Introduction
Curcuma longa Linn. of the family
Zingiberaceae, is a perennial herb, which measures up to 1 m high
with a short stem. The powdered rhizome, turmeric, has been in
continuous use for its flavoring and digestive properties. Turmeric
is used extensively in India and other South Asian cuisines, and is
a significant ingredient in the majority of curry powders. It is
also used as a home remedy for cuts and burns, and is an integral
part of the religious traditions and customs of the Hindu religion
(1). For thousands of years,
turmeric has been used in Ayurveda for a wide range of disorders,
including biliary disorders, anorexia, coryza, cough, wounds,
hepatic disorders, rheumatic disorders, sprains and swellings
caused by injury, and sinusitis (2). The volatile oil obtained from C.
longa also exhibits potent anti-inflammatory and anti-arthritic
activities (3).
The major constituent of turmeric rhizomes is a
yellow-colored phenolic pigment, known as curcumin. In the crude
extract of rhizomes of C. longa, ~75–80% curcumin is
present, along with ~15–20% demethoxycurcumin and 3–5%
bisdemethoxycurcumin (4).
Curcumin is capable of modulating the activities of
a range of transcription factors and signaling pathways, which are
important in several diseases, and include nuclear factor-κB,
activator protein-1, Janus kinase/signal transducer and activator
of transcription, Akt, B cell lymphoma-2, caspases, poly ADP ribose
polymerase, IκB kinase, epidermal growth factor receptor,
platelet-derived growth factor, human epidermal growth factor
receptor 2, nuclear factor erythroid 2-related factor 2,
β-catenin/T-cell factor, c-Jun N-terminal kinase, mitogen-activated
protein kinase, cyclooxygenase and 5-lipoxygenase (5). It also inhibits the undesirable
activities of a number of cytokines, including tumor necrosis
factor-α and interleukins, which are pivotal in inflammation. Its
high antioxidant potential and metal chelating properties further
widen its range of activities (6,7).
Thus, curcumin can act on multiple targets and at multiple levels,
and offer substantial benefits in a number of diseases, including
cancer, heart disease, diabetes, rheumatoid arthritis, Alzheimer's
disease, inflammatory bowel diseases, liver fibrosis and cirrhosis,
HIV, pancreatitis, drug-resistant malaria (8–10).
It is reported to be a safe chemopreventive agent and one the few
molecules capable of preventing cancer metastasis (11).
Curcumin has a poor solubility in water, with the
maximum solubility reported to be 11 ng/ml in aqueous buffer (pH
5.0) (12). The oral
bioavailability of curcumin is low, at only 1% in rats, and higher
doses are required to achieve significant pharmacological effects
(13). In previous clinical
trials, quantifiable serum levels were not achieved until doses of
up to 3.6 g were used (14,15).
To improve the oral bioavailability of curcumin, several approaches
have been investigated. These approaches include loading curcumin
into liposomes or nanoparticles, forming self-microemulsifying drug
delivery systems, complexation with phospholipids, addition of
essential oils and synthesizing structural analogues of curcumin
(16–18).
Turmeric extracts and curcumin have been
investigated for their toxicity in several studies and have been
found to be safe (19,20). Bioavailability investigations with
these regular extracts have shown low serum levels of curcumin
following oral administration in animals (21,22).
Due to poor oral absorption, high doses of turmeric extract are
required to achieve significant pharmacological effects, however,
high doses may induce significant toxicity (23). The emergence of extracts claiming
bioavailable curcumin has led to debate over the presence of
several higher fold levels of curcumin in the blood and tissues and
its safety. In addition, the external bioenhancing agents added
pose additional safety concerns. Although higher bioavailability
leads to the improved delivery of therapeutic levels of curcumin
in vivo, the detailed safety profile of curcumin, as well as
the added bioenhancers, require evaluation. In the present study
the toxicity and safety of one such bioavailable turmeric
formulation, curcuminoid-essential oil complex (CEC), was
investigated in in vitro and in vivo animal models,
as the toxicity profile has not been reported previously.
CEC differs from regular turmeric extracts, as it
contains curcuminoids and essential oil of turmeric containing
turmer-ones in a specific ratio. CEC has been characterized
previously using ultra performance liquid chromatography and gas
chromatography–mass spectrometry methods (24).
Another important reason to assess the toxicity and
safety of curcumin is its wide use as a coloring agent in food and
beverages. The permissible levels of curcumin as a food colorant in
the EU is between 20 and 500 mg/kg, and in beverages is up to 200
mg/l. As turmeric is widely used in cooking, exposure assessment
also requires consideration of dietary exposure to curcumin. The
estimated combined exposure to curcumin from naturally occurring
curcumin in foods and from its use as a food color, ranges between
1.6 and 7.6 mg/kg body weight (bw)/day for children and 2.6 mg/kg
bw/day for adults (25). Curcumin
was evaluated by the Joint Food and Agriculture Organization of the
United Nations and the World Health Organization Expert Committee
on Food Additives in 2004 and established an allocated daily intake
(ADI) of 0–3 mg/kg bw/day (26).
However, this does not take into account the combined dietary
exposure of curcumin. European Food Safety Authority panel noted
that the maximum levels of exposure to curcumin in children are
above the ADI of 3 mg/kg bw/day in certain European countries
(25).
Curcumin is currently widely used as a nutraceutical
product for various indications. The recommended dosage of curcumin
extracts for the general population ranges between 500 and 1,500
mg/day (27), which is higher than
the recommended ADI of 3 mg/kg bw/day.
CEC is one of the commercial curcumin formulations
claimed to have higher levels of bioavailability. The turmerone
rich essential oil of turmeric present in CEC enhances the
bioavailability of curcumin when administered orally. In a pilot
crossover investigation in humans, the relative bioavailability of
CEC was ~6.93-fold higher, compared with normal curcumin, and
~6.3-fold higher, compared with curcumin-lecithin-piperine formula
(28). Previous reports on the
toxicity of regular turmeric extracts or curcumin cannot be
associated with the bioavailable CEC. Therefore, evaluation of the
toxicity of higher curcumin levels in vivo from CEC is
important to confirm its safety for human use. The aim of the
present study was to evaluate the toxicity and safety of CEC in a
well established animal models, including the assessment of acute
toxicity in mice and rats, sub-chronic toxicity in rats and
mutagenicity assessment.
Materials and methods
Drugs and reagents
2-nitro fluorine, sodium azide, mitomycin C,
9-aminoacridine, 2-amino anthracene, cyclophosphamide and
colchicine were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Potassium chloride was purchased from Loba Chemie PVT., Ltd.
(Mumbai, India). Salmonella typhimurium culture discs
(Moltox) were obtained from Krishgen Biosystems (Whittier, CA,
USA). Standard laboratory rat feed was purchased from Pranav Agro
Industries, Ltd. (Banagalore, India). CEC (containing 95%
curcuminoid complex) was provided by Arjuna Natural Extracts Ltd.
(Aluva, India).
Animals
Wistar albino rats, weighing 160–200 g, and Swiss
albino mice, weighing 20–25 g, were used for toxicity and
mutagenicity investigations. The room temperature was maintained at
22±3°C with 30–70% relative humidity. The room was ventilated at a
rate of ~15 air changes/h. Lighting was controlled to provide 12 h
artificial light (8 am–8 pm) each day. Filtered water and
sterilized standard pelleted feed (Pranav Agro Industries Ltd.) was
available ad libitum to the experimental animals. All
experimental protocols were approved by the Institutional Animal
Ethics Committee of Shriram Institute for Industrial Research
(Delhi, India), and guidelines set by the Committee for the Purpose
of Control and Supervision of Experiments on Animals (http://icmr.nic.in/bioethics/final_cpcsea.pdf) were
adhered to.
