Introduction
Acute myocardial infarction is a primary contributor
to rates of mortality and morbidity, which has become a severe
health problem worldwide (1). When
acute myocardial infarction occurs, rapid reperfusion by either
percutaneous coronary intervention or thrombolytic therapy is
important for salvaging myocardial tissue from inevitable necrosis,
and to decrease infarct size (IS). Paradoxically, reperfusion
itself is frequently associated with the exacerbation of tissue
injury and profound inflammatory responses, termed ischemia
reperfusion (I/R) injury (2,3). In
addition, the exposure of a single organ to I/R may subsequently
result in inflammatory activation in other organs, eventually
leading to multi-organ failure and markedly increasing rates of
mortality and morbidity (4).
Therefore, the alleviation of myocardial I/R injury is an important
strategy in the management of acute myocardial ischemia.
Hypoxia inducible factor-1α (HIF-1α) is a central
transcription factor that is key in cellular adaption to hypoxia
and ischemia, which enables cells to survive and differentiate in
low oxygen conditions (5) HIF-1α
is upregulated in hypoxic condition by driving the expression of
>100 genes (6). HIF-1α also
assists in restoring oxygen homeostasis by inducing glycolysis,
erythropoiesis and angiogenesis (7). It is important in cardioprotection
following I/R injury (8). Our
previous study revealed that there is an association between
increased myocardial expression levels of HIF-1α and
cardioprotective effects in rats following acute myocardial
ischemia (9). Therefore, HIF-1α
modulation may reduce tissue injury during acute myocardial
ischemia.
High mobility group box 1 (HMGB1), a ubiquitous and
abundant nuclear protein, can either be passively released into the
extracellular milieu in response to necrotic signals, or actively
secreted in response to inflammatory signals (10,11).
HMGB1 is widely distributed in the liver, brain, spleen, lung,
heart, kidney and lymphatic tissue (12). The first 40 peptide segments of the
B-box can induce the production of tumor necrotic factor-α (TNF-α)
and interleukin (IL)-6 (13).
As a pro-inflammatory cytokine, HMGB1 is important
in several cardiovascular diseases, including atherosclerosis,
myocardial I/R injuries, heart failure and myocardial infarction
(14–18). Clinical studies have shown that
circulating levels of HMGB1 correlate with the severity of coronary
artery disease, and may be a potential and independent predictor of
cardiovascular mortality rates in patients with unstable angina/non
ST segment elevation myocardial infarction (18,19).
Extracellular HMGB1 not only represents an optimal 'necrotic
marker', selected by the innate immune system to recognize tissue
damage and initiate reparative responses, but acts as a potent
pro-inflammatory cytokine, which contributes to the pathogenesis of
diverse inflammatory effects and promotes I/R-induced myocardial
injury (20,21).
Previously, it has been reported that the exogenous
administration of HMGB1 following myocardial infarction or acute
global I/R leads to the recovery of left ventricular function
through the regeneration of cardiomyocytes. However, once the dose
of HMGB1 increases, it can only inhibit inflammatory reactions,
rather than improve the recovery of left ventricular function
(22–24). Furthermore, previous studies have
investigated HMGB1 through direct myocardial injection in various
animal models. However, to date, the effects of intravenously
infused HMGB1 on I/R injury in the myocardium, and the underlying
mechanisms, remain to be fully elucidated.
The aim of the present study was to investigate the
effects of intravenous HMGB1 on the expression of HIF-1α in the
myocardium of rats following acute myocardial ischemia. In
addition, the effects of intravenous HMGB1 on myocardial ischemia
reperfusion injury and the underlying mechanisms were
evaluated.
Materials and methods
Animal groups
All experiments were performed in accordance with
the Guide for the Care and Use of Laboratory Animals, published by
the US National Institutes of Health (no. 85–23, revised 1996)
(25), and were approved by the
Institutional Review Board of Liaocheng People's hospital
(Liaocheng, China). Male Wistar rats, aged 8–11 weeks (n=50; body
weight, 250–300 g) were obtained from the experimental laboratory
of Shandong Lukang Co., Ltd. (Jining, China). The rats were
maintained in a controlled environment under a 12-h light/dark
cycle (humidity, 55±5%; temperature 22±1°C) with free access to
food and water (provided by the experimental laboratory of Shandong
Lukang, Ltd. (Shandong, China). The rats were randomly divided into
five groups, each containing 10 animals: i) sham operation (sham)
group; ii) ischemia reperfusion (I/R) group; iii) I/R rats
pre-treated intravenously with 100 ng/kg recombinant HMGB1 (R&D
Systems, Inc., Minneapolis, MN, USA) 30 min prior to ischemia
(HMGB1 group); (iv) I/R rats pre-treated intravenously with
LY294002 (Sigma-Aldrich, St. Louis, MO, USA), an inhibitor of
phosphoinositide 3-kinases (PI3K), at a dose of 0.3 mg/kg (LY
group); (v) I/R rats pre-treated with HMGB1 (100 ng/kg) and
LY294002 (0.3 mg/kg) intravenously 30 and 40 min prior to ischemia,
respectively) (HMGB1+LY group). HMGB1 (dissolved in phosphate
buffered saline) and LY (dissolved in dimethyl sulfoxide) were
administered intravenously in the tail vein, in a 0.5-ml volume.
The sham group was treated with an intravenous injection of normal
saline (0.5 ml).
Animal model
The rat I/R model was established according to
methods previously reported (26).
All the I/R rats were subjected to left anterior descending
coronary artery (LAD) occlusion via a suture for 30 min, followed
by reperfusion for 4 h. Following general anesthesia via
intraperitoneal injection of sodium pentobarbital (60 mg/kg;
Sigma-Aldrich), the trachea was cannulated for artificial
ventilation with room air, at a rate of 55 breaths/min. The body
temperature of the rats was maintained at 37±0.50°C using an
electrical heating pad (Ruiwode Life Science Inc., Shenzhen,
China). Lead II of an electrocardiogram (ECG) was monitored using
stainless needle electrodes, which were attached to limbs. The ECG
was recorded and analyzed using an ECG-6511 data acquisition system
(Guangdian Medical Device Co. Ltd., Shanghai, China). In the sham
group, the suture was placed at the origin of the LAD, however, no
complete ligation of the artery was performed.
Biochemical analysis
Blood samples (1 ml) were obtained from the femoral
vein following reperfusion for 4 h, and were centrifuged at 1,400 ×
g for 10 min at 4°C. The sera were obtained and then frozen at
−80°C until future analyses. Serum levels of TNF-α were determined,
according to the manufacturer's protocol, using a rat TNF-α
enzyme-linked immunosorbent assay (ELISA) kit (Shanghai Huiying
Biotechnology Ltd., Co., Zhongshan, China). Serum levels of cardiac
troponin (c-TnI) and IL-6 were detected using rat CTn-I (Lengton
Bioscience Co., Ltd., Shanghai, China) and IL-6 (Jingmei Biotech
Co., Ltd., Shenzhen, China) ELISA kits, respectively.
