 20 mg/kg/day) by gavage for 6 weeks as
previously reported (13). All
protocols and procedures were approved by the Animal Care and Use
Committee of Capital Medical University (Beijing, China). The
animals were housed in an air-conditioned room at 23–25°C with a 12
h dark/light cycle for 1 week prior to initiation of the
experiment. All animals received humane care during the study with
unlimited access to chow and water.
Cell culture
The HSC-T6 cell line was provided by Dr Scott L.
Friedman (Mount Sinai School of Medicine, New York, NY, USA) and
was cultured in Dulbecco's modified Eagle's medium (DMEM;
Invitrogen Life Technologies, Carlsbad, CA, USA) with 10% fetal
calf serum (FCS; Gibco Life Technologies, Carlsbad, CA, USA). The
cell viability of HSCs was >90% at 24 h in the presence of 2
µM NIM811 in DMEM under serum-free conditions, thus
concentrations of 0.5, 1 and 2 µM were used. After serum
starvation for 16 h, HSC-T6 cells were treated with NIM811 (0.5, 1
or 2 µM), TGFβ1 (5 ng/ml) (14) or TGFβ1 (5 ng/ml) with NIM811 (0.5,
1 or 2 µM) in serum-free medium for 6, 12 or 18 h.
Determination of collagen I and TGF-β1 in
HSC-T6 cell supernatant
Cultured HSC-T6 cells were plated into 6-well cell
culture plates at a density of 105 cells/well and were
incubated in serum-free medium with 0, 0.5, 1 or 2 µM NIM811
for 18 h. Collagen I and TGF-β1 production was determined in the
culture medium using an ELISA (USCN Life Science Inc., Wuhan,
China) and immunoassay kit (R&D Systems, Minneapolis, MN, USA),
respectively.
mRNA levels of collagen I, α-smooth
muscle actin (SMA), TGF-β1 and TGF-β pathway downstream signaling
molecule-detection by reverse transcription-quantitative polymerase
chain reaction (RT-qPCR)
Total RNA was extracted from snap-frozen rat liver
specimens and HSC-T6 cells using TRIzol reagent (Invitrogen Life
Technologies). The amount of RNA was quantified, and its quality
was verified by ultraviolet absorbance spectrophotometry at 260 and
280 nm (BioPhotometer® D30; Eppendorf North America
Inc., Hauppauge, NY, USA). cDNA was reversed transcribed using the
High-Capacity cDNA Reverse Transcription kit (Applied Biosystems,
Foster City, CA, USA), according to the manufacturer's
instructions. The reaction conditions were as follows: 25°C for 10
min, 37°C for 150 min, 85°C for 5 sec, and 4°C for 5 min, prior to
chilling on ice. cDNA was stored at −20°C for future use.
Collagen I, α-SMA, TGF-β1, TβR-I, Smad1, Smad7 and
Id1 mRNA levels were quantified by RT-qPCR. The sequences of the
primers used are shown in Table I.
GAPDH, a housekeeping gene, was used as an internal control primer
for target genes. All primers were obtained from Invitrogen Life
Technologies (Beijing, China). The expression of mRNA was measured
by SYBR Green real-time PCR using an ABI 7500 instrument (Applied
Biosystems). PCR was performed in 20 µl buffer that
contained 2 µg cDNA, 1 µl each primer, and 10
µl SYBR Green PCR Master mix (Applied Biosystems).
Comparative cycle quantification (Cq) calculations were all
relative to the control group. The expression of mRNA relative to
the control was derived using the equation 2−ΔΔCq.
 | Table IPrimers for reverse
transcription-quantitative PCR analysis. |
Table I
Primers for reverse
transcription-quantitative PCR analysis.