Acute toxicity evaluation in mice
A total of 20 Swiss albino mice (10 males/10
females), weighing 20–25 g and aged 8–12 weeks, were used for the
evaluation of acute toxicity. The investigation was divided into
two steps, as per the guidelines of the Organization for Economic
Cooperation and Development (OECD; no. 420) (29). The animals were acclimatized for 5
and 7 days for steps I and II, respectively, prior to commencement
of each step. The animals were fasted 4 h prior to, and 2 h
following, treatment. In the first step, one female and one male
mouse were used to perform a limit test. The test substance (CEC
dissolved in corn oil; 200 mg/ml w/v) was administered orally at a
dose of 5,000 mg/kg bw (2.5 ml/100 g bw), via a cannula attached to
a syringe. No mortality, or signs and symptoms of toxicity were
observed in any animals, therefore, to confirm these findings, the
second step was performed by assessing four female and male mice,
which were administered the same dose of 5,000 mg/kg bw.
Similarly, a group of five male and female mice were
treated with vehicle (corn oil), in a stepwise manner, and were
designated as the control group, comprising one female and male
mouse in the first step, and four female and male mice in the
second step. The animals were observed for a total period of 14
days. No mortality or treatment-associated signs or symptoms of
toxicity were observed in the animals in either of the steps. As a
result of these findings, no further assessment was required. After
14 days, the animals were sacrificed using CO2
asphyxiation. Gross pathological changes, if any, were
recorded.
Acute toxicity assessment in rats
As it is recommended that acute toxicity is
evaluated in two mammalian species prior to initial human exposure,
according to the FDA Single Dose Acute Toxicity Testing for
Pharmaceuticals; Revised Guidance (61 FR43934 to 43935; August 26,
1996), CEC was also investigated in rats. A total of 20 Wistar
albino rats (10 males and 10 females) weighing 160–200 g, aged 8–12
weeks, were used for this assessment. The investigation was divided
into two steps, as per OECD guidelines (no. 420) (29). The animals were acclimatized for 5
and 7 days for steps I and II, respectively, prior to commencement
of each step. The animals were fasted overnight prior to, and 4 h
following, treatment. In the first step, one female and one male
rat were assessed using a limit test. The test substance (CEC
dissolved in corn oil) was administered orally at a dose of 5,000
mg/kg bw using a cannula attached to a syringe. No mortality, or
signs and symptoms of toxicity were observed in any animals.
Therefore, to confirm these findings, the second step was performed
using four more female and male rats, which were administered with
the same dose of 5,000 mg/kg bw.
Similarly, a group of five male and female rats were
treated with vehicle (corn oil), in a stepwise manner, and were
designated as the control group (one female and male rat in the
first step; four female and male rats in the second step). The
animals were observed for a total period of 14 days. No mortality
or treatment associated signs and symptoms of toxicity were
observed in the animals in either step. As a result, no further
assessment was required. After 14 days, the animals were sacrificed
using CO2 asphyxiation. Gross pathological changes, if
any, were recorded.
Assessment of 90-day repeated dose oral
toxicity in rats
The assessment of repeated dose was performed as per
OECD guidelines for Testing of Chemicals (no. 408). A total of 100
Wistar albino rats (50 males and 50 females) were divided into four
groups of 20 animals, each comprising 10 males and 10 females, and
satellite groups of 10 animals (five males and five females). The
animals were acclimatized for 7 days prior to commencement of
treatment. Subsequently, three of the groups of 20 rats were
administered with CEC orally at a dose of 100 mg/kg bw (low dose),
500 mg/kg bw (intermediate dose) and 1,000 mg/kg bw (high dose),
respectively, each day for 90 days, using a cannula attached to a
syringe. The fourth group of 20 rats were orally administered with
corn oil only (vehicle) for 90 days and was designated the control
group.
The two satellite groups of 10 rats were designated
the satellite control and satellite high dose groups, and were
administered with corn oil (vehicle) and CEC (1,000 mg/kg),
respectively, for 90 days. Homogeneity of the test sample was
maintained on a magnetic stirrer during administration. Following
sacrifice of the experimental and control groups of animals, the
satellite control and satellite high dose groups were maintained
under observation for an additional 28 days to assess for
reversibility, persistence or delayed toxic effects, if any. The
animals were observed daily for behavior, appearance and
toxicological signs and symptoms. The animals were anaesthetized
using anaesthetic ether stabilized with 0.002% propyl gallate
(Sharad Laboratories, Andhra Pradesh, India) and 2 ml Blood was
collected from the retro-orbital sinus under light anesthesia from
all animals prior to sacrifice for detailed hematological and
biochemical evaluation. Urine samples (24 h samples of ~6–8 ml)
were also collected from all animals at the termination of the
experiment, and were analyzed for appearance/colour, specific
gravity, pH, glucose, proteins, ketones, bilirubin, urobilinogen,
nitrite and white blood cells. Additionally, ~3 ml of the urine
samples were centrifuged at 2,000 rpm for 5 min, and sediments were
examined for pus cells, epithelial cells, casts, red blood cells
and crystals. Criteria used to evaluate compound-associated effects
included appearance, behavior, morbidity and mortality rates, body
weights, feed consumption, hematological and biochemical analysis,
urine analysis, organ weights, necropsy and histopathology. All
these assessments and analyses were performed at Shriram Toxicology
Centre, Shriram Institute for Industrial Research (Delhi,
India).
Mutagenicity assessment
A bacterial reverse mutation assay (Ames test) was
performed, in which CEC was assessed against five strains of
Salmonella typhimurium viz. (TA-98, TA-100, TA-102, TA-1535
and TA-1537), with and without metabolic activation (S-9 liver
fraction; Krishgen Biosystems, Whittier, CA, USA), at
concentrations of 1,000, 2,000, 3,000, 4,000 and 5,000
µg/plate. The experiment was performed as per OECD
guidelines (no. 471). For the mutation assessment, sub culture was
prepared in nutrient broth and grown overnight at 37°C in an
incubator. The overnight culture provided ~1×109
cells/ml. The actual number of cells was assessed by cell counting
and was used as the standard bacterial suspension.
The solvent, dimethyl sulfoxide (DMSO; 5,000
µg/plate), was included as a negative control in the same
bacterial cultures, and S-9 mix (10% solution in pH 7.4 PBS) was
used as the test substance (0.5 ml/plate). The quantity of DMSO was
equal to the maximum quantity used in the plates. To ensure strain
integrity and effectiveness of the metabolic activation system,
positive control compounds were included in each assay, with and
without the metabolic activation system. 2-nitro fluorine (10
µg/plate), sodium azide (5 µg/plate), mitomycin C
(2.5 µg/plate), 9-aminoacridine (20 µg/plate), and
2-amino anthracene (2 µg/plate) dissolved in DMSO were used
as positive controls.
Following incubation at 37°C in the dark for 48 h,
the petri dishes were observed under a microscope (Olympus BX46;
Olympus Corporation, Tokyo Japan) for the growth of revertant
bacterial colonies at all five concentrations (1,000, 2,000, 3,000,
4,000 and 5,000 µg/plate), as well as in the negative
control and positive controls against all the bacterial strains
assessed, with and without metabolic activation.
Mammalian bone marrow chromosome
aberration test in rats
A total of 60 albino wistar rats (30 males and 30
females; ~8–12 weeks old) were selected and randomly distributed
into three groups of 10 females and males. The animals were
acclimatized for 5 days prior to commencement of treatment.
