Introduction
As the most important subcellular organelle of
energy metabolism in heart, mitochondria are also important in the
balance of oxidative metabolism, Ca2+ homeostasis and
for formation of reactive oxygen species (1). Abnormal mitochondrial respiration can
result in oxidative stress via oxidative phosphorylation, and a
compromised antioxidant system may lead to excessive oxidative
stress in cardiovascular diseases, including hypertension, cardiac
hypertrophy and atherosclerosis, indicating that mitochondria exert
a fundamental role in disease progression (2–4).
Complex I (NADH:ubiquinone oxidoreductase) is the
largest multimeric enzyme in the mitochondrial respiratory chain,
which is responsible for electron transport and the generation of a
proton gradient across the mitochondrial inner membrane to drive
ATP production. It consists of 45 subunits with a combined mass of
~980 kDa. Of those subunits, seven are encoded by mitochondrial
DNA, whereas the other 38 subunits are encoded by nuclear genes.
Ndufa10, a nuclear-encoded 42 kDa protein, is located in the
hydrophobic membrane (5,6). A previous study in our laboratory
reported that the NADH dehydrogenase 1α subcomplex 10 (Ndufa10)
from the spontaneously hypertensive rat (SHR) strain exhibited a
different profile from that of Wistar Kyoto (WKY) rats on
two-dimensional (2D) fluorescence difference gel electrophoresis
(7). The position of the Ndufa10
protein from the SHR strain demonstrated a horizontal shift to a
lower isoelectric point (pI; ~0.2 pH units), with a slight increase
in molecular weight (MW; ~2 kDa) in the 2D gel compared with
the protein from WKY. Genetic mutations and phosphorylation events
are the most common causes for a pI shift decrease, and an MW
increase, on performing 2D gel electrophoresis. Ndufa10 was
reported to be phosphorylated by the cAMP-dependent protein kinase,
and contains phosphothreonine residues in the steady state in
bovine heart mitochondria (8–10).
In addition to phosphorylation, the increase in MW could also be
caused by other post-translational modifications, including
deamidation, acetylation and methylation. At present, 33
modifications of a distinct chemical nature, targeting 59 specific
residues of Ndufa10, have been found to be common to the acidic and
basic forms (11). Previously, an
A/G nucleotide transition at position 358 of Ndufa10, leading to an
AAT-to-GAT mutation and, consequently, an Asn (N) to Asp (D)
mutation at position 120 of the Ndufa10 amino acid sequence, was
identified in our laboratory. However, the estimated difference in
the MW caused by this mutation was only ~1 Da, which did not match
the apparent MW, as determined from 2D gel electrophoretic and
western blot analyses. It may be speculated that the mutation
already co-existed, although with certain modifications. The aim of
the present study was to determine the unique characteristics of
the Ndufa10 protein from SHRs using 2D gel electrophoresis.
Materials and methods
2D gel electrophoresis
The isolation of the mitochondria from the left
ventricles of 20-week-old WKY and SHR male rats was performed, as
described previously (7). All the
animals were obtained from the Shanghai SLAC Laboratory Animal Co.,
Ltd. (Shanghai, China) and housed in a temperature-controlled
environment (22–24°C) with a 12/12 h light-dark cycle with free
access to food and water. All the procedures were approved by the
local ethics committee of the School of Medicine, Shanghai Jiao
Tong University (Shanghai, China; approval no. 2014069). Aliquots
of 40 µg protein from each sample were diluted in
rehydration solution [8 M urea, 2% (w/v) CHAPS, 65 mM
dithiothreitol (DTT) and 0.5% (v/v) Bio-lyte 3/10 ampholyte;
Bio-Rad Laboratories, Inc., Hercules, CA, USA)], adjusting the
total volume to 125 µl prior to applying the solution to
non-linear IPG strips (7 cm in length; pH 3-10; Bio-Rad
Laboratories, Inc.) using the following rehydration and running
conditions: 200 V for 30 min, 500 V for 1 h, 1,000 V for 2 h and
4,000 V until 10,000 V/h were reached. Following the completion of
the isoelectric focusing procedure, the IPG strips were
equilibrated for 30 min with 50 mM Tris-HCl (pH 8.8), 6 M urea, 30%
glycerol and 2% SDS, followed by reduction with 1% DTT and
alkylation with 2.5% iodoacet-amide (Bio-Rad Laboratories, Inc.).
The 2D separations were performed on 12% SDS-PAGE gels, and
subsequently the proteins were transferred onto nitrocellulose
membranes. The membranes were blocked with 5% nonfat dry milk
solution for 1 h at room temperature. The membranes were blotted
with mouse monoclonal anti-β-tubulin (dilution, 1:1,000; cat. no.
T4026; Sigma-Aldrich, St. Louis, MO, USA) and chicken polyclonal
anti-human Ndufa10 (dilution, 1:2,000; cat. no. ab19131; Abcam,
Cambridge, UK) separately, followed by incubation with horseradish
peroxidase-conjugated horse anti-mouse IgG (H+L) (dilution,
1:2,000; cat. no. 7076; Cell Signaling Technology, Inc., Danvers,
MA, USA) and peroxidase-labeled goat anti-chicken IgG (H+L)
(dilution, 1:2,000; cat. no. 14-24-06; KPL, INC., Gaithersburg, MD,
USA). The membranes were developed using luminol-based enhanced
chemiluminescence reagent (SuperSignal™ West Pico Chemiluminescent
Substrate; Pierce Biotechnology, Inc., Rockford, IL, USA).