| Gene | Primer
sequence | Product size
(bp) | Accession
number |
|---|
| GAPDH | F:
5′-CCTGCCAAGTATGATGACATCAAGA3′ | 75 | BC059110.1 |
| R:
5′-GTAGCCCAGGATGCCCTTTAGT3′ | | |
| Col I | F:
5′-CCTTTCTCCACCCCCTCTT-3′ | 69 | NM_053304.1 |
| R:
5′-TGTGTCTTTGGGGGAGACTT -3′ | | |
| α-SMA | F:
5′-TGCCATGTATGTGGCTATTCA-3′ | 61 | NM_001613.2 |
| R:
5′-ACCAGTTGTACGTCCAGAAGC-3 | | |
| TGF-β1 | F:
5′-CCTGGAAAGGGCTCAACAC-3′ | 100 | NM_021578.2 |
| R:
5′-CTGCCGTACACAGCAGTTCT-3′ | | |
| TβR-I | F:
5′-CACGATGAGCTGAGCCTGTA-3′ | 60 | NM_012775.2 |
| R:
5′-ACCCTGGAGTGCATGGTAAG-3′ | | |
| Smad1 | F:
5′-AGAAAGGGGCCATGGAAG-3′ | 78 | NM_013130.2 |
|
R:5′-AGCGAGGAATGGTGACACA-3′ | | |
| Smad7 | F:
5′-GGAGTCCTTTCCTCTCTC -3′ | 73 | NM_030858.1 |
| R:
5′-GGCTCAATGAGCATGCTCAC -3′ | | |
| Id1 | F:
5′-GCGAGATCAGTGCCTTGG-3′ | 123 | NM_012797.2 |
| R:
5′-TTTTCCTCTTGCCTCCTGAA-3′ | | |
Protein detection of α-SMA, TβR-I and
TGF-β pathway downstream signaling molecules by western
blotting
Protein was extracted from liver samples and HSC-T6
cells using a Protein Extractor IV (DBI, Shanghai, China),
homogenized, and assayed using a Pierce BCA Protein Assay kit
(Thermo Fisher Scientific Inc., Rockford, IL, USA). Protein samples
(40 µg) were subjected to SDS-PAGE (80 V for 40 min on a 5%
acrylamide stacking gel and 120 V for 70 min on a 10 or 15% running
gel), and then transferred (390 MA for 70 min or 80 V for 120 min)
to a nitrocellulose membrane (Hybond-C Extra Membrane 45; Amersham
Biosciences, Uppsala, Sweden). The membranes were soaked in
Tris-buffered saline (10 mmol/l Tris-HCl and 250 mol/l NaCl) that
contained 5% non-fat powdered milk and 0.1% Tween-20 (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) for 2 h to block
non-specific sites. They were then incubated with primary antibody
overnight at 4°C in blocking solution. Antibody dilutions were as
follows: Rabbit anti-rat TβR-I (sc-9048) and Id1 (sc-488)
polyclonal antibodies (1:1,000; Santa Cruz Biotechnology Inc.);
monoclonal mouse anti-rat α-SMA (SAB5500002) and β-actin
(SAB1403520) antibodies (1:1,000, 1:10,000; Sigma-Aldrich, St.
Louis, MO, USA); monoclonal rabbit anti-rat phosph-Smad2 (#3108),
phosph-Smad3 (#9520) and phosph-Smad1/5/8 (#13820) antibodies
(1:500, Cell Signaling Technology Inc., Beverly, MA, USA);
polyclonal rabbit anti-rat Smad7 antibodies (PA1-41506; 1:1,000;
Invitrogen Life Technologies); and horseradish peroxidase
(HRP)-linked anti-rabbit (ZDR-5306) or anti-mouse (ZDR-5307) IgG
secondary antibodies (1:10,000; Beijing Zhongshan Golden Bridge