The first group of 20 wistar rats (10 males and 10
females) were administered orally with CEC (dissolved in corn oil)
at a single dose of 2,000 mg/kg bw via a cannula attached to a
syringe. Similarly, the second group (negative control group) of 20
rats was administered with corn oil (vehicle) only.
Cyclophosphamide was administered as a positive control in the
third group of 20 rats, at a dose of 50 mg/kg bw
intraperitoneally.
Subsequently, 2 h prior to sacrifice (16 and 40 h
following treatment with the respective compound), the animals in
each group were treated with colchicine at a dose of 4 mg/kg bw
intraperitoneally. Subsequently, five male and five female wistar
rats from each group were sacrificed by cervical dislocation at
specified time intervals (18 and 42 h following treatment) and both
femora were removed from each animal. The femora were cleared of
tissue, and the bone marrow cells were aspirated using Hank's
Balanced Salt Solution, followed by centrifugation for 10 min at
581 × g at 4°C. The cells were then treated with hypotonic
potassium chloride solution (0.56% for 30 min at 37°C) to cause
swelling of the cells. Following further centrifugation for 10 min
at 581 × g at 4°C, the cells were fixed in methanol: glacial acetic
acid (3:1 v/v) and a homogenous cell suspension was prepared. Final
cell suspensions were then dropped onto pre-cleaned microscopic
slides, which were maintained at 4°C prior to use. Two slides were
prepared from each animal, and were dried at room temperature,
followed by staining with Giemsa (Sigma-Aldrich) and mounting in
DPX. The slides were then scored for chromosomal aberration.
Standard forms were used to record gaps, breaks and reunion
figures, and 100 metaphase spreads with intact diploid chromosomes
were scored for each animal. A mitotic index was calculated based
on 1,000 cells, which was calculated by scoring the number of cells
in mitosis per 1,000 cells observed.
Mammalian erythrocyte micronucleus test
in mice
A total of 60 mice (30 males and 30 females; ~8–12
weeks old) were selected and randomly distributed into three groups
containing 10 males and females. The females used in the present
study were nulliparous and non-pregnant. The animals were
acclimatized for 5 days prior to commencement of treatment.
The first group of 20 mice was administered with CEC
orally at a dose of 2,000 mg/kg bw (maximum tolerated dose), using
a metallic cannula attached to a syringe. The second group of 20
mice was administered orally with corn oil only (negative control).
As a positive control group, the third group of 20 mice were
treated with cyclophosphamide at a dose of 40 mg/kg bw
intraperitoneally. The animals were sacrificed 24 and 48 h
following treatment by cervical dislocation. At each time point,
five male and five female mice from each group were sacrificed.
Both femur bones were removed, muscle teased and the ends were
carefully shortened using scissors until a small opening to the
marrow canal become visible. Subsequently, 2 ml fetal calf serum
was injected into the mouth of the marrow cavity using a syringe
attached to a needle, and the marrow was flushed into centrifuge
tubes. The bone marrow suspension in the fetal calf serum was
centrifuged at 1,500 rpm for 10 min, and the supernatant was
removed using a Pasteur pipette. The cells in the sediment were
carefully mixed and a small drop of this cell suspension was placed
on a microscopic slide, from which smear slides were prepared. The
smears were fixed in methanol for 15 min and stained with Giemsa
(5% in deionized water). A total of 200 erythrocytes in the bone
marrow cells were scored in each slide, and the total numbers of
immature erythrocytes and mature erythrocytes were counted, from
which the percentage of immature erythrocytes were recorded. The
total number of micronuclei present in the 2,000 immature
erythrocytes was also noted.
Statistical Analysis
The data were statistically analyzed using a
GraphPad Prism software (version 4; GraphPad Software, Inc., San
Diego, CA, USA). Total variation present in a set of data was
analyzed using one-way analysis of variance, followed by Dunnet's
t-test. P<0.05 was considered to indicate a statistically
significant difference. Data are presented in tabular form as the
mean ± standard deviation.
Results
Acute toxicity in mice
In the acute toxicity assessment performed in the
present study, no clinical signs of toxicity were observed in any
of the treated or control mice at the dose of 5,000 mg/kg body
weight. Similarly, no mortality was observed in the animals in
either step I or step II following administration of corn oil or
CEC at 5,000 mg/kg bw. Individual body weights were recorded prior
to oral administration (day 0), and on days 7 and 14 following oral
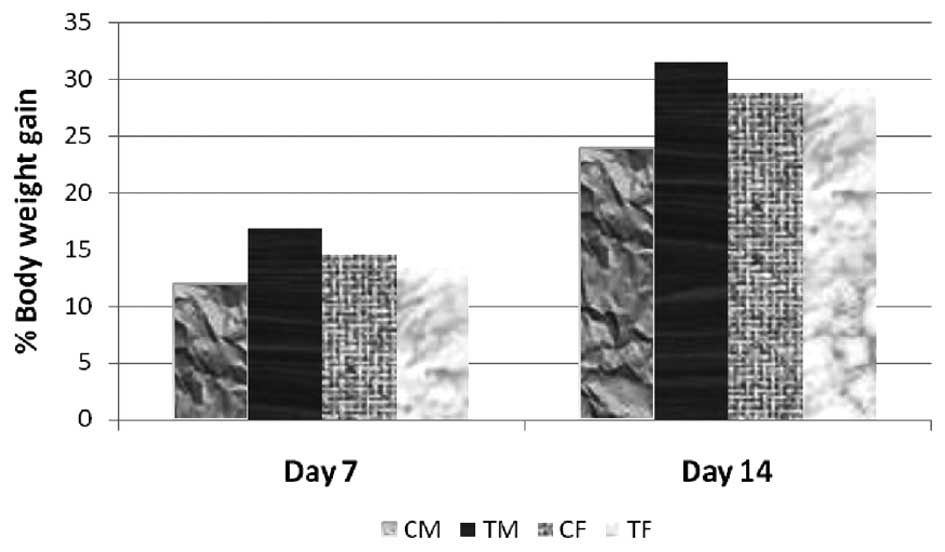
administration. The increase in the body weights (%) of the
CEC-treated animals (5,000 mg/kg bw) were comparable to the weight
gain in the control animals (Fig.
1). The difference in body weights between the control and
respective test groups were not statistically significant. All
animals were sacrificed via CO2 asphyxiation at the end
of the investigation, and no abnormalities of pathological
significance were observed. External examination of the sacrificed
mice also revealed no abnormality of pathological significance.
No mortality or signs and symptoms of toxicity were
observed in any of the animals at the maximum recommended dose of
5,000 mg/kg bw CEC. Therefore, the maximal tolerance dose (MTD),
minimal lethal dose (MLD) and lethal dose, 50% (LD50) of CEC for
mice was >5,000 mg/kg bw.
Acute toxicity in rats
No clinical signs of toxicity were observed in any
of the CEC-treated or control rats at a dose of 5,000 mg/kg bw.