DNA sequencing
The genomic DNA was extracted from blood leukocytes
from WKY and SHR rats aged 20 weeks using a blood genomic DNA
extraction system [Tiangen Biotech (Beijing) Co., Ltd, Beijing,
China], according to the manufacturer's protocol. Exon 3 of the
Ndufa10 gene, where the mutation resides, was PCR-amplified and
sequenced using, as the forward primer,
5′-TGGTTGTAGAGAACGGAGAAGCTC-3′, and as the reverse primer,
5′-CTGCATCAACAGGGATTTTGAGG-3′.
Plasmid construction and the
transformation of prokaryotic cells
The total RNA was extracted from WKY and SHR heart
tissue using Invitrogen TRIzol® reagent (Thermo Fisher
Scientific, Waltham, MA, USA), according to the manufacturer's
protocol. Samples of 1 µg total RNA were used for cDNA
synthesis using the Takara RNA polymerase chain reaction (PCR) kit
(Takara Bio, Dalian, China), according to manufacturer's protocol.
The cDNA encoding Ndufa10 was amplified by PCR using Pyrobest™ DNA
polymerase and the Takara PCR kit (Takara Bio) in a final volume of
50 µl, containing the forward primer,
5′-CACCCATGGCCTTGAGGTTGCTG-3′, and the reverse primer,
5′-CCACTCGAGTCACTTCAGCCAGATCCA-3′. The forward and reverse primers
contained an NcoI site and an XhoI site,
respectively. The thermal cycle for a hot-start PCR was: 95°C for 5
min; 40 cycles of 95°C for 30 sec; 65°C for 30 sec; 72°C for 1 min
30 sec; and, finally, 72°C for 10 min. The amplified DNA fragments
were separated using a 1% agarose gel. Subsequently, bands of the
correct size (1.068 kb) were cut from the gel and recovered using
the GenClean™ agarose gel DNA recovery kit I (Shanghai Generay
Biotech Co., Ltd., Shanghai, China), prior to subcloning into
Novagen pET28a-c (+) plasmids (EMD Millipore, Billerica, MA, USA)
using the NcoI and XhoI sites. The resultant
recombinant plasmids were subsequently transformed into DH5α cells
(Tiangen biotech (Beijing) Co., Ltd.). Following DNA sequencing to
verify the constructs, the wild-type and mutant constructs were
transformed into competent BL21(DE3) cells (Tiangen biotech
(Beijing) Co., Ltd.) for recombinant protein production. Single
colonies were inoculated into Luria-Bertani (LB) media containing
kanamycin (50 µg/ml), and grown overnight at 37°C. The
overnight cultures were diluted 1:100 into fresh LB media
containing 50 µg/ml kanamycin, and grown at 37°C with
agitation until the optical density at 600 nm had reached between
0.6 and 0.8. Protein expression was induced by adding 0.8 M
isopropyl-1-thio-β-D-galactopyranoside at a ratio of 1:1,500 for 3
h. The cultures were centrifuged at 9,600 × g for 1 min at 4°C, the
supernatant was discarded and the cell pellets were dissolved in 5X
SDS PAGE loading buffer. The expression level of the protein was
analyzed using 10% SDS-PAGE, and the proteins were visualized using
Commassie Brilliant Blue staining. The protein expression level of
Ndufa10 was detected using western blotting.
Recombinant protein purification and
measurement of the MW of Ndufa10 variants
For the large-scale production of wild-type and
mutant Ndufa10 proteins, the cell pellets were resuspended in 1X
phosphate-buffered saline and sonicated on ice using an
ultra-sonicator (×l2,000; Misonix sonicator, Farmingdale, NY, USA)
at 40% power, with 60 cycles of 5 sec on/10 sec off. The
suspensions were collected by centrifugation for 30 min at 20,000 ×
g at 4°C. The resulting pellets, containing inclusion bodies, were
extracted twice in lysis buffer [10 mM imidazole, 100 mM
NaH2PO4, 10 mM Tris, 8 M urea (pH 8.0)] for
30 min each at room temperature. The protein extracts were
subsequently centrifuged for 30 min at 20,000 × g at 4°C. The
supernatants were pooled, and equilibrated
Ni2+-nitrilotriacetic acid (NTA) agarose was added
(Qiagen, Manchester, UK) and placed on a rotary shaker at 4°C
overnight. Following five washes with wash buffer [20 mM imidazole,
100 mM NaH2PO4, 10 mM Tris and 8 M urea (pH
8.0)], the proteins were eluted with elution buffer [250 mM
imidazole, 100 mM NaH2PO4, 10 mM Tris and 8 M
urea (pH 8.0)] at 4°C. The purity of the proteins was determined
using 10% SDS-PAGE with Coomassie Blue G-250 staining. The eluted
fractions were desalted and concentrated using
Centricon® YM-10 centrifugal filter devices (EMD
Millipore) prior to the determination of the MW using
matrix-assisted laser desorption/ionization-time of flight-mass
spectrometry (MALDI-TOF-MS). Finally, the Ndufa10-120N and
Ndufa10-120D proteins were mixed and diluted with rehydration
buffer, prior to 2D electrophoresis.