Measurement of myocardial activities of
malondialdehyde (MDA) and superoxide dismutase (SOD) (27)
The hearts were harvested from the rats and washed
with normal saline. Subsequently, 0.5 g of the ischemic heart
tissue was ground at 0–4°C, following which the myocardial
homogenate was centrifuged at 2,500 × g for 30 min. The
supernatants (~0.4 g) were harvested and stored at −80°C until MDA
concentration and SOD activity assays were performed. A
thiobarbituric acid reactive substance assay (DTBA-100; Bioassay
Systems LLC, Hayward, CA, USA) was used to measure the levels of
MDA, by measuring the absorbance value at a wavelength of 532 nm
using a microplate reader (SP-Max 2300A; Molecular Devices, LLC,
Sunnyvale, CA, USA). A xanthine oxide method was used to determine
SOD activity, by measuring the absorbance value at a wavelength of
550 nm. An MDA Assay kit and an SOD Assay kit (Nanjing Jiancheng
Bioengineering Co., Ltd., Nanjing, China) were used to measure MDA
concentration and SOD activity, respectively, according to the
manufacturer's protocol. The conditions of the assays were as
follows: MDA, 90–100°C for 50 min; SOD, 37°C for 20 min.
Histological analysis of the heart
The rats were sacrificed by decapitation, the hearts
were immediately removed and fixed in 10% formalin for 60 min at
room temperature, and then for 24 h at 4°C. Subsequently, the
samples were washed and stored in 70% ethanol at 4°C, prior to
being embedded in paraffin wax (Leica Biosystems Richmond Inc.,
Richmond, IL, USA) and sectioning. For MDA and SOD measurement,
there were five rats per group, and five rats underwent
histological analysis. The paraffin sections (5 µm) were
stained with hematoxylin and eosin, using a Tissue-Tek®
DRS™ 2000 (all obtained from Sakura Finetek Japan Co., Ltd., Tokyo,
Japan). Myocardiocyte and tissue morphology were examined under a
digitalized microscope (Eclipse 80i; Nikon Corporatin, Tokyo,
Japan).
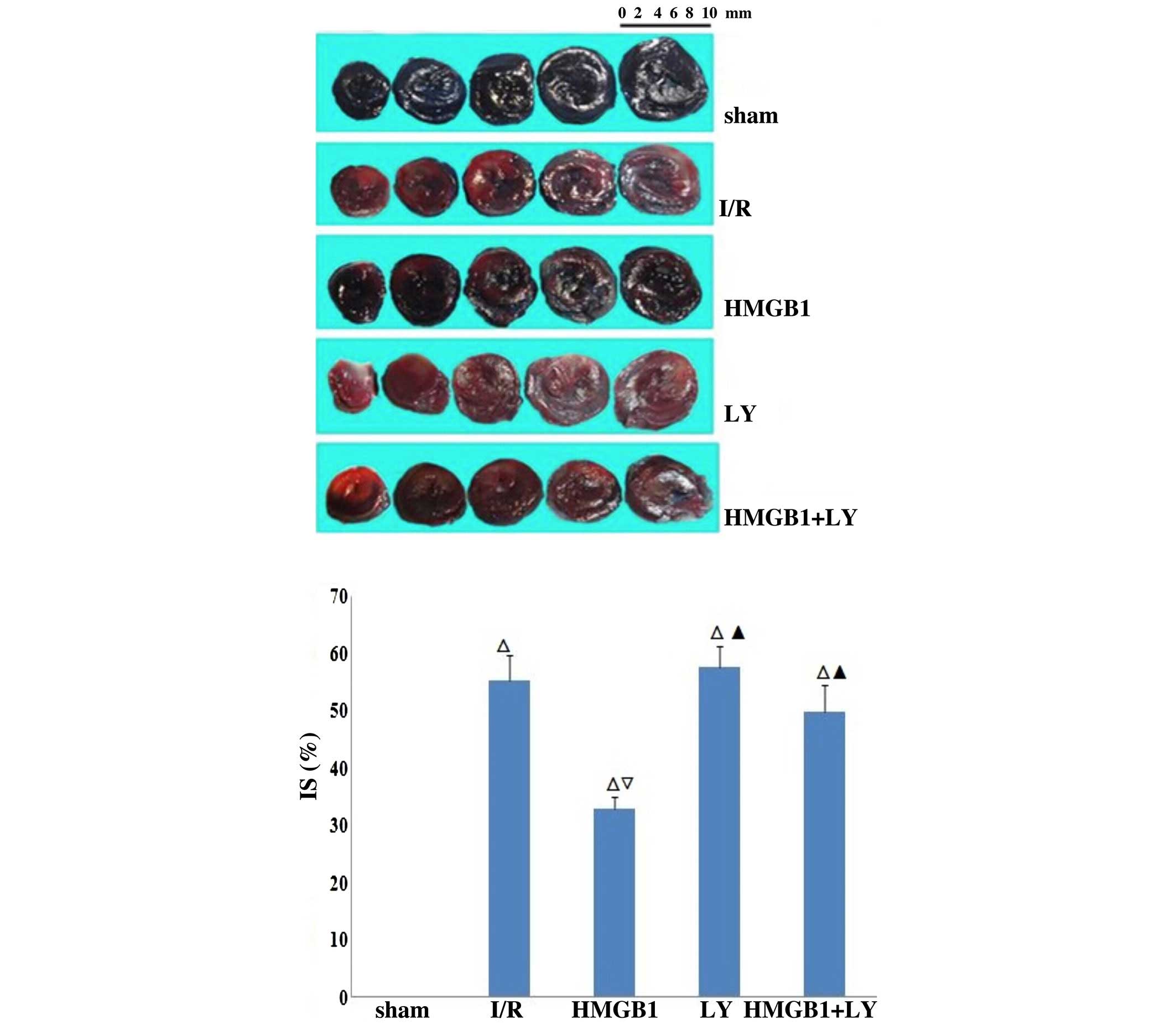
Assessment of infarct size (IS)
The IS was assessed using 2,3,5-triphenyltetrazolium
chloride (TTC; Sigma-Aldrich) staining methods, as previously
reported (26). Following
reperfusion for 4 h, the LAD was occluded again on five rats from
each group to assess the IS, and 1 ml 2.0% Evans blue dye
(Sigma-Aldrich) was injected via the chest aorta. The entire heart
was excised, rinsed of excess blue dye with normal saline, and the
right and left atria were removed. The left ventricle was deep
frozen at −80°C. The frozen left ventricle was then sliced
horizontally to yield five slices between the apex and base. The
slices were incubated in 1% TTC for 15 min at 37°C. The impaired
myocardium was stained red, the infarcted myocardium was stained
white and the normal myocardium was stained blue. Images of the
slices were captured using a Nikon D90 digital camera (Nikon
Corporation, Tokyo, Japan). The borders of the infarct, ischemic
and nonischemic areas of the heart images were traced and measured
using Image-Pro Plus 3.0 (Media Cybernetics, Silver Spring, MD,
USA). Infarct size was calculated as a percentage of the risk area
(infarct size / risk area).