Similarly, no mortality was observed in the animals in step I or
step II, following administration with corn oil or CEC at 5,000
mg/kg bw. Individual body weights were recorded prior to oral
administration (day 0), and on days 7 and 14 following oral
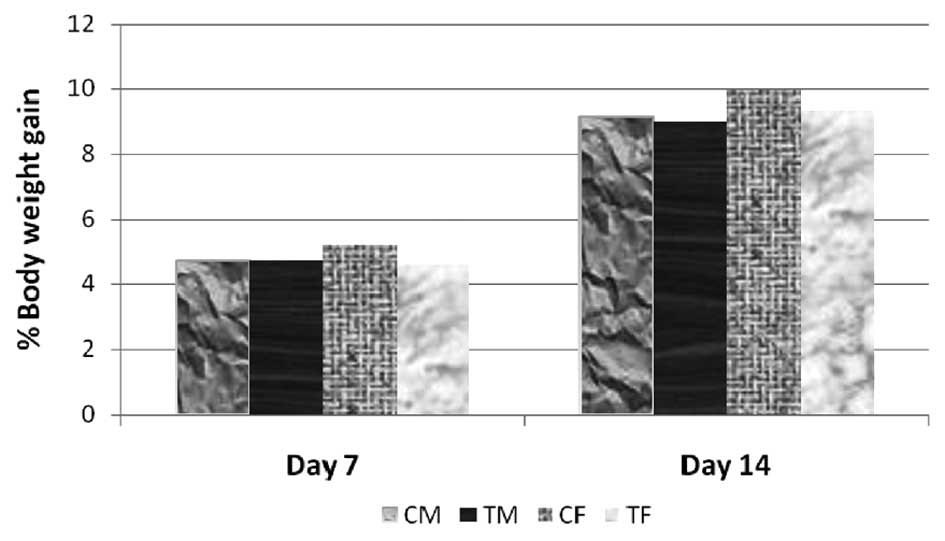
administration. The increase in body weights (%) of the treated
animals (5,000 mg/kg bw) was comparable to the percentage weight
gain of control animals (Fig. 2).
The differences in body weights between the control and respective
test groups, were not statistically significant. All animals were
sacrificed via CO2 asphyxiation, at the end of the
experiment, at which no abnormalities of pathological significance
were observed. External examination of the sacrificed rats also
revealed no abnormality of pathological significance.
No mortality of signs and symptoms of toxicity were
observed in any of the animals at the maximum recommended dose of
5,000 mg/kg bw CEC. Therefore, the MTD, MLD and LD50 of the CEC for
rats was confirmed as >5,000 mg/kg bw.
Toxicity of 90 day-repeated dose in
rats
No treatment-associated signs or symptoms of
toxicity were observed in the low dose (100 mg/kg bw), intermediate
dose (500 mg/kg bw), high dose (1,000 mg/kg bw) or satellite high
dose (1,000 mg/kg bw) groups of animals, compared with their
respective control counterparts. The body weights of all the test
and control group animals were recorded weekly. The increase in
body weight of the rats in the three treatment groups and satellite
high dose group were comparable to those in their respective
control counterparts. The consumption of feed by the animals in the
low dose, intermediate dose, high dose and satellite high dose
groups were comparable to those in the control group and satellite
control group animals (Table
I).
 | Table IConsumption of feed in the 90 day
repeated dose toxicity test in rats. |
Table I
Consumption of feed in the 90 day
repeated dose toxicity test in rats.
| Group | Average daily feed
consumption (g)
|
|---|
| Male | Female |
|---|
| Control (n=10) | 181.87±8.60 | 183.06±8.81 |
| Low dose (n=10) | 183.46±7.71 | 180.52±7.81 |
| Intermediate dose
(n=10) | 184.50±7.22 | 185.69±7.10 |
| High dose (n=10) | 182.79±9.08 | 182.67±8.29 |
| Satellite control
(n=5) | 94.24±3.24 | 93.60±4.02 |
| Satellite high dose
(n=5) | 95.15±2.64 | 94.15±3.25 |
There were no variations in the hematological
parameters of the animals in the low dose, intermediate dose, high
dose or satellite high dose groups, compared with their respective
control groups (Tables II and
III). Similarly, the biochemical
parameters of the animals in the low dose, intermediate dose and
high dose groups were comparable with the biochemical parameters of
their respective control group animals at the point of sacrifice.
The biochemical parameters of the satellite high dose group were
also comparable to those of the satellite control counterpart
(Tables IV and V). Therefore, the hematological and
biochemical parameters of the CEC-treated groups were not
significantly different from their respective control groups.
| Table IIHematological parameters of male rats
in the 90 day repeated dose toxicity assessment. |
Table II
Hematological parameters of male rats
in the 90 day repeated dose toxicity assessment.
| Group | WBC
(×103) | L % | N % | E % | M % | B % | RBC
(×106) |
Reticulocyte
% | Hb
(g %) | HCT
(PCV %) | Protime
(sec) | Platelet
count (×105) |
|---|
Control
(0 mg/kg) | 7.74±1.38 | 74.10±3.87 | 22.90±3.45 | 1.60±0.52 | 1.40±0.52 | 0.00±0.00 | 7.45±0.52 | 7.70±0.48 | 13.63±0.84 | 41.06±1.83 | 17.90±0.74 | 8.04±0.83 |
Low dose
(100 mg/kg) | 8.21±2.17 | 75.20±2.39 | 22.20±2.39 | 1.40±0.52 | 1.20±0.42 | 0.00±0.00 | 8.37±0.36 | 7.30±0.48 | 15.13±0.73 | 43.43±1.88 | 18.40±0.52 | 8.49±0.66 |
Intermediate
dose
(500 mg/kg) | 6.57±1.39 | 73.70±2.50 | 23.40±2.32 | 1.30±0.48 | 1.60±0.52 | 0.00±0.00 | 8.46±0.42 | 7.60±0.52 | 15.61±0.92 | 45.23±2.42 | 17.60±0.84 | 8.68±0.87 |
High dose
(1,000 mg/kg) | 7.94±1.87 | 74.90±2.28 | 22.20±2.62 | 1.50±0.53 | 1.20±0.42 | 0.00±0.00 | 8.51±0.49 | 7.60±0.52 | 15.50±0.83 | 45.20±1.87 | 17.40±0.70 | 8.99±0.45 |
Satellite
control
(0 mg/kg) | 8.68±1.22 | 78.00±0.71 | 19.00±0.71 | 1.80±0.45 | 1.20±0.45 | 0.00±0.00 | 8.12±0.48 | 6.80±0.84 | 14.06±0.80 | 42.04±2.13 | 18.20±1.10 | 8.06±1.10 |
High dose
satellite
(1,000 mg/kg) | 8.26±0.85 | 77.80±1.48 | 19.20±0.84 | 1.60±0.55 | 1.40±0.55 | 0.00±0.00 | 8.59±0.50 | 7.20±0.45 | 15.02±0.79 | 43.40±2.30 | 17.60±0.55 | 8.38±0.62 |
| Table IIIHematological parameters of female
rats in the 90 day repeated dose toxicity assessment. |
Table III
Hematological parameters of female
rats in the 90 day repeated dose toxicity assessment.
