Introduction
Prostate cancer (PCa) is the second leading cause of
cancer-associated mortality in men in the US, and up to 40% of PCa
cases eventually develop metastasis (1). Metastasis remains a significant
clinical challenge in various cancer types, as it is the major
cause of mortality and is responsible for low five-year survival
rates (2). Epithelial-mesenchymal
transition (EMT) is thought to be the initial step of tumor
metastasization, as it renders cancer cells with migratory and
invasive properties required to detach from the primary tumor and
enter vessels (3). Downregulation
of E-cadherin and upregulation of N-cadherin are considered as
hallmarks of EMT (4,5).
Several transcription factors that induce EMT, such
as SNAI1 and SNAI2/SLUG, two members of the Snail super-family of
zinc finger transcriptional repressors (6,7),
have been shown to act as E-cadherin repressors. Furthermore, zinc
finger E-box-binding homeobox 1 (ZEB1) and ZEB2, which belong to
the ZEB family, have emerged as key factors that regulate
E-cadherin and the induction of the EMT (8,9). In
addition, certain basic helix-loop-helix factors, including E47 and
TWIST1, function as EMT regulators affecting tumor cell invasion
and metastasis (10). Vimentin has
been demonstrated to significantly contribute to the invasive
phenotype of PCa cells (11,12).
EMT of PCa cells involves the acquisition of mesenchymal markers
such as vimentin and differentiation into a phenotype with an
increased probability of metastasis formation.
Histone modifications, including methylation,
acetylation, phosphorylation and ubiquitination, are considered to
be epigenetic variations linked with processes of carcinogenesis
and cancer progression, including EMT, and modification of gene
expression; furthermore, they appear to have a prognostic value
(13,14). In these modifications, histone
methylation has been proved to have important roles in various
aspects of chromatin function. Methylation of H3K9 and H3K27 is
associated with gene silencing, whereas methylation of H3K4, H3K36
and H3K79 has been linked to transcriptional activation (15,16).
However, the effects of methylation of H4K20 to form monomethylated
H4K20 (H4K20me1) on gene transcription requires further study.
SET8, also known as PR-SET7/KMT5A, is a histone H4K20-specific
methyltransferase (17), which
specifically monomethylates H4K20 (18). SET8 was reported to exert a dual
role on transcription by the activation of N-cadherin and
repression of E-cadherin (19). Of
note, SET8 has been reported to be involved in activation as well
as repression of transcription (20,21).
SET8, through its methylase activity, has been implicated in a
variety of biological processes, including transcriptional
regulation, genomic stability, heterochromatin formation and
cell-cycle progression, while the role of SET8 in PCa and the
underlying mechanisms have remained poorly understood. The present
study aimed to shed light on the role of SET8 in the cancer
progression and EMT of PCa cells; furthermore, the implication of
ZEB1 and H4K20me1 was evaluated.
Materials and methods
Reagents
Anti-human SET8 antibody (cat. no. sc-135009, rabbit
IgG, 1:500), ZEB1 antibody (cat. no. sc-25388, rabbit IgG,
1:1,000), E-cadherin antibody (cat. no. sc-21791, mouse IgG1,
1:1,000), N-cadherin antibody (cat. no. sc-7939, rabbit IgG,
1:1,000), α-catenin antibody (cat. no. sc-107193, goat IgG,
1:1,000), vimentin antibody (cat. no. sc-373717, mouse IgG1,
1:500), β-actin antibody (cat. no. sc-7210, rabbit IgG, 1:500) and
secondary antibodies (cat. no. sc-2039, goat anti-mouse IgG-B,
1:2,000; and cat. no. sc-2040, goat anti-rabbit IgG-B, 1:2,000)
were all purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA). Anti-H4K20me1, anti-H4K20me2, anti-H4K20me3 and anti-H4 were
obtained from Abcam (Cambridge, MA, USA). Specific small
interfering (si)RNAs targeting SET8 or ZEB1 (SiSET8#1: 5′-CCG GTT
GAA CAG ATG GCC TTA TAT TCT CGA GAA TAT AAG GCC ATC TGT TCA ATT
TTTG-3′; SiSET8#2: 5′-CCG GGC CTA GGA AGA CTG ATC AAT CCT CGA GGA
TTG ATC AGT CTT CCT AGG CTT TTTG-3′; SiZEB1#1: 5′-CCG GGT CTG GGT
GTA ATC GTA AAT TCT CGA GAA TTT ACG ATT ACA CCC AGA CTT TTTG-3′;
and SiZEB1#2: 5′-CCG GCT GAA CCT CAG ACC TAG TAA TCT CGA GAT TAC
TAG GTC TGA GGT TCA GTT TTTG-3′) were purchased from Sigma-Aldrich
(St. Louis, MO, USA). A negative control siRNA used as control was
also from Sigma-Aldrich. Matrigel® was purchased from BD
Biosciences (Franklin Lakes, NJ, USA). The restriction enzymes were
from New England Biolabs, Inc. (Ipswich, MA, USA). Lipofectamine
2000™ and Lipofectamine RNAimax (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) were used for the transfection
experiments. A Renilla-Glo™ Luciferase assay system kit (Promega
Corp., Madison, WI, USA) was used for the luciferase assay.