Site-directed mutagenesis
A series of Ndufa10 mutations at amino acid position
120 were made using a PCR site-directed mutagenesis kit (Toyobo,
Co., Ltd., Osaka, Japan) and pET28a-Ndufa10-120N as the template,
according to the manufacturer's protocol. These mutations included
neutral non-polar leucine (L), basic polar lysine (K) and acidic
polar glutamic acid (E) residues. All the mutants were confirmed
using DNA sequencing. The production of recombinant Ndufa10 mutants
was performed as described above. The primers used were as follows:
120E, forward:
5′-CTGTAGTTTAGAGAAATTTTATGACGAACCCAAAAGCAACGACGGCAACAGCTA-3′;
reverse:
5′-TAGCTGTTGCCGTCGTTGCTTTTGGGTTCGTCATAAAATTTCTCTAAACTACAGC-3′;
120L, forward:
5′-GCTGTAGTTTAGAGAAATTTTATGACCTTCCCAAAAGCAACGACGGCAACAGCTA-3′;
reverse:
5′-TAGCTGTTGCCGTCGTTGCTTTTGGGAAGGTCATAAAATTTCTCTAAACTACAGC-3′;
120K, forward:
5′-GCTGTAGTTTAGAGAAATTTTATGACAAGCCCAAAAGCAACGACGGCAACAGCTA-3′; and
reverse:
5′-TAGCTGTTGCCGTCGTTGCTTTTGGGCTTGTCAAAAATTTCTCTAAACTACAGC-3′.
Results
Detection of ndufa10 variants using 2D
western blot analysis and MALDI-TOF-TOF tandem MS
The Ndufa10 variant proteins from the cardiac tissue
of SHR and WKY rats were detected using 2D western blot analysis,
by which proteins were sequentially separated on the basis of their
pI and size. As shown in Fig. 1A,
the Ndufa10 protein from the SHR rats was identified to have lower
pI value (~0.2 pH unit difference) and a higher MW (~2 kDa
difference) compared with that from the WKY rats. Using
MALDI-TOF-TOF tandem MS led to the detection of two different
peptides, FYDN120PK and FYDD120PK, in samples from the WKY and SHR
rats, respectively (Fig. 1B). The
D/N substitution at amino acid position 120 resulting from an A/G
transition at position 358 in the coding region of Ndufa10 from the
SHR rat was further confirmed by genomic DNA sequencing (RefGene_