Western blot analysis
The protein expression levels of HIF-1α and p-Akt
were analyzed using western blot analysis. Myocardial tissues were
dissected from each of the treatment groups, and the rodent tissue
homogenates were prepared in Tris-HCl buffer (pH 7.5), containing
150 mM NaCl, 1% NP-40, 1 mM EDTA, 1 µg/ml leupeptin, 1
µg/ml pepstatin, 3.8 µg/ml aprotinin, 1 mM PMSF, 1 mM
Na3VO4 and 2 mM NaF (all Beyotime Institute
of Biotechnology, Haimen, China). The extracts were clarified by
centrifugation at 4°C (14,000 × g for 20 min). Supernatants were
collected and eluted with SDS sample buffer (Beyotime Institute of
Biotechnology), and the proteins were resolved using 10% SDS-PAGE
(Beyotime Institute of Biotechnology). Bicinchoninic acid protein
assay reagent (Sigma-Aldrich) was used to determine the protein
expression levels. Proteins were separated by PAGE and transferred
onto polyvinilidene fluoride membranes (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) with 50 µg of lysate per lane. The
membranes were blocked with 5% non-fat milk in 400 ml Tris-buffered
saline (1X) with 200 µl Tween-20 (Beyotime Institute of
Biotechnology) at room temperature for 60 min. The membranes were
incubated with the following primary antibodies at 4°C for 24 h:
Rabbit anti-rat HIF-1α polyclonal antibody (cat. no. sc-10790;
Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA; 1:2,000),
rabbit anti-rat β-actin monoclonal antibody (cat. no. ab119716;
Abcam, Cambridge, UK; 1:5,000), rabbit anti-rat p-Akt monoclonal
antibody (cat. no. 4060; Cell Signaling Technology, Inc., Beverly,
MA, USA; 1:1,000) and rabbit anti-rat t-Akt monoclonal antibody
(cat. no. 4691; Cell Signaling Technology, Inc.; 1:1,000). The
membranes were then incubated with secondary antibody, IRDye680
goat anti-rabbit IgG (cat. no. 926–68071, 925–68071; LI-COR
Biosciences, Lincoln, NE, USA; 1:2,000) at room temperature for 2
h. Following incubation, the membranes were exposed to
chemiluminescence and a ChemiDoc imaging system (Bio-Rad
Laboratories, Inc.,) to analyze the protein bands. The relative
content of each sample was repeated at least three times.
Statistical analysis
Data are expressed as the mean ± standard deviation
or percentages, where appropriate. SAS 6.12 software (SAS
Institute, Inc., Cary, NC, USA) was used for statistical analyses.
One way analysis of variance was used to compare the means between
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
Serum levels of c-TnI and TNF-α
As shown in Table
I, serum levels of IL-6, c-TnI and TNF-α in the I/R group were
significantly higher, compared with those in the sham group
(P<0.01). Pre-treatment with HMGB1 significantly decreased serum
levels of IL-6, c-TnI and TNF-α, compared with the I/R group
(P<0.01). However, compared with the HMGB1 group, the decreased
serum levels of IL-6, c-TnI and TNF-α in the LY and LY+HMGB1 groups
were significantly reversed (P<0.01). Serum levels of IL-6,
c-TnI and TNF-α in the LY group were significantly higher, compared
with those in the LY+HMGB1 group (P<0.01).
 | Table ISerum levels of IL-6, TNF-α, cTnI,
SOD and MDA, and IS. |
Table I
Serum levels of IL-6, TNF-α, cTnI,
SOD and MDA, and IS.
| Variable | Sham | I/R | HMGB1 | LY | HMGB1+LY |
|---|
| IL-6 (pg/ml) | 160.48±12.03 |
385.44±15.34a |
215.66±18.11a,b |
376.86±17.05a,c,d |
355.32±20.53a,c |
| TNF-α (pg/ml) | 13.72±5.18 | 76.98±6.89a | 52.53±3.87a,b | 77.22±5.01a,c,d | 70.32±4.99a,c |
| cTnI
(µg/l) | 0.09±0.22 | 73.47±8.11a | 47.33±3.16a,b | 70.42±7.08a,c,d | 63.56±4.71a,c |
| SOD (u/mg) | 142.18±27.67 | 64.67±19.91a | 101.79±31.6a,b | 64.42±13.37a,c,d | 75.13±16.65a,c |
| MDA (nmol/mg) | 1.27±0.23 | 8.45±0.47a | 3.55±0.67a,b | 8.56±0.96a,c,d | 8.14±0.76a,c |
| IS (%) | 0 | 55.18±4.43a | 32.89±2.12a,b | 57.60±3.59a,c | 49.73±4.78a,c |
MDA levels and SOD activity
The results of the MDA and SOD anaylses, shown in
Table I, indicated that the
myocardial level of MDA in the I/R group was significantly
increased, whereas the activity of SOD was significantly decreased,
compared with the sham group (P<0.0). The increase in the level
of MDA and reduction of SOD activity were significantly inhibited
by HMGB1 pretreatment (P<0.01). However, the decreased levels of
MDA and increased SOD activities were reversed significantly in the
LY and LY+HMGB1 groups, compared with the HMGB1 group (P<0.01).
The myocardial level of MDA increased and SOD activity decreased
significantly in the LY group, compared with the LY+HMGB1 group
(P<0.01).