Cell culture
The PC-3 androgen-non-responsive PCa cell line and
the PZ-HPV-7 non-transformed prostate epithelial cell line were
obtained from the American Type Culture Collection (Manassas, VA,
USA). The cells were cultured in Dulbecco's modified Eagle's medium
(DMEM; HyClone; GE Healthcare, Little Chalfont, UK) supplemented
with 10% fetal bovine serum (FBS; HyClone). The LNCaP
androgen-responsive PCa cell line (also obtained from the American
Type Culture Collection) was cultured in RPMI-1640 with 10% FBS,
100 units of penicillin/streptomycin (Invitrogen; Thermo Fisher
Scientific, Inc.) and maintained at 37°C in a humidified atmosphere
containing 5% CO2.
Reverse-transcription quantitative
polymerase chain reaction (RT-qPCR)
Total RNA of cell lysates was extracted with TRIzol
solution (Invitrogen) at 48 h post-transfection and
reverse-transcribed into cDNA using 1 µg total RNA with
Moloney murine leukemia virus reverse transcriptase (Beijing
TransGen Biotech Co., Ltd., Beijing, China), according to the
manufacturer's protocol. Real-time PCR primers (Invitrogen; Thermo
Fisher Scientific, Inc.) were as follows: SET8 forward, 5′-TAT CAC
TCT GTT TCA CGCCA-3′ and reverse, 5′-ACC ATT CCT CCA TCT CATCC-3′;
β-actin forward, 5′-TGG CAC CCA GCA CAA TGAA-3′ and reverse, 5′-CTA
AGT CAT AGT CCG CCT AGA AGCA-3′. Real-time PCR was performed using
SYBR Green (Roche Diagnostics, Basel, Switzerland) in an ABI 7500
sequence detection system (Applied Biosystems; Thermo Fisher
Scientific). The PCR reaction conditions were as follows: An
initial stage of 95°C for 2 mins; then 35 cycles of 95°C, 1 min,
55°C, 1 min, 72°C, 1 min; and finally, 72°C for 10 min, with a 4°C
pause. The 2−ΔΔCq method was used. All experiments were
performed in triplicate with β-actin used as a normalization
control.
Western blot analysis
Cells were lysed in 50 ml lysis buffer [100 mM
Tris-HCl (pH 7.4), 10 mM ethylenediaminetetraacetic acid (EDTA), 4%
sodium dodecyl sulfate (SDS) and 10% glycine; Sigma-Aldrich] on ice
for 45 min. Following centrifugation at 12,000 × g for 30 min at
4°C, the supernatants were collected and the protein concentration
was determined using the bicinchoninic acid (BCA) method, with a
commercially available BCA kit (GE Healthcare Life Sciences, Logan,
UT, USA). Equal amounts (40 µg protein) of lysate were
separated by 8–10% SDS-polyacrylamide electrophoresis (PAGE) on an
SDS-PAGE gel (Invitrogen; Thermo Fisher Scientific, Inc.). The
proteins were electro-transferred onto a nitrocellulose membrane
(GE Healthcare Life Sciences) using an electro-blotting apparatus
(Biorad Mini-Protean Tetra Electrophoresis system; Bio-Rad
Laboratories, Inc., Hercules, CA, USA). For Western blot analysis,
membranes were blocked with milk and then incubated with the
appropriate antibodies for 1 h at room temperature, followed by
five washes with washing buffer (Tris-buffered saline/Tween 20
buffer; Sigma-Aldrich), prior to incubation with a secondary
antibody. Immunoreactive bands were visualized using Western
blotting Luminal reagent (Santa Cruz Biotechnology, Inc.) according
to the manufacturer's protocol, using Kodak X-OMAT BT film (Kodak,
Rochester, NY, USA). β-actin was used as a loading control.