NM_199495 range=chr9: 91655135-91689653; Fig. 1C).
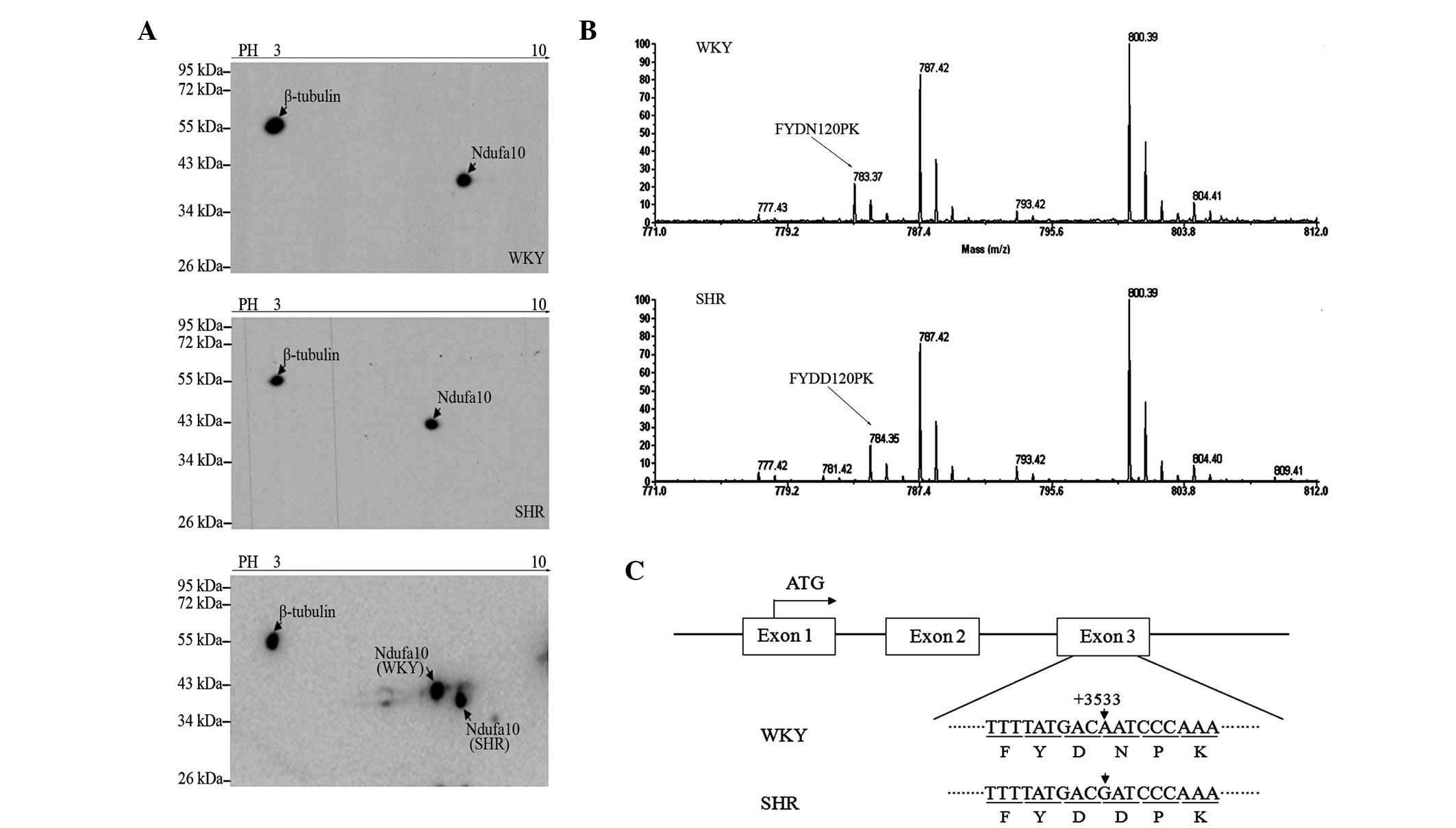 | Figure 1Ndufa10 variants in a 2D western blot
and MALDI-TOF-TOF tandem MS. (A) A 2D-western blot of Ndufa10 in
the left ventricle of WKY and SHR rats is shown. β-tubulin was used
as an internal control. (B) The matched peptide, FYDNPK, obtained
using MALDI-TOF-TOF tandem MS is shown in capital letters for the
WKY protein, and the peptide, FYDDPK, of MALDI-TOF-TOF tandem MS
shown in capital letters in for the SHR protein. The D/N
substitution was at amino acid position 120 of the Ndufa10 amino
acid sequence. (C) The expression of the Ndufa10 gene is shown,
highlighting the genetic point mutation which is modulated in the
SHR protein. MALDI-TOF-TOF tandem MS, matrix-assisted laser
desorption/ionization-time of flight time-of-flight tandem mass
spectrometry; Ndufa10, NADH dehydrogenase 1α subcomplex 10; SHR,
spontaneously hypertensive rat; WKY, Wistar Kyoto. |
Expression of the Ndufa10 variants in
vitro
In order to determine whether the apparent
difference in MW between the two Ndufa10 variants was solely caused
by the N-to-D substitution or post-translational modifications,
initially, the MW of these two bacterially expressed recombinant
Ndufa10 variants were compared. As shown in Fig. 2, the expression of both recombinant
Ndufa10-120N and Ndufa10-120D Ndufa10 proteins was markedly
induced, as revealed by Coomassie Brilliant Blue staining (the
bottom panel) and western blotting (the top panel). Similarly to
what was previously observed with rat tissue samples in our
laboratory, the apparent MW of the Ndufa10-120D protein waŝ2 kDa
higher compared with that of Ndufa10-120N, suggesting that the
N-to-D substitution was sufficient to cause the shift in its
MW.
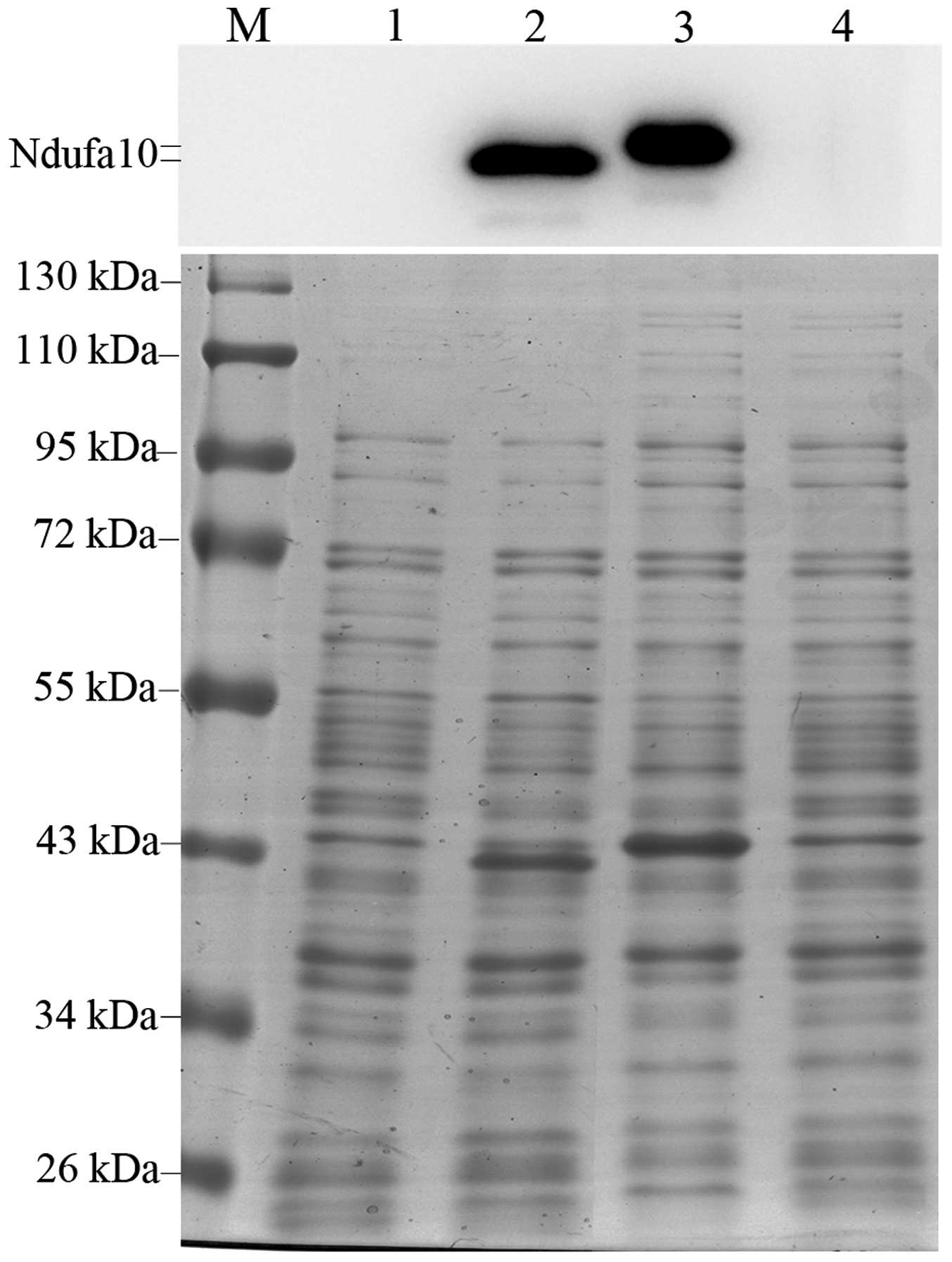 | Figure 2Detection of IPTG-induced expression