Biotechnology Co., Ltd., Beijing, China). The resultant blots were
washed and incubated with HRP-linked goat anti-rabbit IgG secondary
antibody for 2 h at room temperature. Immunoreactivity was
visualized using an enhanced chemiluminescence kit (Thermo Fisher
Scientific Inc.). Films (Kodak, Beijing, China) were scanned using
the Bio-Rad imaging system. Protein expression levels were
normalized to β-actin.
Statistical analysis
The results are expressed as the mean ± standard
deviation. Statistical analysis was performed using one-way
analysis of variance and unpaired Student's t-test as appropriate.
P<0.05 was considered to indicate a statistically significant
difference. Statistical analyses were performed using SPSS version
17.0 (SPSS Inc., Chicago, IL, USA).
Results
Effect of NIM811 on collagen I, α-SMA,
TGF-β1 and TβR-I expression in rats
RT-qPCR analysis showed that after treatment with
CCl4 for 6 weeks, collagen I mRNA expression increased
compared with that in normal rats, whereas NIM811 inhibited the
expression significantly (10 and 20 mg/kg, respectively; P<0.01;
Table II).
| Table IILevel of mRNA expression in rat liver
in 6 weeks. |
Table II
Level of mRNA expression in rat liver
in 6 weeks.
| mRNA | Normal (n=7) | CCl4
(n=8) | NIM811 (10 mg/kg)
(n=6) | NIM811 (20 mg/kg)
(n=7) |
|---|
| Col I | 1.0±0.4 | 92.0±4.6b | 38.6±7.8a | 40.9±2.4a |
| α-SMA | 1.0±0.3 | 60.5±4.6b | 18.3±2.2a | 16.3±3.0a |
| TGF-β1 | 1.0±0.1 | 8.6±0.2b | 3.0±0.6a | 3.4±0.5a |
| TβR-I | 1.0±0.2 | 1.9±0.3b | 1.0±0.2a | 0.9±0.2a |
| Id1 | 1.0±0.4 | 2.9±0.2b | 1.0±0.2a | 1.1±0.2a |
| Smad1 | 1.0±0.3 | 2.1±0.1b | 1.5±0.2a | 1.2±0.3a |
| Smad2 | 1.0±0.2 | 2.9±0.3b | 1.6±0.2a | 1.2±0.4a |
| Smad7 | 1.0±0.1 | 1.9±0.2b | 2.7±0.3a | 3.7±0.3a |
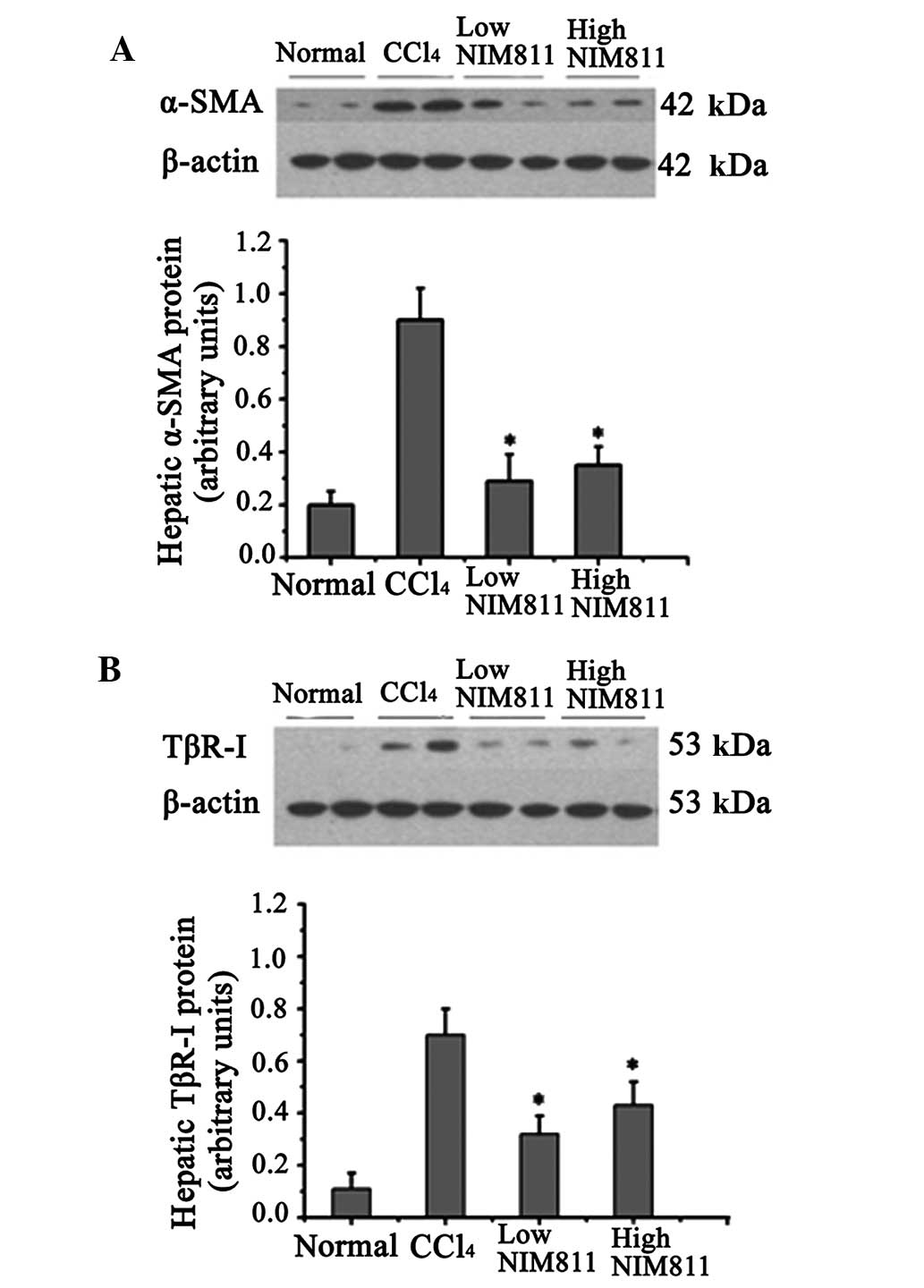
α-SMA mRNA expression in rats with liver fibrosis
was significantly increased compared with that in the normal
controls, and decreased significantly in the two NIM811-treated
groups (P<0.01) (Table II). In
the liver of rats with CCl4-induced liver fibrosis,
α-SMA protein expression increased significantly compared with
normal rats, whereas its expression was inhibited by NIM811
treatment (Fig. 1A).
The level of TGF-β1 and TβR-I mRNA increased after
CCl4 injection for 6 weeks and was blocked by NIM811
treatment (Table II). Similar to
mRNA expression, TβR-I protein was also reduced in the liver of
NIM811-treated rats compared with rats with CCl4-induced
liver fibrosis (Fig. 1B). These
results indicate that NIM811 inhibited liver fibrosis through the
TGF-β pathway.