Immunoprecipitation (IP)
Cells were washed with cold phosphate-buffered
saline and lysed with cold lysis buffer (Sigma-Aldrich; as detailed
above) at 4°C for 45 min. Whole-cell lysates were incubated with
appropriate primary antibodies (the SET8 or the ZEB1 antibody) or
normal rabbit/mouse immunoglobulin G (IgG; Santa Cruz
Biotechnology, Inc.) with agitation overnight at 4°C, followed by
addition of protein A/G Sepharose CL-4B beads (GE Healthcare Life
Sciences) for 2 h at 4°C. Beads were then washed five times with
lysis buffer (50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1%
Nonidet P-40, 0.25% sodium deoxycholate and protease inhibitor
mixture; Sigma-Aldrich)). The immune complexes were subjected to
10% SDS-PAGE, followed by immunoblotting with secondary
antibodies.
Construction of E-cadherin or vimentin
reporter plasmid and luciferase reporter assay
For vimentin reporter plasmid construction, a
sequence of the vimentin promoter and partial first exon (-736 to
+164 bp) was obtained by PCR using specific primers; the E-cadherin
reporter was generated as described previously (19). These PCR products were ligated into
the pGL3 basic vector to generate pGL3-vimentin or pGL3-E-cadherin
luciferase reporter constructs (Promega Corp., Madison, WI, USA)
according to the manufacturer's protocol. Cells in 96-well plates
were transfected with vimentin or E-cadherin promoter luciferase
reporter, Renilla plasmid and the indicated expression constructs
using Lipofectamine LTX-Plus (Invitrogen). The amount of DNA was 2
µg in each transfection. Forty-eight hours after
transfection, the firefly and Renilla luciferases were assayed
according to the manufacturer's protocol (Promega) with Renilla
luciferase plasmid as a transfection efficiency normalization
control. Each experiment was repeated in triplicate.
Chromatin IP (ChIP)
ChIP assays were performed according to the
manufacturer's protocol (Upstate Biotechnology, Inc., Lake Placid,
NY, USA). The chromatin was obtained according to the
manufacturer's protocol (Upstate Biotechnology, Inc.). Chromatin
was incubated with 3 µg SET8 antibody, ZEB1 antibody,
H4K20me1 antibody, H4K20me2 antibody, H4K20me3 antibody, anti-H4 or
normal rabbit IgG as a negative control, at 4°C overnight.
Immunoprecipitated DNA was purified using the Qiagen PCR
purification kit (Qiagen, Hilden, Germany). qChIP was performed by
qPCR using specific primers as stated below. For common ChIP
assays, the final target DNA sequence was amplified and resolved on
standard agarose DNA gels (Invitrogen; Thermo Fisher Scientific,
Inc.). For the DNA amplification, the PCR reaction conditions were:
An initial stage of 95°C for 2 mins; then 35 cycles of 95°C, 1 min,
55°C, 30 sec, 72°C, 1 min; and finally, 72°C for 10 min, with a 4°C
pause.