| Group | WBC
(×103) | L % | N % | E % | M % | B % | RBC
(×106) |
Reticulocyte
% | Hb
(g %) | HCT
(PCV %) | Protime
(sec) | Platelet
count
(×105) |
|---|
Control
(0 mg/kg) | 7.92±2.55 | 72.90±2.73 | 24.50±2.55 | 1.50±0.53 | 1.10±0.32 | 0.00±0.00 | 7.37±0.34 | 7.60±0.52 | 14.49±1.07 | 42.03±2.76 | 17.80±0.79 | 8.58±0.49 |
Low dose
(100 mg/kg) | 7.99±2.06 | 73.50±2.51 | 23.30±2.31 | 1.60±0.52 | 1.60±0.52 | 0.00±0.00 | 7.65±0.40 | 7.50±0.53 | 14.70±0.75 | 42.86±2.07 | 18.00±0.67 | 7.73±0.73 |
Intermediate
dose
(500 mg/kg) | 7.24±2.36 | 73.10±2.81 | 24.40±2.80 | 1.30±0.48 | 1.20±0.42 | 0.00±0.00 | 8.26±0.29 | 7.50±0.53 | 14.85±0.54 | 43.60±1.68 | 17.60±0.52 | 8.41±0.79 |
High dose
(1,000 mg/kg) | 7.56±2.03 | 74.80±2.82 | 22.70±2.45 | 1.30±0.48 | 1.20±0.42 | 0.00±0.00 | 7.17±0.67 | 7.50±0.53 | 14.30±0.61 | 42.72±1.92 | 18.00±0.67 | 8.28±0.63 |
Satellite
control
(0 mg/kg) | 8.34±1.27 | 73.60±1.95 | 24.00±1.22 | 1.40±0.55 | 1.40±0.55 | 0.00±0.00 | 7.67±0.38 | 7.40±0.55 | 13.72±0.53 | 41.02±1.75 | 17.60±0.89 | 8.08±1.20 |
High dose
satellite
(1,000 mg/kg) | 8.24±1.04 | 75.00±1.58 | 22.40±1.14 | 1.20±0.45 | 1.40±0.55 | 0.00±0.00 | 8.05±0.27 | 7.60±0.55 | 14.18±0.22 | 43.30±2.22 | 18.60±0.55 | 8.18±0.70 |
| Table IVBiochemical parameters of male rats
in the 90 day repeated dose toxicity assessment. |
Table IV
Biochemical parameters of male rats
in the 90 day repeated dose toxicity assessment.
| Group | ALB
(g/dl) | CHO
(mg/dl) | GLU
(mg/dl) | TG
(mg/dl) | GOT
(U/I) | GPT
(U/I) | PHOS
(mM/l) | BUN
(mg/dl) | CRE
(mg/dl) | ALP
(U/I) | T-BIL
(mg/dl) | Ca
(mm/l) | TP
(g/dl) |
Na+
(mEq/l) |
K+
(mEq/l) |
Cl−
(mEq/l) |
|---|
Control
(0 mg/kg) | 4.10±0.32 | 53.87±5.76 | 90.97±3.70 | 50.67±2.78 | 141.71±9.41 | 66.09±5.11 | 4.66±0.79 | 16.13±1.30 | 0.42±0.08 | 162.43±18.97 | 0.15±0.01 | 8.9±0.43 | 7.55±0.48 | 132.16±0.59 | 4.20±0.72 | 92.19±4.38 |
Low dose
(100 mg/kg) | 4.46±0.26 | 54.41±2.25 | 95.60±3.10 | 59.51±4.78 | 142.33±2.37 | 65.76±2.99 | 4.85±0.40 | 15.82±1.97 | 0.38±0.06 | 161.15±5.98 | 0.16±0.02 | 8.0±0.6 | 7.54±0.54 | 132.37±0.22 | 4.80±0.32 | 93.55±1.47 |
Intermediate
dose
(500 mg/kg) | 4.20±0.28 | 55.56±5.17 | 95.47±1.29 | 63.39±3.42 | 138.54±7.78 | 62.70±5.81 | 4.37±0.49 | 18.51±1.44 | 0.43±0.07 | 159.89±18.54 | 0.16±0.03 | 8.19±0.47 | 7.25±0.36 | 132.12±0.25 | 5.20±0.37 | 94.38±2.33 |
High dose
(1,000 mg/kg) | 4.13±0.27 | 60.65±2.46 | 99.19 ± 3.65 | 70.10±3.23 | 140.03±5.66 | 63.07±2.97 | 5.16±0.26 | 16.96±1.51 | 0.39±0.03 | 151.46±7.7 | 0.20±0.02 | 8.69±0.40 | 7.31±0.29 | 132.18±0.72 | 5.40±0.36 | 95.33±1.97 |
Satellite
control
(0 mg/kg) | 4.03±0.42 | 53.58±6.06 | 101.30±1.01 | 75.24±1.22 | 157.32±1.28 | 75.02±2.34 | 4.88±0.55 | 19.25±1.88 | 0.35±0.09 | 145.86±3.64 | 0.17±0.02 | 8.31±0.40 | 7.44±0.47 | 133.36±0.17 | 5.66±0.39 | 93.34±1.55 |
High dose
satellite
(1,000 mg/kg) | 4.55±0.09 | 60.40±3.79 | 104.04±2.65 | 80.42±5.14 | 153.16±5.45 | 74.82±4.26 | 5.00±0.16 | 19.38±0.84 | 0.40±0.04 | 152.70±7.51 | 0.16 ± 0.02 | 8.14±0.59 | 7.38±0.41 | 133.64±0.15 | 5.89±0.80 | 96.50±1.58 |
| Table VBiochemical parameters of female rats
in the 90 day repeated dose toxicity assessment. |
Table V
Biochemical parameters of female rats
in the 90 day repeated dose toxicity assessment.
| Group | ALB
(g/dl) | CHO
(mg/dl) | GLU
(mg/dl) | TG
(mg/dl) | GOT
(U/I) | GPT
(U/I) | PHOS
(mM/l) | BUN
(mg/dl) | CRE
(mg/dl) | ALP
(U/I) | T-BIL
(mg/dl) | Ca
(mm/l) | T.P
(g/dl) |
Na+
(mEq/l) |
K+
(mEq/l) |
Cl−
(mEq/l) |
|---|
Control
(0 mg/kg) | 4.36±0.59 | 66.91±3.60 | 94.54±3.43 | 64.94±2.80 | 122.58±5.11 | 47.99±2.69 | 5.11
0.57 | 15.82±2.25 | 0.36±0.04 | 100.60±4.77 | 0.18±0.01 | 8.59±0.33 | 7.24±0.12 | 133.20±1.48 | 4.92±0.41 | 95.75±3.72 |
Low dose
(100 mg/kg) | 4.54±0.22 | 61.28±3.45 | 95.61±2.11 | 62.90±4.06 | 123.31±4.25 | 40.37±6.88 | 4.90
0.43 | 15.98±1.89 | 0.43±0.08 | 102.53±3.45 | 0.15±0.02 | 8.24±0.53 | 7.26±0.34 | 131.97±0.64 | 4.63±0.25 | 97.32±2.42 |
Intermediate
dose
(500 mg/kg) | 5.10±0.28 | 61.39±4.51 | 89.22±2.38 | 75.06±3.63 | 124.05±2.53 | 41.52±2.07 | 4.38
0.44 | 15.19±1.80 | 0.42±0.11 | 103.08±3.95 | 0.16±0.02 | 8.40±0.51 | 7.43±0.29 | 132.12±0.25 | 5.16±0.58 | 96.43±2.65 |
High dose
(1,000 mg/kg) | 4.13±0.27 | 60.65±2.46 | 99.19±3.65 | 70.10±3.23 | 140.03±5.66 | 63.07±2.97 | 5.16
0.26 | 16.96±1.51 | 0.39±0.03 | 151.46±7.79 | 0.20±0.02 | 8.69±0.40 | 7.31±0.29 | 132.18±0.72 | 5.40±0.36 | 95.33±1.97 |
Satellite
control
(0 mg/kg) | 4.74±0.67 | 69.41±5.02 | 102.64±3.82 | 75.60±2.89 | 132.82±3.58 | 50.32±2.24 | 4.12
0.38 | 17.00±1.93 | 0.36±0.03 | 111.36±1.49 | 0.16±0.01 | 9.73±0.61 | 7.75±0.36 | 133.44±0.26 | 5.48±0.37 | 96.48±1.51 |
High dose
satellite
(1,000 mg/kg) | 4.93±0.52 | 63.90±2.95 | 104.88±1.08 | 80.06±1.38 | 141.02±1.82 | 69.82±3.94 | 4.86
0.51 | 14.28±0.81 | 0.46±0.04 | 124.04±3.78 | 0.18±0.01 | 8.83±0.77 | 7.30±0.25 | 133.62±0.18 | 5.76±0.22 | 100.56±1.09 |
Urine samples were collected from all animals at
termination of the experiment. No significant changes were noted in
the urine parameters of any of the CEC groups, compared with the
control groups. The animals in the satellite groups were sacrificed
28 days post-treatment. No animals succumbed to morality during the
experiment in any of the CEC-treated groups or satellite groups.