Effect of NIM811 on TGF-β/anaplastic
lymphoma kinase (ALK)5 and TGF-β/ALK1 signaling pathway in rat
liver
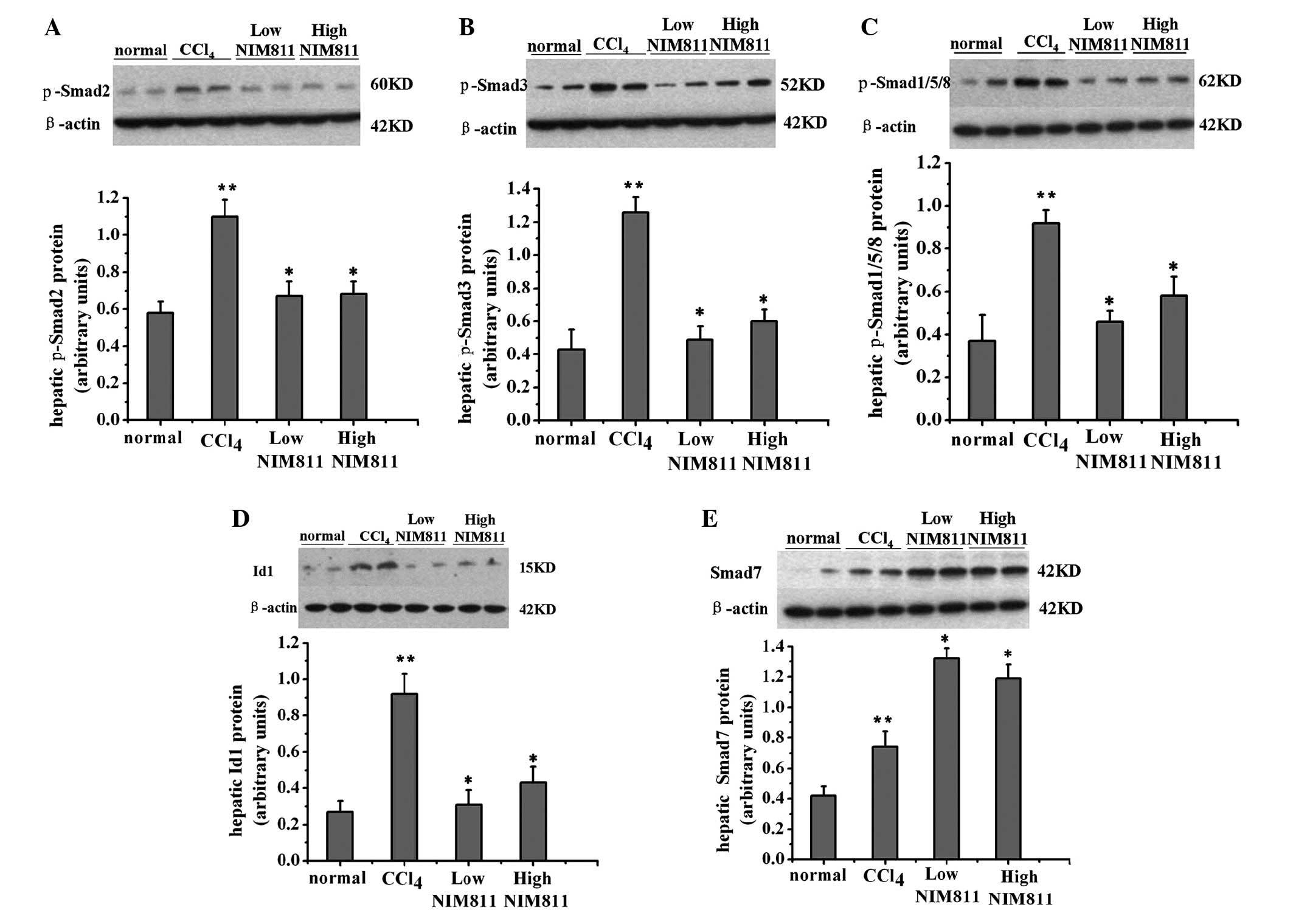
p-Smad2, p-Smad3, Smad7, p-Smad1/5/8 and Id1 were
detected by western blotting. p-Smad2, p-Smad3 and p-Smad1/5/8
protein levels increased following treatment with CCl4,
and decreased in NIM811-treated rats (Fig. 2A–C). The level of Smad7 increased
in rats with liver fibrosis and further increased in NIM811-treated
rats (Fig. 2D). Id1 is the
downstream signaling molecule of the TGF-β/ALK1/Smad1/5/8 pathway
and was decreased following NIM811 treatment (Fig. 2E). Thus, NIM811 downregulated the
TGF-β pathway in CCl4-treated rats.
Effect of NIM811 on collagen I expression
in HSC-T6 cells
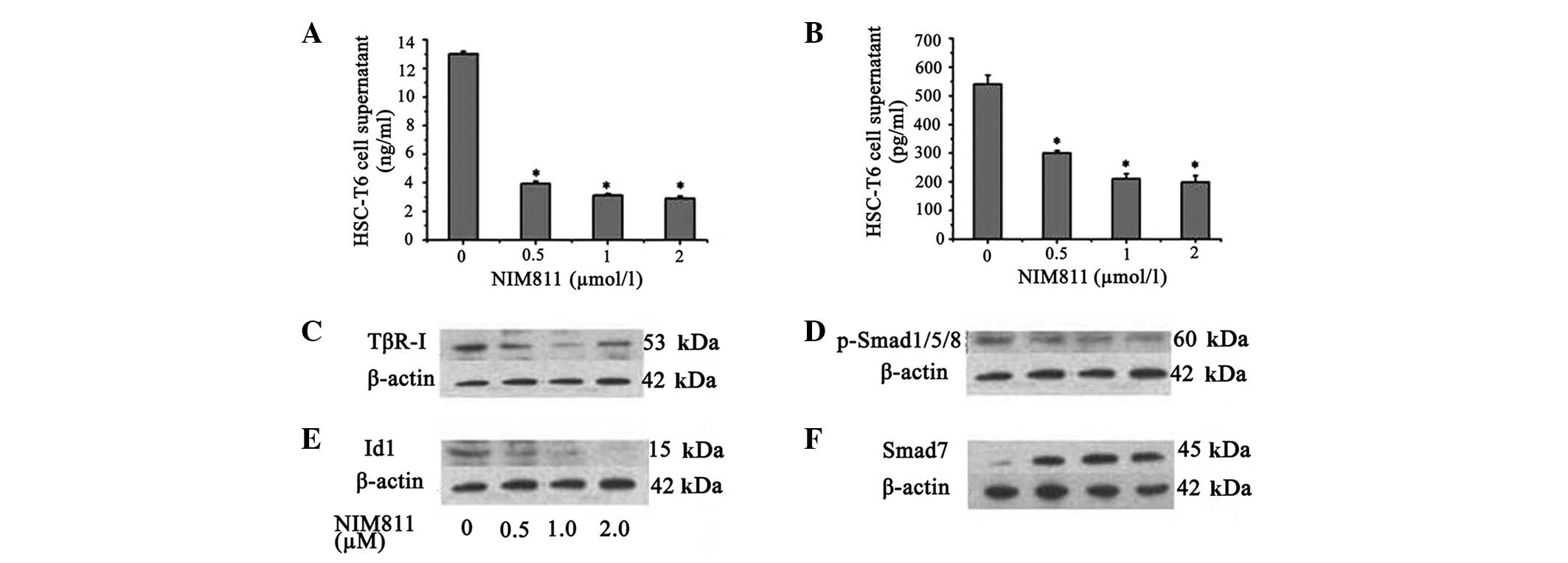
To assess the effect of NIM811 on ECM production by
HSCs, collagen I mRNA and protein levels were determined in HSCs
and culture medium. It was demonstrated that the collagen I mRNA
level decreased in the presence of NIM811 compared with normal
control HSC-T6 cells (P<0.05; Table III). This effect was most
pronounced in the presence of NIM811 after 18 h, thus, 18 h was
selected as the time point for the follow-up experiments. Treatment
of the cells with increasing concentrations of NIM811 led to a
concentration-dependent suppression of collagen I production; 0.5
µM NIM811 reduced collagen I accumulation by ~70% and 2
µM NIM811 reduced collagen I accumulation by ~77% in HSC-T6
cell supernatant after treatment for 18 h (Fig. 3A).