Primers (Invitrogen; Thermo Fisher Scientific, Inc.)
used for common ChIP were as follows: E-cadherin forward, 5′-GAG
ACT GGC ACA GTA ATC TTC-3′ and reverse, 5′-TGG CTA ACG CAG TGA
AAC-3′; α-catenin forward, 5′-TGG TCC TAT TGC CCT TTG-3′ and
reverse, 5′-GCC CTT ACC GTG TTT ACC-3′; N-cadherin forward, 5′-GCA
CTC AGA ACA GGC ACAT-3′ and reverse, 5′-GCC CAG GAG TTC GAG
ACCA-3′; vimentin forward, 5′-CCC TGC CTT AGT CTC CCA-3′ and
reverse, 5′-CTC CTC CTT CCA ACC TGTC-3′.
qChIP primers: E-cadherin forward, 5′-AAC CCA GTG
GAA TCA GAAC-3′ and reverse, 5′-ATA GAC GCG GTG ACC CTC-3′;
α-catenin forward, 5′-AGA GGG TAA ACA CGG TAAGG-3′ and reverse,
5′-TCA CGG GAT GAT GAA TAAGA-3′; N-cadherin forward, 5′-AAT CCT CCC
ACT TCA GCC-3′ and reverse, 5′-AGC CCA GGA GTT CGA GAC-3′; vimentin
forward, 5′-AGC CTA TCA CAG CCC AGAG-3′ and reverse, 5′-CCC ATA GCC
GAT TCC TCA-3′.
Transwell invasion assay
The invasive ability of the cells was investigated
using Transwell inserts for 24-well plates (8 µm pore size;
EMD-Millipore, Billerica, MA, USA). First, the surfaces of the
membranes were coated with 100 µl Matrigel® (50
µg/ml) for 1 h. A total of 5×104 cells in 500
µl serum-free medium was added to each upper chamber, while
500 µl DMEM containing 10% FBS was added to the lower
compartment. Following incubation for 24 h, the cells on the upper
surface of the membrane were removed using cotton swabs, while the
cells that had transgressed through the Matrigel-coated membrane to
the lower surface were fixed with 2% paraformaldehyde and stained
with crystal violet (Sigma-Aldrich). Images were captured using an
inverted microscope (Olympus BX46; Olympus Corp., Tokyo, Japan) at
×100 magnification. Quantification of invaded cells in each well
was performed in three randomly selected fields. Each experiment
was performed in triplicate and representative images are
shown.
Statistical analysis
Values are expressed as the mean ± standard
deviation of three independent experiments, using the paired t-test
to compare the mean values (± standard deviation). SPSS version
17.0 software was used for the statistical analysis (SPSS, Inc.,
Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Knockdown of SET8 in PC-3 cells
As shown in Fig.
1A, PC-3 cells were transfected with control siRNA, siSET8#1 or
siSET8#2. Knockdown of SET8 was confirmed at the mRNA and protein
level. RT-qPCR showed that the mRNA levels of SET8 were
significantly reduced following transfection with siSET8#1 or
siSET8#2 (P<0.05) (Fig. 1A,
left panel). Concomitantly, western blot analysis showed that in
siSET8-transfected PC-3 cells, SET8 was markedly reduced as
compared with that in the the control siRNA-transfected cells
(Fig. 1A, middle panel). While the
results confirmed that the two SET8-specific siRNA-containing
vectors were successfully constructed and transfected into PC-3
cells, the knockdown efficiency of siSET8#2 was higher than that of
siSET8#1. Furthermore, western blot analysis confirmed that SET8
was markedly overexpressed in PC-3 cells transfected with specific
overexpression vector (Fig. 1A,
right panel).