of Ndufa10 protein in Escherichia coli BL21(DE3) cells. The
pET28a-Ndufa10-120N and -120D plasmids were transformed into the
BL21(DE3) strain of E. coli, and induced by the addition of
0.8 mM IPTG for 3 h at 37°C. The protein expression levels were
investigated using 10% SDS-PAGE with western blotting (upper panel)
and Commassie Brilliant Blue staining (lower panel). Lane 1,
Ndufa10-120N, uninduced; lane 2, Ndufa10-120N, induced with IPTG;
lane 3, Ndufa10-120D, induced with IPTG; lane 4, Ndufa10-120D,
uninduced; lane M, standard protein size markers (kDa). IPTG,
isopropyl-1-thio-β-D-galactopyranoside; Ndufa10, NADH dehydrogenase
1α subcomplex 10. |
Purification and MW measurement of the
Ndufa10 variants
To measure the MW more precisely, bacterially
expressed, histidine-tagged recombinant Ndufa10 proteins from the
two groups were assessed using Ni2+-NTA agarose gel
electrophoresis under denaturing conditions. The purification
processes were revealed to yield high-purity proteins for both
Ndufa10-120N and Ndufa10-120D (Fig.
3A). Purified, recombinant Ndufa10-120N and Ndufa10-120D
proteins, and a mixture of equal quantities of them, were subjected
to 2D electrophoretic separation. As shown in Fig. 3B, the recombinant Ndufa10-120N and
Ndufa10-120D proteins appeared as two distinct bands, although a
shift in the MW was observed with the Ndufa10-120D protein,
similarly to what was observed previously in our laboratory with
SHR rats. Further analysis using MALDI-TOF-TOF tandem MS revealed
an experimental MW for the Ndufa10-120N protein of 41,618.4688 Da
(theoretical MW, 41,655.49 Da), whereas the experimental MW of the
Ndufa10-120D protein was 41,618.6172 Da (theoretical MW, 41,656.47
Da), further confirming that the difference in mobility in SDS-PAGE
was caused by the N/D substitution (Fig. 3C).