Histological analysis of the heart
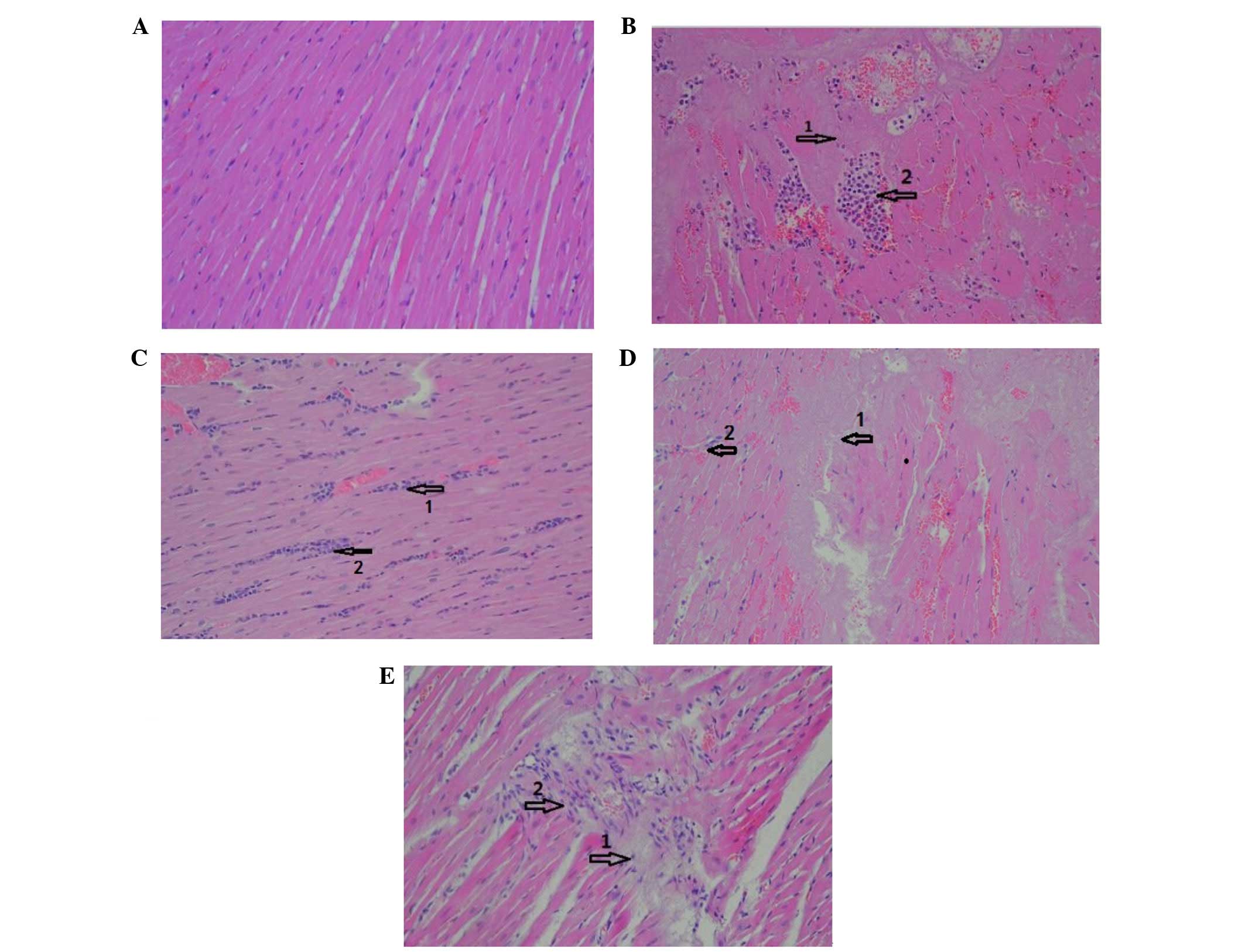
In the sham group, myocardial fibers were arranged
regularly with clear boundaries, and no inflammatory infiltration
was present (Fig. 1A). In the I/R
group (Fig. 1B), morphological
changes in the affected cardiomyocytes primarily comprised
different degrees of swelling, necrosis, myocytolysis and
myofibrillar loss. The myocardial fibers were disrupted and
arranged irregularly. The foci of myocardial infarction were
infiltrated with an increased number of neutrophils. In the HMGB
group (Fig. 1C), the myocardial
fiber arrangement was relatively regular. Infiltration of an
increased number of neutrophils were also observed. In the LY group
(Fig. 1D), myocardial fibers were
disrupted and dissolved, and the stripes were no longer visible. An
increased number of inflammatory cells were observed to have
infiltrated surrounding the myocardial infarction foci and
capillaries. In the HMGB1+LY group (Fig. 1E), myocardial fibers were arranged
irregularly, and were dissolved and disrupted in places. The number
of myocardiocytes were reduced and the intermuscular spaces were
widened. Myocardial infarction foci and capillaries were surrounded
by inflammatory cell infiltration.
Comparison of IS in each group
As shown in Fig. 2
and Table I, the IS in the I/R
group was significantly higher, compared with those in the sham
group (P<0.01). The IS in the HMGB1-treated group was
significantly decreased, compared with those in the I/R group
(P<0.01). However, the IS was significantly increased in the LY
group and LY+HMGB1, compared with the HMGB1-treated group
(P<0.01).
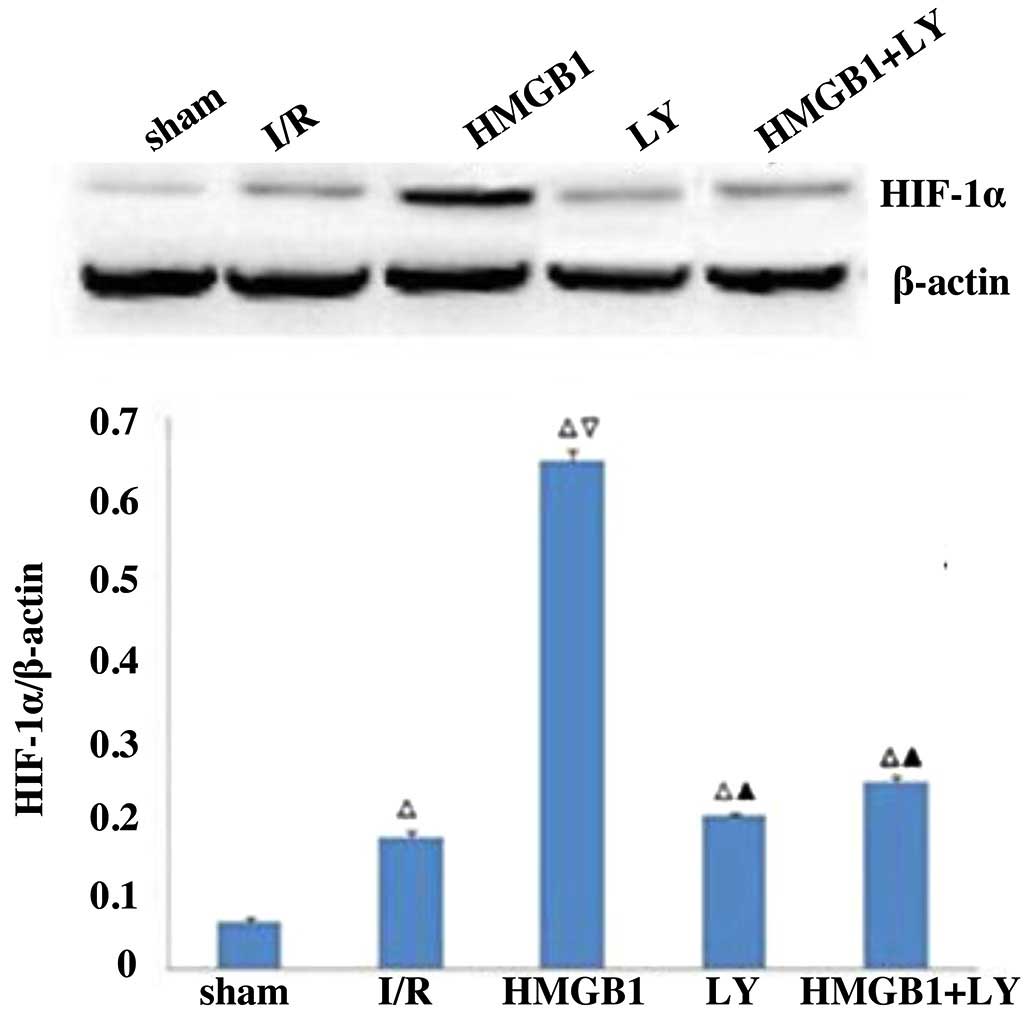
Protein expression of HIF-1α
As shown in Fig. 3,
the protein expression of HIF-1α was significantly increased in the
I/R group, compared with the sham group (P<0.05). Treatment with
HMGB1 significantly increased the protein expression of HIF-1α,
compared with the I/R group (P<0.05). However, the LY group and
LY+HMGB group were found to have significantly lower protein
expression levels of HIF-1α, compared with the HMGB group
(P<0.05).