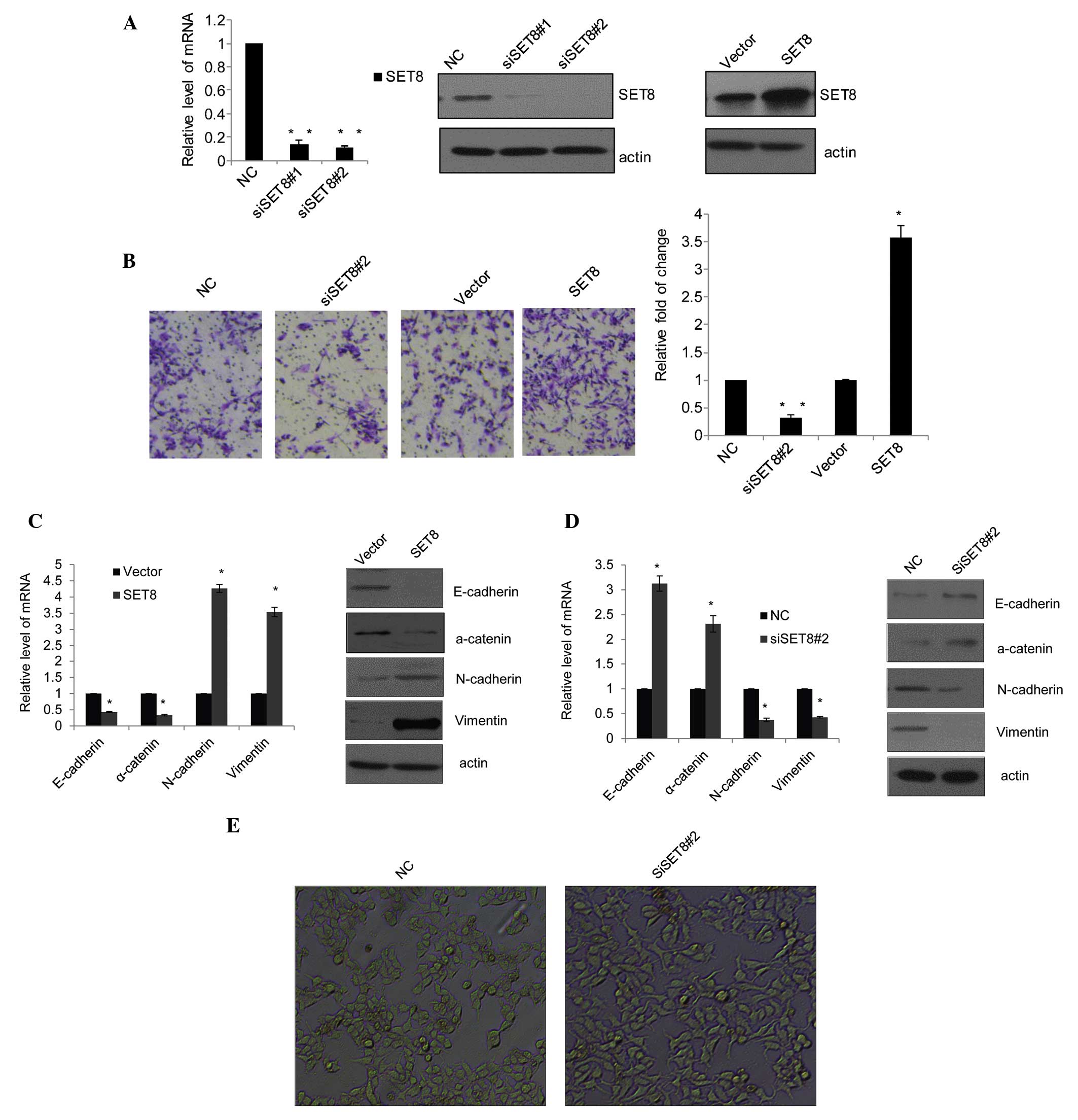 | Figure 1SET8 promotes prostate cancer cell
metastasis and epithelial-mesenchymal transition. (A) The SET8
knockdown efficiency was confirmed by qPCR (left panel) and western
blot analysis (middle panel), and the SET8 overexpression
efficiency was determined by western blot analysis (right panel) in
PC-3 cells. (B) PC-3 cells were transfected with control siRNA,
siSET8#2, empty vector or SET8 overexpression vector for 48 h.
Cells were then starved for 18 h prior to cell invasion assays. The
invaded cells were stained with crystal violet and counted under a
microscope (magnification, ×100). Representative images for each
group are shown and quantitative results are presented as the fold
change compared with the vector group. (C) Effects of SET8
overexpression in PC-3 cells on the mRNA and protein expression of
the epithelial markers E-cadherin and a-catenin as well as
mesenchymal markers N-cadherin and vimentin as examined by RT-qPCR
(left) and western blot analysis (right), respectively. (D) Effects
of SET8 knockdown in PC-3 cells on the expression of the indicated
epithelial or mesenchymal markers as examined by RT-qPCR (left) and
western blot analysis (right), respectively. Values are expressed
as the mean ± standard deviation for triplicate measurements.
*P<0.05; **P<0.01 vs. vector/NC group.