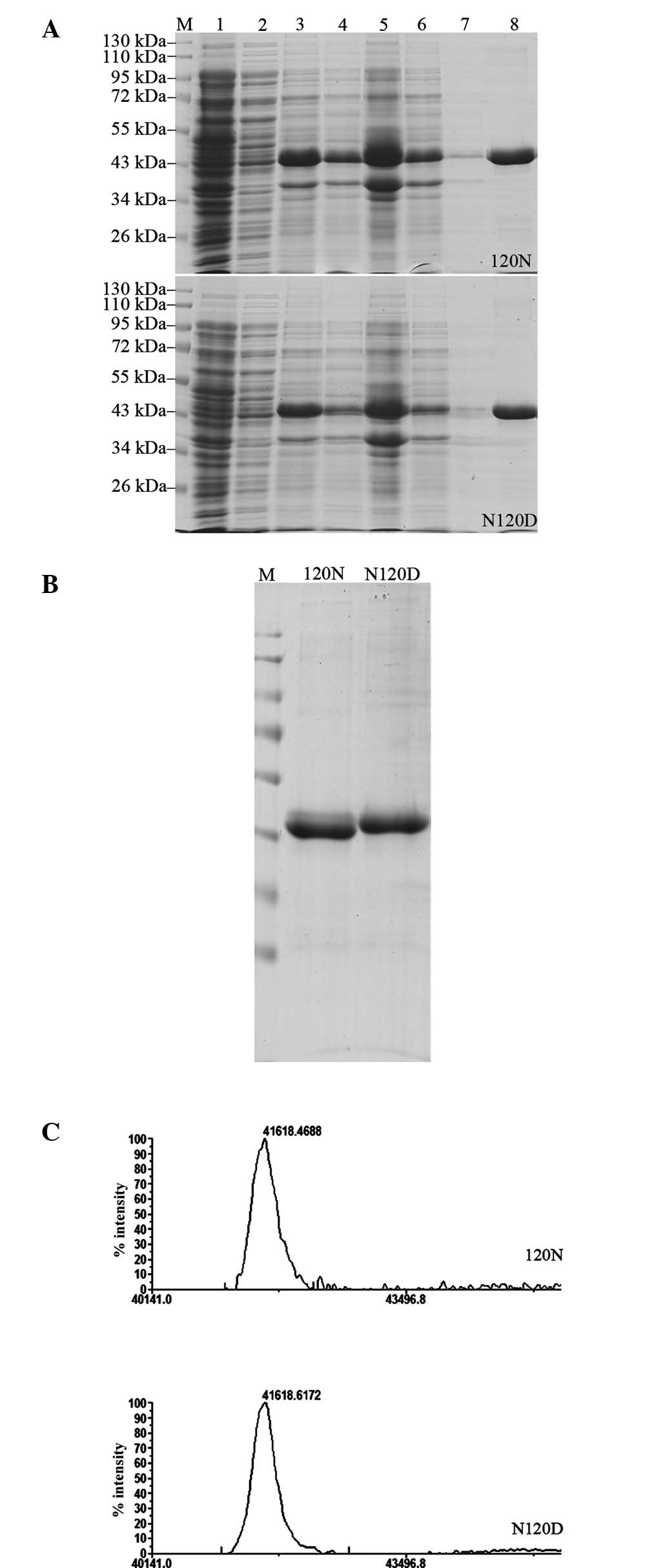 | Figure 3Purification of the Ndufa10 fusion
proteins, and the practical determination of the MW of the
Ndufa10-120N and -120D proteins. (A) Protein purification of the
Ndufa10-120N (upper panel) and the Ndufa10-120D (lower panel)
proteins is shown. Lane 1, uninduced cells; lane 2, supernatant of
the IPTG-induced cells; lane 3, the IPTG-induced cells after a
duration of 3 h; lane 4, cell lysate; lane 5, precipitate following
centrifugation; lane 6, supernatant of Ni2+-NTA agarose
combined protein; lane 7, wash with lysis buffer; lane 8, 250 mM
imidazole elution; lane M, standard protein size markers (kDa). (B)
Separation of the proteins using 10% SDS-PAGE, and detection of the
protein bands by Commassie Brilliant Blue staining (C)
Matrix-assisted laser desorption/ionization-time of
flight/time-of-flight tandem mass spectrometric analysis of the
purified recombinant Ndufa10 protein variants. IPTG,
isopropyl-1-thio-β-D-galactopyranoside; Ndufa10, NADH dehydrogenase
1α subcomplex 10; NTA, nitrilotriacetic acid. |
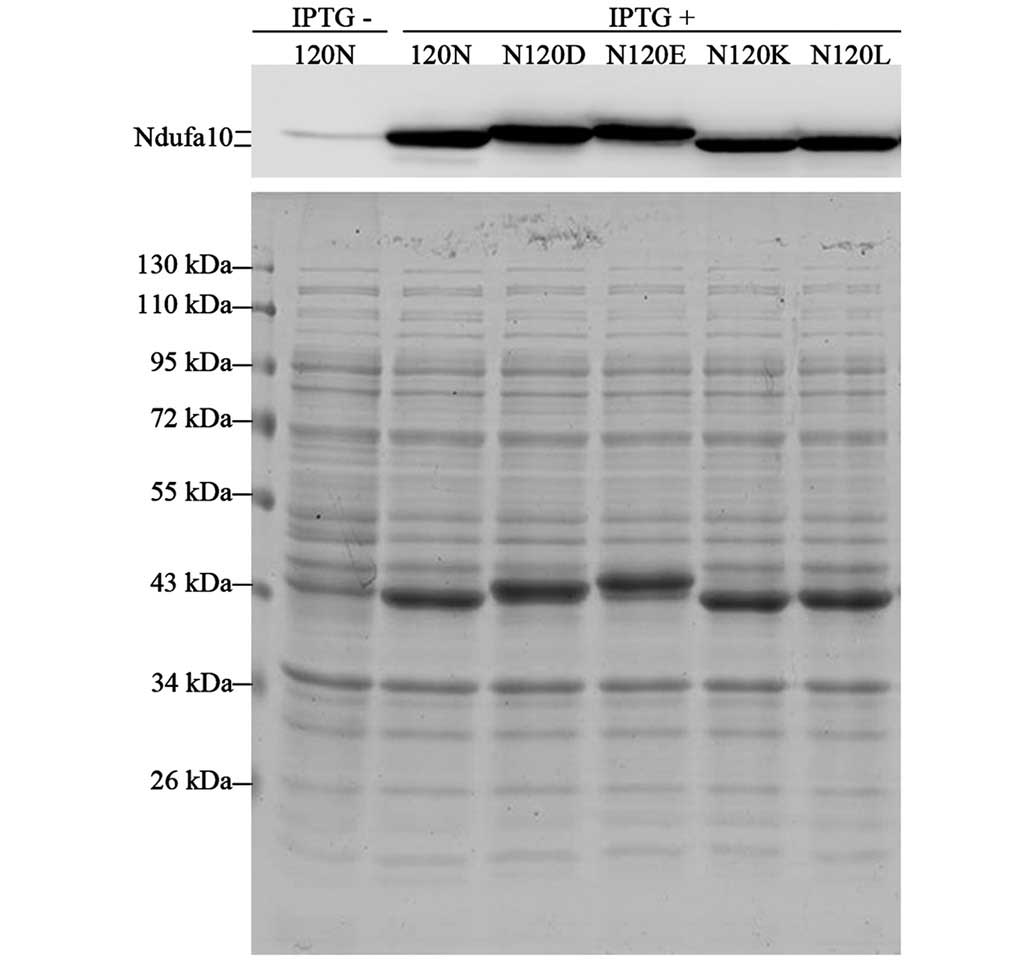
Effect of acidic amino acid substitutions
on the apparent MW of the Ndufa10 protein
To further explore whether the reported phenomena
were attributable solely to the N120D substitution, the influence
of different amino acids at the position 120 on the mobility of the
Ndufa10 proteins on SDS-PAGE was subsequently investigated. To this
end, several Ndufa10 mutant proteins were generated at residue
position 120 by replacing non-polar Asn (N; MW, 114.11 Da) with
neutral, non-polar Leu (L; MW, 113.16 Da), with basic, polar Lys
(K; MW, 128.17 Da), or with acidic, polar Glu (E; MW, 129.12 Da),
in addition to Asp (D; MW, 115.09 Da). As shown in Fig. 4, the Ndufa10-120D protein
demonstrated a similar shift, as described above. The Ndufa10-120K
and Ndufa10-120L proteins exhibited the same mobility as
Ndufa10-120N, whereas the Ndufa10-120E mutant revealed the highest
MW of all. Even after having taken into consideration the MW of the
amino acids, the negative charge of the amino acid in position 120
is therefore revealed to be the major factor accounting for the
mobility of the protein in the SDS-PAGE gel.
Discussion
The predominant findings of the present study were:
(i) that WKY and SHY rats express different variants of the
mitochondria complex I subunit Ndufa10 protein, Ndufa10-120N and