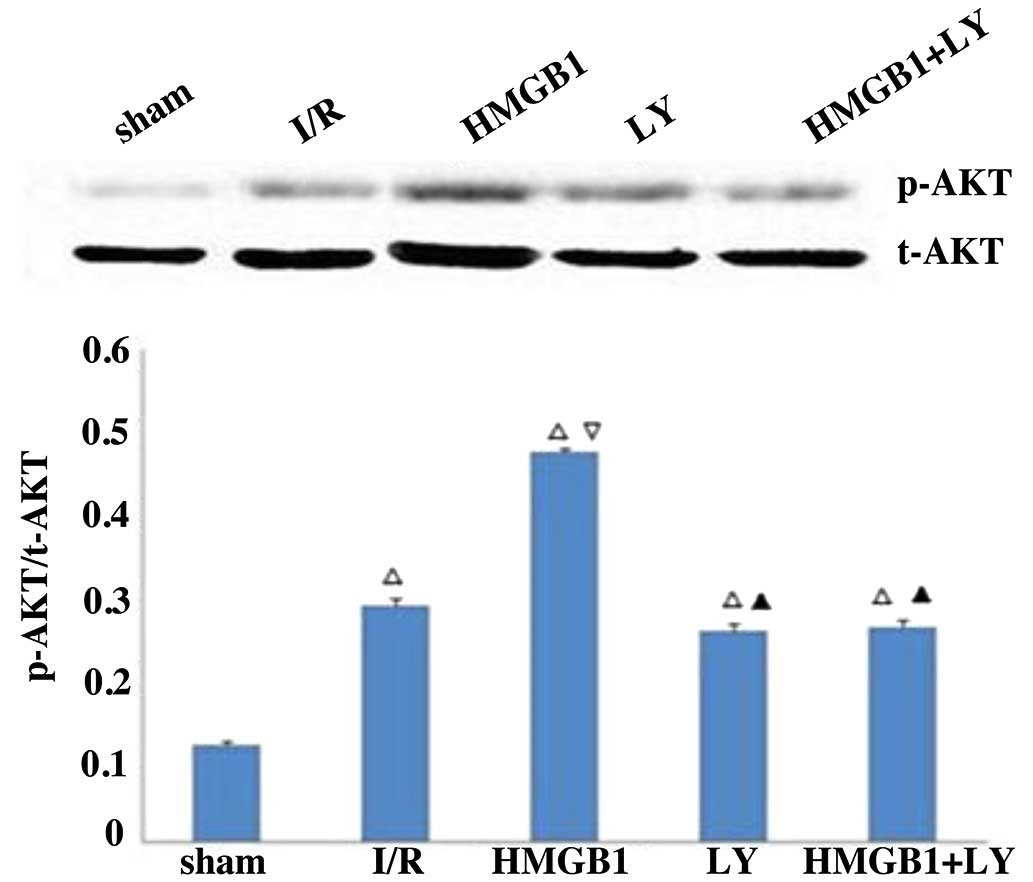
Protein expression of p-Akt
As shown in Fig. 4,
the protein expression of p-Akt was significantly increased in the
I/R group, compared with the sham group (P<0.05). Treatment with
HMGB1 significantly increased the protein expression of p-Akt,
compared with the I/R group (P<0.05). However the LY group and
LY+HMGB group were found to have significantly lower protein
expression levels of p-Akt, compared with the HMGB group
(P<0.05).
Discussion
In the present study, the results demonstrated that:
i) myocardial expression of HIF-1α increased significantly in I/R
rats; ii) pretreatment with HMGB1 led to significant
anti-inflammatory effects, reducing infarct size and increasing the
myocardial expression of HIF-1α; iii) cardioprotective effects
induced by intravenous HMGB1 may be associated with enhancement of
the protein expression of HIF-1α. This protective effect involved a
reduction in oxidative stress, neutrophil infiltration and
pro-inflammatory effects; iv) treatment with the PI3K inhibitor,
LY294002, inhibited Akt phosphorylation and the cardioprotective
effects of intravenous HMGB1. These results suggested that
intravenous HMGB1 may alleviate myocardial I/R injury through an
increase in the protein expression of HIF-1α, involving the
PI3K/Akt/HIF signaling pathway.
The rapid restoration of blood flow in the occluded
coronary artery following acute myocardial ischemia is the most
important aspect in protecting myocardial tissue from inevitable
necrosis. However, the early opening of an occluded coronary artery
may cause myocardial I/R injury (2,3).
Therefore, the attenuation of myocardial I/R injury is an important
strategy in the treatment of acute myocardial ischemia. The present
study hypothesized that HMGB1 pretreatment may protect heart
against I/R injury, thus decreasing infarct size. I/R itself can
cause sterile inflammation, which is characterized by the
accumulation of inflammatory cells. The infiltration of leukocytes
may, in turn, initiate an inflammatory response, which results in
cardiomyocyte damage (2). These
cells may also promote collateral tissue injury (28). Neutrophils are the predominant type
of cells to aggregate in the ischemic myocardium following
reperfusion, to mediate direct injury via the release of toxic
products, including reactive oxygen species (ROS) and photolytic
enzymes (29). It has also been
reported that liver I/R injury is characterized by neutrophil
recruitment and activation (30).
Therefore, agents limiting the infiltration and activity of
leukocytes, and inflammatory reaction have been suggested as a
treatment strategy for attenuating myocardial I/R injury. In the
present study, it was demonstrated that intravenous administration
of HMGB1 prior to inducing myocardial ischemia inhibited the
accumulation of leukocytes following myocardial I/R injury.
Accordingly, pretreatment with HMGB1 attenuated I/R-induced
necrosis, myocardial infarct size and the expression of
proinflammatory cytokines, including TNF-α. These results suggested
that intravenous administration of HMGB1 may attenuate myocardial
I/R injury through suppressing the recruitment of leukocytes and
inhibiting the inflammatory reaction.
Several studies have shown that the process of
myocardial I/R injury is associated with the increased generation
of ROS and oxidative stress (31,32).