Following completion of the 90 day treatment period, the animals in
all groups, with the exception of the satellite groups, were
sacrificed and were examined for gross pathological features. The
organs of all the animals (treatment and control) were trimmed of
any adherent fat tissue and their weights were measured. The organ
weights of the animals in all treatment groups were comparable to
those of their respective control counterparts. No significant
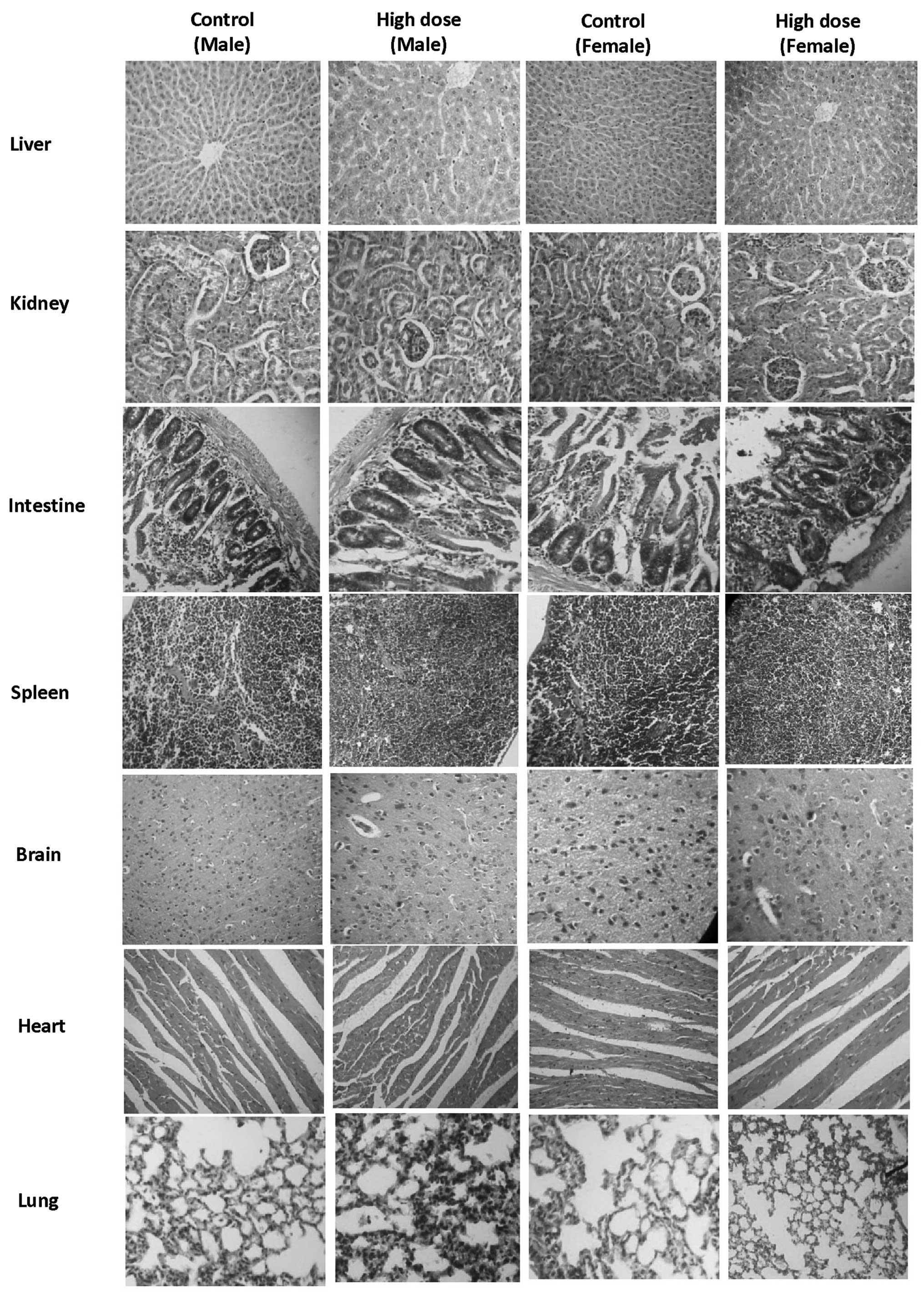
histopathological changes were identified in the animals of the low
dose group, intermediate dose group, high dose group or satellite
high dose group, compared with their control counterparts (Fig. 3).
Therefore, the repeated oral administration of CEC
for 90 days to wistar rats at a dose of 1,000 mg/kg bw did not
induce any observable toxic effects, compared with the
corresponding control group, and this dose may be considered a 'no
observed adverse effect level'.
Ames test
CEC was subsequently assessed against five strains
of Salmonella typhimurium viz. (TA-98, TA-100, TA-102,
TA-1535 and TA-1537), with and without metabolic activation, at
concentrations of 1,000, 2,000, 3,000, 4,000 and 5,000
µg/plate. The test was negative in all strains, with or
without metabolic activation (Table
VI), as no growth was observed at any of the test
concentrations, compared with that in the untreated control. No
significant differences were observed in the number of colonies in
the CEC groups, compared with the negative control group. However,
growth was observed in the positive control group, confirming the
sensitivity of the Salmonella typhimurium strains. The
numbers of colonies in the positive control groups were
significantly different from those in the negative control groups.
Therefore, under the conditions of the present study, CEC was found
to be non-mutagenic.
| Table VIAmes test of the number of colonies
in the negative control, CEC and positive control groups. |
Table VI
Ames test of the number of colonies
in the negative control, CEC and positive control groups.
| Salmonella
typhimurium strain | Colony count
|
|---|
Negative control
| CEC (5,000
µg/plate)
| Positive control
|
|---|
| +S-9 | −S-9 | +S-9 | −S-9 | +S-9 | −S-9 |
|---|
| TA-98 | 10.33±1.52 | 9.00±1.00 | 16.33±0.57b | 15.66±1.52b | 196.00±4.35a | 188.66±2.51a |
| TA-100 | 15.00±1.00 | 11.66±0.57 | 12.00±1.00b | 12.66±1.52b | 207.33±3.05a | 196.33±6.43a |
| TA-102 | 11.33±0.57 | 10.66±1.15 | 9.33±1.53b | 12.33±0.57b | 190.33±2.08a | 179.00±2.64a |
| TA-1535 | 8.33±1.52 | 10.66±1.52 | 9.66±2.08b | 11.00±1.00b | 198.66±2.51a | 188.33±4.51a |
| TA-1537 | 9.66±2.08 | 10.00±2.00 | 11.00±2.00b | 11.66±1.15b | 192.33±1.52a | 183.33±5.03a |
Mammalian bone marrow chromosome
aberration test
The mammalian bone marrow chromosome aberration test
was performed in the present study to detect any structural
chromosomal aberrations induced by CEC in the bone marrow cells.