Ndufa10-120D, respectively; (ii) that the Ndufa10-120D protein is
shifted to a higher MW and a lower pI compared with the
Ndufa10-120N protein; (iii) that Ndufa10-120D is specifically
expressed in the SHR rats, whereas the WKY rats express
Ndufa10-120N; and (iv) that the in-gel MW shift of the Ndufa10-120D
protein is not caused by additional post-translational
modifications, since an in-gel mobility difference exists between
bacterially expressed recombinant Ndufa10-120N and Ndufa10-120D
proteins. However, the two recombinant variants exhibited an almost
identical experimental MW, as determined by the MALDI-TOF MS
analysis.
The in-gel mobility is influenced by various
properties of the protein. Among them, hydrophobicity has the
greatest effect. Statistical analysis revealed that hydrophilic
proteins tend to migrate more slowly (12). In general, SDS binds to a protein
at a ratio of approximately one molecule/two amino acid residues.
However, because this binding is mediated via hydrophobic
interactions, SDS preferentially binds to the hydrophobic region of
proteins. As a result, it is likely that hydrophobic proteins bind
larger quantities of SDS than hydrophilic ones, and therefore
migrate faster on SDS-PAGE analysis. Furthermore, the pI values of
proteins also influence gel mobility to a certain extent. Acidic
proteins exhibited a lower mobility than predicted, which may be
due to the presence of a negative charge repulsion effect with SDS
(12). Negatively charged, acidic
aspartic acid residues are more hydrophilic than uncharged, polar
asparagines, and therefore the Ndufa10-120D protein exhibited a
lower pI, and slower migration, compared with the Ndufa10-120N
protein. Furthermore, the Ndufa-120K or the Ndufa-120L mutated
proteins migrated very similarly to the Ndufa10-120N protein due to
their hydrophobic properties. It is notable that the Ndufa-120E
mutant migrated differently, although the difference was comparable
with that exhibited by Ndufa10-120D, since the negatively charged,
acidic glutamic acid residue also has hydrophilic properties.
Unlike the other complex I components, which are
mostly hydrophobic, the Ndufa10 subunit is relatively hydrophilic,
which suggests that it interacts less strongly with other
components of complex I and it is more accessible to external
interactions with other non-complex I proteins, including
cAMP-dependent protein kinase or phosphatase (8,10,13).