Oxidative stress can modify proteins and phospholipids, resulting
in lipid peroxidation and oxidation of thiol groups, which may lead
to alteration of cell membrane configuration, and permeability and
modification of various cellular proteins (33). I/R may cause arrhythmias,
abnormality in gene expression, loss of adrenergic pathways and
depression in contractile function (34). When the heart is pretreated with
either cardiac subcellular organelles or various ROS-generating
systems, similar changes to those mentioned above are found
(3). However, when treated with
SOD and catalase, the heart is protected from these changes
(35). Therefore, it can be
concluded that oxidative stress may result, at least in part, in
these alterations in the myocardium during I/R. In the present
study, it was found that HMGB1 reduced the myocardial content of
MDA and increased the activity of SOD induced by I/R, whereas
LY294002 eliminated these effects. It has been reported that ROS
can directly injure the cell membrane and cause cell death during
myocardial I/R (36). Furthermore,
ROS-mediated apoptosis and necrosis can eventually be a determinant
in infarct size (37). Therefore,
HMGB1 may exert its cardioprotective through antioxidative stress
in myocardial I/T injury, which may be regulated by the PI3K/Akt
signaling pathway.
HIF-1α is a heterodimeric DNA-binding complex, which
is composed of oxygen sensitive, HIF-1α, and aryl hydrocarbon
nuclear translocator, HIF-1β (38). HIF-1α is continuously undergoing
proteasomal degradation, resulting in a short half-life under
normoxic conditions (39). By
contrast, HIF-1α can translocate to the nucleus and dimerize with
HIF-1β, as HIF-1α evades proteasomal degradation under hypoxic
conditions. The complex then binds to hypoxic response elements,
driving the expression of >100 genes (6,7).
HIF-1α regulates the cellular response to hypoxia. Studies have
show that the expression of HIF-1α increases significantly in
different ischemic organs and tissues, including the retina
(40), myocardium (9,41)
and nervous system (42). It has
also been reported that HIF-1α can prevent hypoxia or I/R-induced
myocardial injury in the heart (8,43,44).
In accordance with these previous studies, the present study found
that basic fibroblast growth factor enhanced the myocardial mRNA
expression of HIF-1α, thus decreasing infarct size and improving
left ventricular function in rats following acute MI (9). As mentioned above, HIF-1α
overexpression confers protective effects against myocardial I/R
injury by promoting the transcription of a number of
cardioprotective genes, including those involved in glucose
metabolism, mitochondrial function, cell apoptosis, erythropoietin,
vascular endothelial growth factor, inducible nitric oxide
synthase, hemeoxygenase, cardiotropin and resistance to oxidative
stress (45,46). Other mechanisms associated with the
cardioprotective effects of HIF-1α include prolyl hydroxylase 2
inhibition (8), the upregulation
of cardiotrophin-1 (47) and the
promotion of angiogenesis (48). A
previous study showed that partial deficiency of HIF-1α is
associated with a complete loss of cardioprotection against I/R
injury (49).
Cardiomyocyte-specific HIF-1α gene deletion causes reductions in
contractility and vascularization, and alters the expression of
multiple genes in the heart during normoxia (50).
The present study demonstrated that I/R
significantly increased the myocardial expression of HIF-1α, and
that the expression of HIF-1α was markedly elevated by HMGB1.
Furthermore, consistent with the increased expression of HIF-1α,
the myocardial injury induced by I/R was inhibited by HMGB1. It was
also found that intravenous HMGB1 decreased the levels of MDA and
increased SOD activity in the I/R myocardium, which suggested that
these changes may be located downstream of alterations in HIF-1α
overexpression. Thus, the results from the present study indicated
that intravenous HMGB1 may exert its cardioprotective effects
through increased myocardial expression of HIF-1α. However, the
signaling pathway, which mediates the upregulation of HIF-1α
induced by HMGB1 in I/R myocardium, remains to be elucidated.
PI3K and the downstream effector Akt are a conserved
family of signal transduction enzymes, which are involved in
regulating cellular activation, inflammatory responses and
apoptosis (51). Several previous
studies have reported that the PI3K/Akt signaling pathway is key in
myocardial protection against I/R injury (52–55).
However, whether the PI3K/Akt pathway mediates the cardioprotective
effects of intravenous HMGB1 remains to be fully elucidated. In the
present study, it was found that HMGB1 markedly enhanced Akt
phosphorylation, leading to the subsequent upregulation of HIF-1α
and attenuation of myocardial I/R injury. Treatment with LY294002
inhibited the HMGB1-induced expression of HIF-1α. Furthermore, the
cardioprotective effects exerted by intravenous HMGB1 in an I/R
rats model were eliminated by in vivo administration of PI3K
inhibitor. Taken together, the present study hypothesized that
HMGB1 upregulated the myocardial expression of HIF-1α, which was
dependant, at least partially, on Akt phosphorylation, suggesting
that the PI3K/Akt signaling pathway may be involved in the
cardioprotective effects of intravenous HMGB1 during myocardial I/R
injury. Although the possibility of other signaling pathways also
contributing to HMGB1-induced HIF-1α expression, the results of the
present study were consistent with previous studies, which
suggested that the phosphorylation of Akt protected organs from I/R
injury (53–58). Although multiple mechanisms are
likely to be involved, the present study provided additional
evidence that the protective effects of intravenous HMGB1 on
myocardial I/R injury are, at least in part, regulated by the
PI3K/Akt signaling pathway. The major mechanisms underlying these
effects may include direct inhibition of leukocyte migration,
production of ROS and inflammatory cytokines and increasing
myocardial expression of HIF-1α, thus decreasing infarct size.
In conclusion, the present study involving an acute
I/R rat model demonstrated that the intravenous administration of
HMGB1 was associated with a reduction in infarct size and increased
myocardial expression levels of HIF-1α. The intravenous
administration of HMGB1 may exert its cardioprotective effect by
upregulating the protein expression of HIF-1α in the ischemic
myocardium via the PI3K/Akt signaling pathway.
Acknowledgments
This study was supported by the Natural Science
Foundation of Shandong Province (grant no. ZR2013HL017), the
Natural Science Foundation of Liaocheng City (grant no. 2012NS13)
and the Science and Technology Developing Project of Liaocheng City
(grant no. 2014GJH26).
References
|
1
|
Zweiter JL and Talukder MA: The role of
oxidants and free radicals in reperfusion injury. Cardiolvasc Res.
70:181–190. 2006. View Article : Google Scholar
|
|
2
|
Yellon DM and Hausenloy DJ: Myocardial
refusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion-from mechnism to translation. Nat Med. 17:1391–1401.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Park SW, Kim M, Brown KM, D'Agati VD and
Lee HT: Paneth cell-derives IL-17A causes multiorgan dysfunction
after hepatic ischemia and reperfusion injury. Hepatology.