Bone marrow metaphase preparations were made following formation of
a hypotonic solution and fixation. Metaphase preparations were
stained with Giemsa, and aberrations were classified and scored. No
evidence of numerical or structural aberrations were observed at
the maximum tolerated dose of CEC at any time point of bone marrow
harvest (Table VII). The total
number of cells with aberration in the CEC group were not
significantly different from the negative control group. In the
positive control group, data was significantly different, compared
with that of in negative control group. Therefore, CEC was
confirmed as non-mutagenic at 2,000 mg/kg bw.
| Table VIICytogenetic analysis with the
mammalian bone marrow chromosome aberration test. |
Table VII
Cytogenetic analysis with the
mammalian bone marrow chromosome aberration test.
| Group | Time of sacrifice
(h) | Gender | Number of
animals | Total aberrations
(n) | Cells with
aberration (n) | Mitotic index
(%) |
|---|
| CEC (2,000
mg/kg) | 18 | M | 5 | 1.20±1.10 | 1.00±1.00 | 6.06±0.20 |
| | F | 5 | 2.00±0.71 | 1.40±0.55 | 6.08±0.24 |
| 42 | M | 5 | 1.00±1.00 | 0.60±0.55 | 5.80±0.19 |
| | F | 5 | 1.20±1.30 | 0.80±0.84 | 5.82±0.68 |
| Positive control
(cyclophosphamide) | 18 | M | 5 | 240.60±5.32a | 52.80±6.46a | 1.90±0.27a |
| | F | 5 | 237.40±7.09a | 51.60±6.27a | 1.80±0.32a |
| 42 | M | 5 | 216.00±9.27a | 48.00±7.58a | 1.98±0.36a |
| | F | 5 | 212.00
±4.45a | 54.40±5.68a | 2.14±0.18a |
| Negative
control | 18 | M | 5 | 2.20±0.84 | 1.80±0.84 | 6.38±0.28 |
| | F | 5 | 2.40±0.55 | 1.80±0.84 | 6.06±0.21 |
| 42 | M | 5 | 2.00±0.71 | 1.20±0.45 | 6.82±0.40 |
| | F | 5 | 1.40±0.89 | 1.20±0.45 | 5.78±0.49 |
Mammalian erythrocyte micronucleus test
in mice
The mammalian erythrocyte micronucleus test was
performed in the present study to investigate the mutagenic effects
of CEC in Swiss albino mice. Following the administration of drugs
in the respective groups, the animals were monitored daily for
signs and symptoms of toxicity or mortality prior to sacrifice. No
mortality of signs and symptoms of toxicity were observed in any of
the animals in the CEC groups. Following sacrifice of five males
and five females from each group, at 24 and 48 h post-dose
administration, the femora were removed and the bone marrow
collected to produce smears, which were then stained and examined
microscopically. No significant changes were observed in the
numbers of immature and mature erythrocytes in the animals treated
with 2,000 mg/kg bw CEC at 24 and 48 h, compared with their
respective negative control animals (Table VIII). Therefore, the percentage
of immature erythrocytes in the treated animals at a dose of 2,000
mg/kg bw at 24 and 48 h, were comparable with the negative control.
However, the numbers of immature and mature erythrocytes were
significantly higher in the positive control group (40 mg/kg wt) at
24 and 48 h, compared with the negative control group.
| Table VIIIBone marrow values determined using a
mammalian erythrocyte micronucleus test in mice. |
Table VIII
Bone marrow values determined using a
mammalian erythrocyte micronucleus test in mice.
| Parameter | 24 h following
treatment
| 48 h following
treatment
|
|---|
Negative control
(corn oil)
| CEC (20,00 mg/kg)
| Positive control
(cyclophosphamide) 40 mg/kg
| Negative control
(corn oil)
| CEC (20,00 mg/kg)
| Positive control
(cyclophosphamide) 40 mg/kg
|
|---|
| M | F | M | F | M | F | M | F | M | F | M | F |
|---|
| Total erythrocytes
counted (n) | 200.00±0.00 | 200.00±0.00 | 200.00±0.00 | 200.00±0.00 | 200.00±0.00 | 200.00±0.00 | 200.00±0.00 | 200.00±0.00 | 200.00±0.00 | 200.00±0.00 | 200.00±0.00 | 200.00±0.00 |
| Immature
erythrocytes obtained (n) | 106.00±6.56 | 104.00±7.58 | 106.40±6.23 | 106.40±4.51 | 98.80±2.59 | 97.20±2.59 | 107.20±5.59 | 100.40±2.88 | 104.40±5.77 | 104.40±4.28 | 99.20±4.09 | 99.60±3.91 |
| Mature erythrocytes
obtained (n) | 94.00±0.56 | 96.00±7.58 | 93.60±6.23 | 93.60±4.51 | 101.20±2.59 | 104.80±2.77 | 92.80±5.59 | 99.60±2.88 | 95.60±5.97 | 95.60±4.28 | 100.80±4.09 | 100.40±3.91 |
| Immature
erythrocytes in total erythrocytes (%) | 53.00±3.28 | 52.00±3.79 | 53.20±3.11 | 53.20±2.25 | 49.40±1.29 | 48.60±1.29 | 53.60±2.79 | 50.20±1.44 | 52.20±2.89 | 52.20±2.14 | 49.60±2.04 | 49.10±1.24 |
| Total immature
erythrocytes counted (n) | 2,000.00±0.00 | 2,000.00±0.00 | 2,000.00±0.00 | 2,000.00±0.00 | 2,000.00±0.00 | 2,000.00±0.00 | 2,000.00±0.00 | 2,000.00±0.00 | 2,000.00±0.00 | 2,000.00±0.00 | 2,000.00±0.00 | 2,000.00±0.00 |
| Total micronuclei
present in immature erythrocytes (n) | 1.00 0.00 | 1.20±0.45 | 1.00±0.71b | 1.00±0.71b | 56.20±2.95a | 56.20±2.95a | 0.60±0.54 | 1.33±0.58 | 0.80±0.49b | 1.33±0.5b | 45.80±4.44a | 45.40±8.29a |
No significant changes in the micronuclei were
induced by CEC at either 24 or 48 h, compared with their negative
control counterparts. As no significant effect was seen in the
CEC-treated animals, it was concluded that CEC exhibited no
mutagenic potential.
Discussion
Curcumin has wide spectrum of therapeutic activity,
however the major limitation with curcumin is its poor oral
absorption and, thus low bioavailability. CEC is a curcumin
formulation, exhibiting increased bioavailability (28), however, it is important to ensure
that increased bioavailability does not increase toxicity, which
was the focus of investigation in the present study.
In the assessment and evaluation of the toxic
characteristics of a test substance, determination of acute oral
toxicity in mice and rats is usually an initial step, and
evaluation in two species (rats and mice) is advantageous to
confirm the findings (30). The
results of acute toxicity obtained in the present study indicated
that CEC was safe up to 5,000 mg/kg in rats and mice. The LD50 for
mice and rats was >5,000 mg/kg, which confirms CEC as a nontoxic
material. The 90 day repeated dose toxicity test in rats further
confirmed the non-toxicity of CEC, as the repeated administration
of CEC for 90 days in rats at a dose of 1,000 mg/kg bw caused no
observable toxic effects, compared with the corresponding control
group.
The Ames test is commonly used as an initial screen
for genotoxic activity and, in particular, for point mutation
activity (31). The results of the
Ames test in the present study indicated that CEC was non-mutagenic
up to 5,000 µg/plate, compared with the untreated control.
Similarly, the results of the chromosomal aberration test indicated
that CEC was non-mutagenic at a dose of 2,000 mg/kg bw in rats.