Thus far, many mutations in complex I subunits have been associated
with human neurodegenerative diseases (14). Mutations in exons 1 and 3 of the
Ndufa10 gene were previously found to cause complex I deficiency in
a patient with Leigh disease (15). Ndufa10 has a phylogenetically
conserved GAT codon, coding for Asp at position 120. A previous
report demonstrated that animals homozygous for AAT or GAT only
exhibited the Ndufa10-120N or Ndufa10-120D isoforms, respectively,
whereas the presence of the two alleles was associated with the
detection of both variants using SDS-PAGE and 2D gel
electrophoresis (7). All the
animals (normal and SHY rats) used in the present study were
homozygous for A/A or G/G in both alleles, respectively. It appears
that the presence of the G/G genotype leads to a greater
susceptibility for rats to develop hypertension. Whether A/G
heterozygous rats were susceptible to developing hypertension was
not known during the course of the present study, since
heterozygous rats were not obtained from the Shanghai SLAC
Laboratory Animal Co., Ltd. Therefore, it would be interesting to
know whether the N/D substitution influences the function of
Ndufa10 and complex I, and, consequently, is associated with the
pathophysiology of spontaneous hypertension in rats. Most
importantly, it remains to be elucidated whether such an amino acid
replacement occurs in humans. Further studies are required to
demonstrate whether the Ndufa10 DNA polymorphism at position 358 is
a disease-causing mutation, and to determine its potential role as
a biomarker to predict the risk of developing hypertension.
Acknowledgments
This study was supported by a grant from the
Shanghai Natural Science Foundation (grant no. 11ZR1421500), and by
sponsorship received from a Shanghai Pujiang Programa grant (grant
no. 14PJ1406300). This study was also supported in part by grants
from the Ministry of Science and Technology of China (grant no.
2013CB910903).
References
|
1
|
Di Lisa F, Canton M, Menabò R, Kaludercic
N and Bernardi P: Mitochondria and cardioprotection. Heart Fail
Rev. 12:249–260. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lopez-Campistrous A, Hao L, Xiang W, Ton
D, Semchuk P, Sander J, Ellison MJ and Fernandez-Patron C:
Mitochondrial dysfunction in the hypertensive rat brain:
Respiratory complexes exhibit assembly defects in hypertension.
Hypertension. 51:412–419. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Takimoto E and Kass DA: Role of oxidative
stress in cardiac hypertrophy and remodeling. Hypertension.
49:241–248. 2007. View Article : Google Scholar
|
|
4
|
Bernal-Mizrachi C, Gates AC, Weng S,
Imamura T, Knutsen RH, DeSantis P, Coleman T, Townsend RR, Muglia
LJ and Semenkovich CF: Vascular respiratory uncoupling increases
blood pressure and atherosclerosis. Nature. 435:502–506. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Smeitink JA, van den Heuvel LW, Koopman
WJ, Nijtmans LG, Ugalde C and Willems PH: Cell biological
consequences of mitochondrial NADH: Ubiquinone oxidoreductase
deficiency. Curr Neurovasc Res. 1:29–40. 2004. View Article : Google Scholar
|
|
6
|
Efremov RG and Sazanov LA: Structure of
the membrane domain of respiratory complex I. Nature. 476:414–420.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Meng C, Jin X, Xia L, Shen SM, Wang XL,
Cai J, Chen GQ, Wang LS and Fang NY: Alterations of mitochondrial
enzymes contribute to cardiac hypertrophy before hypertension
development in spontaneously hypertensive rats. J Proteome Res.
8:2463–2475. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sardanelli AM, Technikova-Dobrova Z,
Scacco SC, Speranza F and Papa S: Characterization of proteins
phosphorylated by the cAMP-dependent protein kinase of bovine heart
mitochondria. FEBS Lett. 377:470–474. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schulenberg B, Aggeler R, Beechem JM,
Capaldi RA and Patton WF: Analysis of steady-state protein
phosphorylation in mitochondria using a novel fluorescent
phosphosensor dye. J Biol Chem. 278:27251–27255. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schilling B, Aggeler R, Schulenberg B,
Murray J, Row RH, Capaldi RA and Gibson BW: Mass spectrometric
identification of a novel phosphorylation site in subunit NDUFA10
of bovine mitochondrial complex I. FEBS Lett. 579:2485–2490. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Muñoz J, Fernández-Irigoyen J, Santamaría
E, Parbel A, Obeso J and Corrales FJ: Mass spectrometric
characterization of mitochondrial complex I NDUFA10 variants.
Proteomics. 8:1898–1908. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shirai A, Matsuyama A, Yashiroda Y,
Hashimoto A, Kawamura Y, Arai R, Komatsu Y, Horinouchi S and
Yoshida M: Global analysis of gel mobility of proteins and its use
in target identification. J Biol Chem. 283:10745–10752. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lazarou M, McKenzie M, Ohtake A, Thorburn
DR and Ryan MT: Analysis of the assembly profiles for
mitochondrial- and nuclear-DNA-encoded subunits into complex I. Mol
Cell Biol. 27:4228–4237. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Distelmaier F, Koopman WJ, van den Heuvel
LP, Rodenburg RJ, Mayatepek E, Willems PH and Smeitink JA:
Mitochondrial complex I deficiency: From organelle dysfunction to
clinical disease. Brain. 132:833–842. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hoefs SJ, van Spronsen FJ, Lenssen EW,
Nijtmans LG, Rodenburg RJ, Smeitink JA and van den Heuvel LP:
NDUFA10 mutations cause complex I deficiency in a patient with
Leigh disease. Eur J Hum Genet. 19:270–274. 2011. View Article : Google Scholar :
|