| Table IIICol I mRNA expression in the HSC-T6
cell line with NIM811 treatment after 6, 12 and 18 h. |
Table III
Col I mRNA expression in the HSC-T6
cell line with NIM811 treatment after 6, 12 and 18 h.
| Col I expression
following NIM811 (µM) treatment
|
|---|
| Incubation time
(h) | 0 | 0.5 | 1 | 2 |
|---|
| 6 | 1.00±0.05 | 0.95±0.09 | 0.89±0.05a | 0.82±0.05a |
| 12 | 1.00±0.06 | 0.92±0.06a | 0.84±0.07a | 0.76±0.08a |
| 18 | 1.00±0.08 | 0.90±0.04a | 0.75±0.06a | 0.64±0.05a |
Furthermore, the level of collagen I increased after
TGFβ1 treatment and was reduced by NIM811 or TGFβ1 and NIM811
co-treatment (Table IV).
| Table IVmRNA expression in HSC-T6 cells
following treatment with TGF-β1, NIM811 or a combination of the
two. |
Table IV
mRNA expression in HSC-T6 cells
following treatment with TGF-β1, NIM811 or a combination of the
two.
| mRNA | Normal | TGF-β1(5
ng/ml) | NIM811(2
µM) | TGF-β1+NIM811 |
|---|
| Col I | 1.00±0.05 | 1.78±0.08a | 0.64±0.05a | 0.66±0.10a |
| α-SMA | 1.00±0.04 | 2.34±0.15a | 1.46±0.09a | 1.44±0.06a |
| TβR-I | 1.00±0.05 | 1.18±0.09a | 0.56±0.08a | 0.60±0.11a |
| Smad1 | 1.00±0.09 | 1.78±0.13a | 0.68±0.06a | 0.72±0.14a |
| Id1 | 1.00±0.06 | 1.46±0.10a | 0.58±0.05a | 0.62±0.08a |
| Smad7 | 1.00±0.04 | 1.05±0.10 | 1.64±0.14a | 1.48±0.10a |
Effect of NIM811 on TGF-β1 and TβR-I and
TGF-β/ALK1 signaling pathway molecules in cultured HSC-T6
cells
It was demonstrated that NIM811 suppressed the
expression of TGF-β1 and TβR-I mRNA in a concentration-dependent
manner (P<0.05; Table V).