53:1662–1675. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maes C, Carmeliet G and Schipani E:
Hypoxia-driven pathways in bone development, regeneration and
disease. Nat Rev Rheumatol. 8:358–366. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kasivisvanathan V, Shalhoub J, Lim CS,
Shepherd AC, Thapar A and Davies AH: Hypoxia-inducible factor-1 in
arterial disease: A putative therapeutic target. Curr Vasc
Pharmacol. 9:333–349. 2011. View Article : Google Scholar
|
|
7
|
Weidemann A and Johnson RS: Biology of
HIF-1alpha. Cell Death Differ. 15:621–627. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Poynter JA, Manukyan MC, Wang Y, Brewster
BD, Herrmann JL, Weil BR, Abarbanell AM and Meldrum DR: Systemic
pretreatment with dimethyloxalylglycine increases myocardial HIF-1α
and VEGF production and improves functional recovery after acute
ischemia/reperfusion. Surgery. 150:278–283. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yao HC, Liu T, Meng XY, Han QF, Zhang M
and Wang LX: Effect of basic fibroblast growth factor on the
myocardial expression of hypoxia-inducible factor- 1α and vascular
endothelial growth factor following acute myocardial infarction.
Heart Lung Circ. 22:946–951. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lakhan SE, Kirchgessner A and Hofer M:
Inflammatory mechnisms in ischemic stroke: Therapeutic approaches.
J Transl Med. 7:972009. View Article : Google Scholar
|
|
11
|
Oozawa S, Mori S, Kanke T, Takahashi H,
Liu K, Tomono Y, Asanuma M, Miyazaki I, Nishibori M and Sano S:
Effects of HMGB1 on ischemia-reperfusion injury in the rat heart.
Circ J. 72:1178–1184. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Deneke SM and Fanburg BL: Normobaric
oxygen toxicity of the lung. N Engl J Med. 303:76–86. 1980.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li J, Kokkola R, Tabibzadeh S, Yang R,
Ochani M, Qiang X, Harris HE, Czura CJ, Wang H, Ulloa L, et al:
Structural basis for the proinflammatory cytokine activity of high
mobility group box 1. Mol Med. 9:37–45. 2003.PubMed/NCBI
|
|
14
|
Kohno T, Anzai T, Naito K, Miyasho T,
Okamoto M, Yokota H, Yamada S, Maekawa Y, Takahashi T, Yoshikawa T,
et al: Role of high-mobility group box 1 protein in post-infarction
healing process and left ventricular remodelling. Cardiovasc Res.
81:565–573. 2009. View Article : Google Scholar
|
|
15
|
Yan XX, Lu L, Peng WH, Wang LJ, Zhang Q,
Zhang RY, Chen QJ and Shen WF: Increased serum HMGB1 level is
associated with coronary artery disease in nondiabetic and type 2
diabetic patients. Atherosclerosis. 205:544–548. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Avalos AM, Kiefer K, Tian J, Christensen
S, Shlomchik M, Coyle AJ and Marshak-Rothstein A: Rage-independent
autore-active B cell activation in response to chromatin and
HMGB1/DNA immune complexes. Autoimmunity. 43:103–110. 2010.
View Article : Google Scholar :
|
|
17
|
Ding HS and Yang J: High mobility group
box-1 and cardiovascular diseases. Saudi Med J. 31:486–489.
2010.PubMed/NCBI
|
|
18
|
Yao HC, Zhao AP, Han QF, Wu L, Yao DK and
Wang LX: Correlation between serum high-mobility group box-1 levels
and high-sensitivity C-reactive protein and troponin I in patients
with coronary artery disease. Exp Ther Med. 6:121–124.
2013.PubMed/NCBI
|
|
19
|
Hashimoto T, Ishii J, Kitagawa F, Yamada
S, Hattori K, Okumura M, Naruse H, Motoyama S, Matsui S, Tanaka I,
et al: Circulating high-mobility group box 1 and cardiovascular
mortality in unstable angina and non-ST-segment elevation
myocardial infarction. Atherosclerosis. 221:490–495. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ulloa L and Messmer D: High-mobility group
box 1 (HMGB1) protein: Friend and foe. Cytokine Growth Factor Rev.
17:189–201. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu H, Yao Y, Su Z, Yang Y, Kao R, Martin
CM and Rui T: Endogenous HMGB1 contributes to
ischemia-reperfusion-induced myocardial apoptosis by potentiating
the effect of TNF-α/JNK. Am J Physiol Heart Circ Physiol.
300:H913–H921. 2011. View Article : Google Scholar
|
|
22
|
Biscetti F, Ghirlanda G and Flex A:
Therapeutic potential of high mobility group box-1 in ischemic
inury and tissue regeneration. Curr Vasc Pharmacol. 9:677–681.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Abarbanell AM, Hartley JA, Herrmann JL,
Weil BR, Wang Y, Manukyan MC, Poynter JA and Meldrum DR: Exogenous
high-mobility group box 1 improves myocardial recovery after acute
global ischemia/reperfusion injury. Surgery. 149:329–335. 2011.
View Article : Google Scholar
|
|
24
|
Hu X, Jiang H, Cui B, Xu C, Lu Z and He B:
Preconditioning with high mobility group box 1 protein protects
against myocardial ischemia-reperfusion injury. Int J Cardiol.
145:111–112. 2010. View Article : Google Scholar
|
|
25
|
National Research Council: Guide for the
Care and Use of Laboratory Animals. National Academic Press;
1996
|
|
26
|
Yao HC, Yang LJ, Han QF, Wang LH, Wu L,
Zhang CY, Tian KL and Zhang M: Postconditioning with simvastatin
decreases the myocardial injury in rats following acute myocardial
ischemia. Exp Ther Med. 9:1166–1170. 2015.PubMed/NCBI
|
|
27
|
Wu YB, Shi LL, Wu YJ, Xu WH, Wang L and
Ren MS: Protective effect of gliclazide on diabetic peripheral
neuropathy through Drp-1 mediated-oxidative stress and apoptosis.
Neurosci Lett. 523:45–49. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Swirski FK, Nahrendorf M, Etzrodt M,
Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler
RH, Chudnovskiy A, Waterman P, et al: Identification of splenic
reservoir monocytes and their deployment to inflammatory sites.
Science. 325:612–616. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kaminski KA, Bonda TA, Korechi J and
Musial WJ: Oidative stress and nuetrophil activation-the two
keystones of ischemia/reperfusion injury. Int J Cardiol. 86:41–59.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhai Y, Busuttil RW and Kupiec-Weglinski
JW: Liver ischemia and reperfusion injury: New insights into
mechnisms of innate-adaptive immune-mediated tissue inflammation.
Am J Transplant. 11:1563–1569. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jahangiri A, Leifert WR, Kind KL and
McMurchie EJ: Dietary fish oil alters cardiomyocyte Ca2+ dynamics
and antioxidant status. Free Radic Bio Med. 40:1592–1602. 2006.
View Article : Google Scholar
|
|
32
|
Wang Y, Sun J, Liu C and Fang C:
Protective effects of crocetin pretreatment on myocardial injury in
an ischemia/reperfusion rat model. Eur J Pharmacol. 741:290–296.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hool LC: Mechnism for mediating pathology.