The mammalian micronucleus test is used for the
detection of damage induced by a test substance towards the mitotic
apparatus of erythroblasts, by analyzing erythrocytes sampled from
bone marrow (32). In the present
study, no effects were observed in the micronuclei at either 24 or
48 h following treatment with CEC, compared with their negative
control counterparts. As no significant effect was seen in the
treated animals, it was concluded that CEC did not exhibit any
mutagenic potential.
In conclusion, the results of the present study
indicated that CEC had no toxic effects in the animal models used,
as per OECD guidelines. The results of the in vitro
assessment also confirmed the non-toxicity and safety of CEC. Taken
together, CEC was observed to be non-toxic and safe, and was
supported by evidence from investigations of acute toxicity,
repeated dose toxicity and mutagenicity.
Acknowledgments
The authors would like to thank the Spices Board,
Ministry of Commerce and Industry, Government of India (Cochin,
India) for financial support and Arjuna Natural Extracts Ltd.
(Aluva, India) for providing the sample of CEC
(BCM-95®/Biocurcumin™).
References
|
1
|
Ravindran PN, Nirmal Babu K and Sivaraman
K: Turmeric. The golden spice of life. Turmeric The Genus Curcuma
Boca Raton, FL, USA: CRC Press; 2007, pp. 1–14
|
|
2
|
Maheshwari RK, Singh AK, Gaddipati J and
Srimal RC: Multiple biological effects of curcumin: A short review.
Life Sci. 78:2081–2087. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chandra D and Gupta SS: Anti-inflammatory
and anti-arthritic activity of volatile oil of Curcuma longa
(Haldi). Indian J Med Res. 60:138–142. 1972.PubMed/NCBI
|
|
4
|
Aggarwal BB, Sundaram C, Malani N and
Ichikawa H: Curcumin: The Indian solid gold. Adv Exp Med Biol.
595:1–75. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Aggarwal BB and Shishodia S: Molecular
targets of dietary agents for prevention and therapy of cancer.
Biochem Pharmacol. 71:1397–1421. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sharma RA, Gescher AJ and Steward WP:
Curcumin: The story so far. Eur J Cancer. 41:1955–1968. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Duvoix A, Blasius R, Delhalle S,
Schnekenburger M, Morceau F, Henry E, Dicato M and Diederich M:
Chemopreventive and therapeutic effects of curcumin. Cancer Lett.
223:181–190. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Joe B, Vijayakumar M and Lokesh BR:
Biological properties of curcumin-cellular and molecular mechanisms
of action. Crit Rev Food Sci Nutr. 44:97–111. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Menon LG, Kuttan R and Kuttan G:
Inhibition of lung metastasis in mice induced by B16F10 melanoma
cells by polyphenolic compounds. Cancer Lett. 95:221–225. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Menon LG, Kuttan R and Kuttan G:
Anti-metastatic activity of curcumin and catechin. Cancer Lett.
141:159–165. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen HW, Yu SL, Chen JJ, Li HN, Lin YC,
Yao PL, Chou HY, Chien CT, Chen WJ, Lee YT and Yang PC:
Anti-invasive gene expression profile of curcumin in lung
adenocarcinoma based on a high throughput microarray analysis. Mol
Pharmacol. 65:99–110. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tønnesen HH, Másson M and Loftsson T:
Studies of curcumin and curcuminoids. XXVII. Cyclodextrin
complexation: Solubility, chemical and photochemical stability. Int
J Pharm. 244:127–135. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pan MH, Huang TM and Lin JK:
Biotransformation of curcumin through reduction and glucuronidation
in mice. Drug Metab Dispos. 27:486–494. 1999.PubMed/NCBI
|
|
14
|
Johnson JJ and Mukhtar H: Curcumin for
chemoprevention of colon cancer. Cancer Lett. 255:170–181. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sharma RA, Euden SA, Platton SL, Cooke DN,
Shafayat A, Hewitt HR, Marczylo TH, Morgan B, Hemingway D, Plummer
SM, et al: Phase I clinical trial of oral curcumin: Biomarkers of
systemic activity and compliance. Clin Cancer Res. 10:6847–6854.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Anand P, Kunnumakkara AB, Newman RA and
Aggarwal BB: Bioavailability of curcumin: Problems and promises.
Mol Pharm. 4:807–818. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cui J, Yu B, Zhao Y, Zhu W, Li H, Lou H
and Zhai G: Enhancement of oral absorption of curcumin by
self-micro-emulsifying drug delivery systems. Int J Pharm.
371:148–155. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Marczylo TH, Verschoyle RD, Cooke DN,
Morazzoni P, Steward WP and Gescher AJ: Comparison of systemic
availability of curcumin with that of curcumin formulated with
phosphatidylcholine. Cancer Chemother Pharmacol. 60:171–177. 2007.
View Article : Google Scholar
|
|
19
|
Ganiger S, Malleshappa HN, Krishnappa H,
Rajashekhar G, Ramakrishna Rao V and Sullivan F: A two generation
reproductive toxicity study with curcumin, turmeric yellow, in
Wistar rats. Food Chem Toxicol. 45:64–69. 2007. View Article : Google Scholar
|
|
20
|
Shankar TN, Shantha NV, Ramesh HP, Murthy
IA and Murthy VS: Toxicity studies on turmeric (Curcuma longa):
Acute toxicity studies in rats, guineapigs and monkeys. Indian J
Exp Biol. 18:73–75. 1980.PubMed/NCBI
|
|
21
|
Ravindranath V and Chandrasekhara N:
Absorption and tissue distribution of curcumin in rats. Toxicology.
16:259–265. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wahlström B and Blennow G: A study on the
fate of curcumin in the rat. Acta Pharmacol Toxicol (Copenh).
43:86–92. 1978. View Article : Google Scholar
|
|
23
|
Burgos-Morón E, Calderón-Montaño JM,
Salvador J, Robles A and López-Lázaro M: The dark side of curcumin.
Int J Cancer. 126:1771–1775. 2010.
|
|
24
|
Kizhakkedath R: Clinical evaluation of a
formulation containing Curcuma longa and Boswellia serrata extracts
in the management of knee osteoarthritis. Mol Med Rep. 8:1542–1548.
2013.PubMed/NCBI
|
|
25
|
EFSA Panel on Food Additives and Nutrient
Sources added to Food (ANS): Scientific Opinion on the reevaluation
of curcumin (E 100) as a food additive. EFSA Journal.
8:16792010.
|
|
26
|
Joint FAO/WHO Expert Committee on Food
Additives (JECFA): Summary and conclusions of the sixty-first
meeting. JECF; Rome, Italy: pp. 1–22. 2010
|
|
27
|
Henrotin Y, Priem F and Mobasheri A:
Curcumin: a new paradigm and therapeutic opportunity for the
treatment of osteoarthritis: curcumin for osteoarthritis
management. Springerplus. View Article : Google Scholar
|
|
28
|
Antony B, Merina B, Iyer VS, Judy N,
Lennertz K and Joyal S: A pilot cross-over study to evaluate human
oral bioavailability of BCM-95CG (Biocurcumax), a novel bioenhanced
preparation of Curcumin. Indian J Pharm Sci. 70:445–449. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
OECD Guidelines for Testing of Chemicals.
(Acute oral toxicity - fixed dose procedure, test no. 420).
Organisation for Economic Co-operation and Development; Paris,
France: pp. 1–14. 2002
|
|
30
|
Parasuraman S: Toxicological screening. J
Pharmacol Pharmacother. 2:74–79. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maron DM and Ames BN: Revised methods for
the Salmonella mutagenicity test. Mutat Res. 113:173–215. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schmid W: The micronucleus test. Mutat
Res. 31:9–15. 1975. View Article : Google Scholar : PubMed/NCBI
|