Production of TGF-β1 in HSC-T6 cell supernatant was also decreased
to ~40% by 2 µM NIM811 treatment (P<0.01; Fig. 3B). It was additionally identified
that TβR-I protein expression was suppressed by NIM811 (P<0.05;
Fig. 3C). It was also found that
NIM811 suppressed Smad1 and Id1 transcription, which was
accompanied by decreased phosphorylation of Smad1/5/8 and decreased
Id1 protein levels (Table V,
Fig. 3D and E). Smad7, which acts
as an inhibitory Smad, increased following NIM811 treatment
(Table V, Fig. 3F).
| Table VmRNA expression in the HSC-T6 cell
line in the presence of NIM811 for 18 h. |
Table V
mRNA expression in the HSC-T6 cell
line in the presence of NIM811 for 18 h.
| NIM811 (µM)
|
|---|
| mRNA | 0 | 0.5 | 1 | 2 |
|---|
| TGF-β1 | 1.00±0.06 | 0.91±0.05a | 0.72±0.04a | 0.50±0.07a |
| TβR-I | 1.00±0.08 | 0.58±0.04a | 0.58±0.06a | 0.57±0.05a |
| Smad1 | 1.00±0.09 | 0.84±0.04a | 0.72±0.07a | 0.64±0.06a |
| Id1 | 1.00±0.12 | 0.77±0.04a | 0.58±0.06a | 0.44±0.05a |
| Smad7 | 1.00±0.09 | 1.49±0.10a | 1.57±0.06a | 1.69±0.12a |
To determine whether the addition of exogenous TGF-β
may restore fibrosis in the presence of NIM811, HSC-T6 cells were
stimulated with TGF-β. Results showed that the levels of TβR-I,
Smad1 and Id1 increased following TGFβ1 treatment and were reduced
by treatment with NIM811 alone or TGFβ1 in combination with NIM811
(Table IV).
Discussion
It was demonstrated that NIM811 attenuated collagen
type I expression in the liver of rats with CCl4-induced
liver fibrosis and cultured HSC-T6 cells. This effect may have been
associated with the suppression of TGF-β1 and the TGF-β1 signaling
pathway.
Collagen type I is the predominant constituent of
the ECM in fibrotic liver, and its expression is regulated
transcriptionally and post-transcriptionally by a number of stimuli
and pathways (15,16). TGF-β1 is derived from paracrine and
autocrine sources, and is the key cytokine in the control of tissue
repair. Deregulation of production and degradation of ECM induced
by TGF-β1 is a cause of liver fibrosis (17). TGF-β1 binds to two different
serine/threonine kinase receptors, TβR-I and TβR-II (18). Upon ligand binding, TβR-I
specifically activates intracellular Smad proteins (19,20),
which are a family of bifunctional molecules that are known TGF-β1
downstream signals. Following Smad activation, numerous
extracellular and intracellular signals converge to fine-tune and
enhance the effects of TGF-β1 during fibrogenesis (1,21,22).
Smad1, 2 and 3 are stimulatory, whereas Smad7 is inhibitory and is
antagonized by Id1 (23,24). Therefore, the TGF-β1 signaling
pathway, including TGF-β/ALK5 and TGF-β/ALK1 branches, has an
important role in liver fibrogenesis.
TGF-β1 signal cascades through Smad2 and Smad3
strongly regulate the expression of the type I collagen gene
(20,25). In the present study, decreased
liver collagen type I mRNA expression was accompanied by
downregulation of phosphorylation of Smad2 and Smad3 in rats with
liver fibrosis treated with NIM811. NIM811 also suppressed p-Smad2
and p-Smad3 in HSCs in a previous study, which indicates that
NIM811 decreases collagen accumulation through downregulation of
TGF-β1/ALK5/Smad2/3 branch (11).
It was also demonstrated that NIM811 inhibited the
protein levels of phospho-Smad1/5/8 and Id1 in the liver of
NIM811-treated rats. Recent research has demonstrated that the
TGF-β1/ALK1/Smad1 pathway and its downstream factor Id1 are
important in liver fibrosis. Id1 is the helix-loop-helix (HLH)
protein inhibitor of differentiation 1, and is hypothesized to
affect the balance between cell growth and differentiation by
negatively regulating the function of basic HLH transcription
factors (26). Recently, Id1 has
been found to be a novel TGF-β1 target gene in HSCs. Id1 expression
and Smad1 phosphorylation are co-induced during hepatic
fibrogenesis. The existence of ALK1 and the TGF-β1/ALK1/Smad1
pathway is crucial during the transdifferentiation of primary rat
HSCs to myofibroblasts (23).
Activation of the TGF-β1/ALK1/Smad1 pathway may be enhanced by
TGFβ1 in LX-2 cells (27).
Treatment of HSCs with TGF-β1 led to increased Id1 protein
expression, which was not directly mediated by the ALK5/Smad2/3,
but by the ALK1/Smad1 pathway (27).
The two branches of the TGF-β1 signaling pathway,
TGF-β1/ALK1/Smad1 and TGF-β1/ALK5/Smad2, are not isolated, they act
together to provide feedback via Id1. It has been proposed that
R-Smads exert their signal transduction effect through protein
phosphorylation. Smad7 acts as a general inhibitor of the TGF-β
family, and inhibits TGF-β1 signaling by preventing activation of
R- and Co-Smads (19,28,29).
Moreover, ectopic Id1 overexpression is sufficient to overcome the
inhibitory effects of Smad7 on HSC activation and α-SMA fiber
formation in vitro (23),
which leads to aggravation of liver fibrosis.
HSCs are perisinusoidal cells in the subendothelial
space between hepatocytes and sinusoidal endothelial cells, and are
key in collagen accumulation (30). They can be identified by their
vitamin A autofluorescence, perisinusoidal orientation, and
expression of the cytoskeletal proteins desmin and α-SMA. The
present study showed that NIM811 suppressed α-SMA protein
expression in the adjacent sinusoids in CCl4-treated
rats, which indicated that NIM811 decreased the extent of HSC
activation in vivo, and that HSCs are one of the targets of
NIM811. Therefore, HSCs were selected to conduct an in vitro
study to confirm these in vivo results, which demonstrated
that NIM811 inhibited TGF-β1 and its downstream signaling
molecules.