Clin Exp Phamacol Physiol. 35:229–234. 2008.
|
|
34
|
Rensing H, Bauer I, Kubulus D, Wolf B,
Winning J, Ziegeler S and Bauer M: Heme oxygenase-1 gene expression
in pericentral hepatocytes through beta-1 adrneoceptor stimulation.
Shock. 21:376–387. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xu Y, Liu B, Zweier JL and He G: Formation
of hydrogen peroxide and reduction of peroxynitrite via dismutation
of superoxide at reperfusion enhances myocardial blood flow and
oxygen consumption in postischemic mouse heart. J Pharmacol Exp
Ther. 327:402–410. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
van Dijk A, Krijnen PA, Vermond RA, Pronk
A, Spreeuwenberg M, Visser FC, Berney R, Paulus WJ, Hack CE, van
Milligen FJ and Niessen HW: Inhibition of type 2A secretory
phospholipase A2 reduces death of cardiomyocytes in acute
myocardial infarction. Apoptosis. 14:753–763. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Matsui Y, Takagi H, Qu X, Abdellatif M,
Sakoda H, Asano T, Levine B and Sadoshima J: Distinct roles of
autophagy in heart during ischemia and reperfusion: Roles of
AMP-activated protein kinase and Beclin 1 in mediating autophagy.
Circ Res. 15:914–922. 2007. View Article : Google Scholar
|
|
38
|
Wang GL, Jiang BH, Rue EA and Semenza GL:
Hypoxia-inducible factor-1 is a basichelix-loop-helix-PAS
heterodimer regulated by cellular tension. Proc Natl Aacad Sci USA.
92:5510–5514. 1995. View Article : Google Scholar
|
|
39
|
Jewell UR, Kvietikova I, Scheid A, Bauer
C, Wenger RH and Gassmann M: Induction of HIF-1alpha in response to
hypoxia is instantaneous. FASEB J. 15:1312–1314. 2001.PubMed/NCBI
|
|
40
|
Zarbin MA: Current concepts in the
pathogenesis of age related macular degeneration. Arch Ophthalmol.
122:598–614. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
AI-Salam S and Hashmi S: Galectin-1 in
early acute myocardail infarction. PloS One. 9:e869942014.
View Article : Google Scholar
|
|
42
|
Rapino C, Bianchi G, Di Giulio C,
Centurione L, Cacchio M, Antonucci A and Cataldi A: HIF-1alpha
cytoplasmic accumulation is associated with cell death in old rat
cerebral cortex exposed to intermittent hypoxia. Aging Cell.
4:177–185. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Siddqi A, Aminova LR and Ratan RR: Hypoxia
inducible factor proly 4-hydroxylase encimes: Center stage in the
battle against hypoxia, metabolic compomise and oxidative stress.
Neurochem Res. 32:931–946. 2007. View Article : Google Scholar
|
|
44
|
Wang Z and Si LY: Hypoxia inducible
factor-1α and vascular endothelial growth factor in the
cardioprotective effects of intermittent hypoxia in rats. Ups J Med
Sci. 118:65–74. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ke Q and Costa M: Hypoxia-inducible
factor-1 (HIF-1). Mol Pharmacol. 70:1469–1480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hashmi S and Al-Salam S: Hypoxia-inducible
factor-1 alpha in the heart: A double agent? Cardiol Rev.
20:268–273. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Robador PA, San José G, Rodríguez C,
Guadall A, Moreno MU, Beaumont J, Fortuño A, Díez J,
Martínez-González J and Zalba G: HIF-1 mediated up-regulation of
cardiotrophin-1 is involved in the survival response of
cardiomyocytes to hypoxia. Cardiovasc Res. 92:247–255. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Blanco Pampín J, García Rivero SA, Otero
Cepeda XL, Vázquez Boquete A, Forteza Vila J and Hinojal Fonseca R:
Immunohistochemical expression of HIF-1alpha in response to early
myocardial ischemia. J Forensic Sci. 51:120–124. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cai Z, Zhong H, Bosch-Marce M, Fox-Talbot
K, Wang L, Wei C, Trush MA and Semenza GL: Complete loss of
ischaemic preconditioning-induced cardioprotection in mice with
partial deficiency of HIF-1 alpha. Cardiovasc Res. 77:463–470.
2008. View Article : Google Scholar
|
|
50
|
Huang Y, Hickey RP, Yeh JL, Liu D, Dadak
A, Young LH, Johnson RS and Giordano FJ: Cardiac myocyte-specific
HIF-1alpha deletion alters vascularization, energy availability,
calcium flux and contractility in the normoxic heart. Faseb J.
18:1138–1140. 2004.PubMed/NCBI
|
|
51
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wu QL, Shen T, Ma H and Wang JK:
Sufentanil postconditioning protects the myocardium from
ischemia-reperfusion via PI3k/Akt-GSK-3β pathway. J Surg Res.
178:563–570. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yu Y, Jia XJ, Zong QF, Zhang GJ, Ye HW, Hu
J, Gao Q and Guan SD: Remote ischemic postconditioning protects the
heart by upregulating ALDH2 expression levels through the PI3K/Akt
signal pathway. Mol Med Rep. 10:536–542. 2014.PubMed/NCBI
|
|
54
|
Chen K, Li G, Geng F, Zhang Z, Li J, Yang
M, Dong L and Gao F: Berberine reduces ischemia/reperfusion-induced
myocardial appoptosis via activating AMPK and PI3K-Akt signaling in
diabetic rats. Appoptosis. 19:946–957. 2014. View Article : Google Scholar
|
|
55
|
Zhou Y, Wang D, Gao X, Lew K, Richards AM
and Wang P: MTORC2 phosphorylation of Akt1: A possible mechnism for
hydrogen sulfide-induced cardioprotection. PloS One. 9:e996652014.
View Article : Google Scholar
|
|
56
|
Lu C, Ha T, Wang X, Liu L, Zhang X,
Kimbrough EO, Sha Z, Guan M, Schweitzer J, Kalbfleisch J, et al:
The TLR9 ligand, CpG-ODN, induces protection against cerebral
ischemia/reper-fusion injury via activation of PI3K/Akt signaling.
J Am Heart Assoc. 3:e0006292014. View Article : Google Scholar
|
|
57
|
Zuo W, Chen J, Zhang S, Tang J, Liu H,
Zhang D and Chen N: M-H004 prevents toxicity induced by delayed
treatment of tPA in a rat model of focal cerebal ischemia involving
PKA-and PI3K- independent Akt activation. Eur J Neurosci.
39:2107–2118. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chen W, Zheng G, Yang S, Ping W, Fu X,
Zhang N, Wang DW and Wang J: CYP2J2 and EETs protect against
oxidative stress and apopotosis in vivo and in vitro Following Lung
Ischemia/Reperfusion. Cell Physio Biochem. 33:1663–1680. 2014.
View Article : Google Scholar
|