The results suggest another possible antifibrotic
mechanism of NIM811, which inhibits the transcription of Smad1 and
Id1, accompanied by a decrease in phospho-Smad1/5/8 and Id1 protein
expression. NIM811 can attenuate the fibrogenic signaling of HSCs
by upregulating Smad7 expression. The results also showed that the
TGFβ1/ALK1/Smad1 pathway may represent a potential target for
antifibrotic therapy. However, which components in the compound
exhibit the inhibitory effects requires further investigation.
Fibrosis is caused by increased matrix production by
HSCs and an increase in HSC numbers. It has been shown that NIM811
suppressed HSC proliferation, but not apoptosis in vitro
(7). Platelet-derived growth
factor (PDGF) is the most potent stellate cell mitogen identified
thus far (31,32). Downstream pathways of PDGF
signaling have been carefully characterized in stellate cells, and
induction of PDGF receptors early in stellate cell activation
increases responsiveness to this potent mitogen (33–36).
Therefore, our next study will investigate the effect of NIM811 on
cultured HSCs isolated from CCl4-induced rats and the
PDGF signaling pathway.
In conclusion, the results further implicate the
involvement of the TGFβ1/ALK1/Smad1 and TGF-β1/ALK5/Smad2 pathways
during the development of hepatic fibrosis, and suggest that NIM811
may emerge as a novel option for hepatic fibrosis therapy through
downregulation of the TGF-β1 pathway.
Abbreviations:
|
TGF-β
|
transforming growth factor-β
|
|
HSC
|
hepatic stellate cell
|
|
SMA
|
α-smooth muscle actin
|
References
|
1
|
Friedman SL: Mechanisms of hepatic
fibrogenesis. Gastroenterology. 134:1655–1669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Moreno M and Bataller R: Cytokines and
reninangiotensin system signaling in hepatic fibrosis. Clin Liver
Dis. 12:825–852. 2008. View Article : Google Scholar
|
|
3
|
Gressner OA, Lahme B, Demirci I, Gressner
AM and Weiskirchen R: Differential effects of TGF-beta on
connective tissue growth factor (CTGF/CCN2) expression in hepatic
stellate cells and hepatocytes. J Hepatol. 47:699–710. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wynn TA: Cellular and molecular mechanisms
of fibrosis. J Pathol. 214:199–210. 2008. View Article : Google Scholar
|
|
5
|
Parsons CJ, Takashima M and Rippe RA:
Molecular mechanisms of hepatic fibrogenesis. J Gastroenterol
Hepatol. 22(Suppl 1): S79–S84. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Casini A, Pinzani M, Milani S, Grappone C,
Galli G, Jezequel AM, Schuppan D, Rotella CM and Surrenti C:
Regulation of extracellular matrix synthesis by transforming growth
factor beta 1 in human fat-storing cells. Gastroenterology.
105:245–253. 1993.PubMed/NCBI
|
|
7
|
Kohjima M, Enjoji M, Higuchi N, Kotoh K,
Kato M, Takayanagi R and Nakamuta M: NIM811, a nonimmunosuppressive
cyclosporine analogue, suppresses collagen production and enhances
collagenase activity in hepatic stellate cells. Liver Int.
27:1273–1281. 2007.PubMed/NCBI
|
|
8
|
Waldmeier PC, Feldtrauer JJ, Qian T and
Lemasters JJ: Inhibition of the mitochondrial permeability
transition by the nonimmunosuppressive cyclosporin derivative
NIM811. Mol Pharmacol. 62:22–29. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Theruvath TP, Zhong Z, Pediaditakis P,
Ramshesh VK, Currin RT, Tikunov A, Holmuhamedov E and Lemasters J:
Minocycline and N-methyl-4-isoleucine cyclosporin (NIM811) mitigate
storage/reperfusion injury after rat liver transplantation through
suppression of the mitochondrial permeability transition.
Hepatology. 47:236–246. 2008. View Article : Google Scholar
|
|
10
|
Zhong Z, Theruvath TP, Currin RT,
Waldmeier PC and Lemasters JJ: NIM811, a mitochondrial permeability
transition inhibitor, prevents mitochondrial depolarization in
small-for-size rat liver grafts. Am J Transplant. 7:1103–1111.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kohjima M, Enjoji M, Higuchi N, Kotoh K,
Kato M, Takayanagi R and Nakamuta M: NIM811, a nonimmunosuppressive
cyclosporine analogue, suppresses collagen production and enhances
collagenase activity in hepatic stellate cells. Liver Int.
27:1273–1281. 2007.PubMed/NCBI
|
|
12
|
Wang H, Zhang Y, Wang T, You H and Jia J:
N-methyl-4-isoleucine cyclosporine attenuates CCl-induced liver
fibrosis in rats by interacting with cyclophilin B and D. J
Gastroenterol Hepatol. 26:558–567. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang H, Zhang Y, Wang T, You H and Jia J:
N-methyl-4-isoleucine cyclosporine attenuates CCl-induced liver
fibrosis in rats by interacting with cyclophilin B and D. J
Gastroenterol Hepatol. 26:558–567. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dooley S, Delvoux B, Lahme B,
Mangasser-Stephan K and Gressner AM: Modulation of transforming
growth factor beta response and signaling during
transdifferentiation of rat hepatic stellate cells to
myofibroblasts. Hepatology. 31:1094–1106. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Friedman SL: Hepatic fibrosis-overview.
Toxicology. 254:120–129. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Breitkopf K, Godoy P, Ciuclan L, Singer MV
and Dooley S: TGF-beta/Smad signaling in the injured liver. Z
Gastroenterol. 44:57–66. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Border WA and Noble NA: Transforming
growth factor beta in tissue fibrosis. N Engl J Med. 331:1286–1292.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kolodziejczyk SM and Hall BK: Signal
transduction and TGF-beta superfamily receptors. Biochem Cell Biol.
74:299–314. 1996. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Miyazawa K, Shinozaki M, Hara T, Furuya T
and Miyazono K: Two major Smad pathways in TGF-beta superfamily
signalling. Genes Cells. 7:1191–1204. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Attisano L and Wrana JL: Signal
transduction by the TGF-beta superfamily. Science. 296:1646–1647.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Higashiyama R, Inagaki Y, Hong YY, Kushida
M, Nakao S, Niioka M, Watanabe T, Okano H, Matsuzaki Y, Shiota G
and Okazaki I: Bone marrow-derived cells express matrix
metalloproteinases and contribute to regression of liver fibrosis
in mice. Hepatology. 45:213–222. 2007. View Article : Google Scholar
|
|
22
|
Inagaki Y and Okazaki I: Emerging insights
into transforming growth factor beta Smad signal in hepatic
fibrogenesis. Gut. 56:284–292. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wiercinska E, Wickert L, Denecke B, Said
HM, Hamzavi J, Gressner AM, Thorikay M, ten Dijke P, Mertens PR,
Breitkopf Ka and Dooley S: Id1 is a critical mediator in
TGF-beta-induced transdifferentiation of rat hepatic stellate
cells. Hepatology. 43:1032–1041. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Miyazono K, Kusanagi K and Inoue H:
Divergence and convergence of TGF-beta/BMP signaling. J Cell
Physiol. 187:265–276. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Friedman SL, Yamasaki G and Wong L:
Modulation of transforming growth factor beta receptors of rat
lipocytes during the hepatic wound healing response. Enhanced
binding and reduced gene expression accompany cellular activation
in culture and in vivo. J Biol Chem. 269:10551–10558.
1994.PubMed/NCBI
|
|
26
|
Zebedee Z and Hara E: Id proteins in cell
cycle control and cellular senescence. Oncogene. 20:8317–8325.
2001. View Article : Google Scholar
|
|
27
|
Li L, Zhao XY and Wang BE: Down-regulation
of transforming growth factor beta 1/activin receptor-like kinase 1
pathway gene expression by herbal compound 861 is related to
deactivation of LX-2 cells. World J Gastroenterol. 14:2894–2899.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Itoh S, Itoh F, Goumans MJ and Ten Dijke
P: Signaling of transforming growth factor-beta family members
through Smad proteins. Eur J Biochem. 267:6954–6967. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Itóh S, Landström M, Hermansson A, Itoh F,
Heldin CH, Heldin NE and ten Dijke P: Transforming growth factor
beta1 induces nuclear export of inhibitory Smad7. J Biol Chem.
273:29195–29201. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Friedman SL: Hepatic stellate cells:
Protean, multifunctional and enigmatic cells of the liver. Physiol
Rev. 88:125–172. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pinzani M: PDGF and signal transduction in
hepatic stellate cells. Front Biosci. 7:d1720–d1726. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Borkham-Kamphorst E, van Roeyen CR,
Ostendorf T, Floege J, Gressner AM and Weiskirchen R:
Pro-fibrogenic potential of PDGF-D in liver fibrosis. J Hepatol.
46:1064–1074. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pinzani M and Marra F: Cytokine receptors
and signaling in hepatic stellate cells. Semin Liver Dis.
21:397–416. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lechuga CG, Hernandez-Nazara ZH, Hernández
E, Bustamante M, Desierto G, Cotty A, Dharker N, Choe M and Rojkind
M: PI3K is involved in PDGF-beta receptor upregulation post-PDGF-BB
treatment in mouse HSC. Am J Physiol Gastrointest Liver Physiol.
291:G1051–G1061. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Harder KW, Owen P, Wong LK, Aebersold R,
Clark-Lewis I and Jirik FR: Characterization and kinetic analysis
of the intracellular domain of human protein tyrosine phosphatase
beta (HPTP beta) using synthetic phosphopeptides. Biochem J.
298:395–401. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wong L, Yamasaki G, Johnson RJ and
Friedman SL: Induction of beta-platelet-derived growth factor
receptor in rat hepatic lipocytes during cellular activation in
vivo and in culture. J Clin Invest. 94:1563–1569. 1994. View Article : Google Scholar : PubMed/NCBI
|