(E) PZ-HPV-7 cells (magnification, ×10) were treated with specific
siRNA against SET8, and the morphological alterations of these
cells were examined by phase-contrast microscopy. siRNA, small
interfering RNA; NC, negative control; RT-qPCR,
reverse-transcription quantitative polymerase chain reaction. |
SET8 promotes PCa-cell metastasis and
EMT
To assess the effects of SET8 on cell invasion, a
Transwell assay was performed. The number of migrated cells
transfected with siSET8#2 accounted for one third of those
transfected with control siRNA, while SET8 overexpression increased
cell invasion to 3.5 times of that of the control cells (Fig. 1B). These results indicated that
SET8 is involved in metastasis of PCa. Considering EMT as the
initiation step of metastasis, the present study further examined
the role of SET8 in this process. In SET8-overexpressing PC-3
cells, the expression of the epithelial markers E-cadherin and
α-catenin was downregulated, while the mesenchymal markers
N-cadherin and vimentin were elevated as indicated by RT-qPCR
(Fig. 1C, left panel) and western
blot analysis (Fig. 1C, right
panel). Conversely, following SET8 knockdown in PC-3 cells,
E-cadherin and α-catenin were upregulated, while N-cadherin and
vimentin were decreased (Fig. 1D).
In addition, observation of PZ-HPV-7 prostate epithelial cells by
phase-contrast microscopy showed that depletion of SET8 led to
obvious morphological alterations (Fig. 1E).
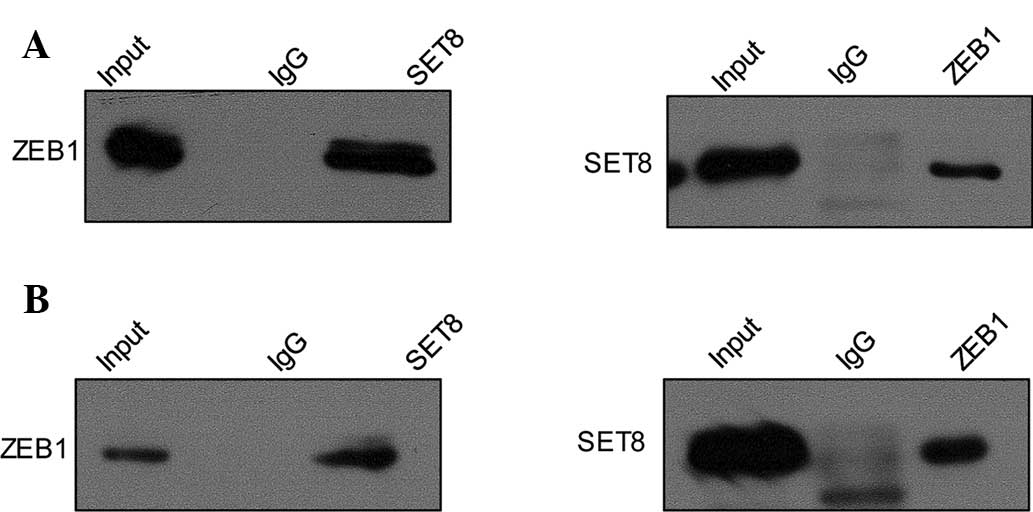
SET8 is physically associated with ZEB1
but not ZEB2 in PCa cell lines
In order to investigate the mechanism of the role of
SET8 in EMT and metastasis, a co-IP assay was performed to assess
the interaction between SET8 and various transcription factors,
which have been confirmed to have pivotal roles in tumor metastasis
by promoting EMT. Total protein extracts from PC-3 cells were
prepared and co-IP experiments were performed with specific
antibodies against target proteins. First, IP with anti-SET8
followed by immunoblotting (IB) with the anti-ZEB1 in PC-3 cells
indicated that SET8 co-immunoprecipitated with ZEB1 (Fig. 2A, upper panel), and similar results
were obtained for IP with anti-ZEB1 followed by IB with anti-SET8
(Fig. 2A, lower panel). This in
vitro interaction of SET8 and ZEB1 was also demonstrated in the
LNCaP cell line (Fig. 2B). Co-IP
experiments using antibodies against other transcription factors,
including SNAI1 and ZEB2, showed no interaction with SET8 (data not
shown).
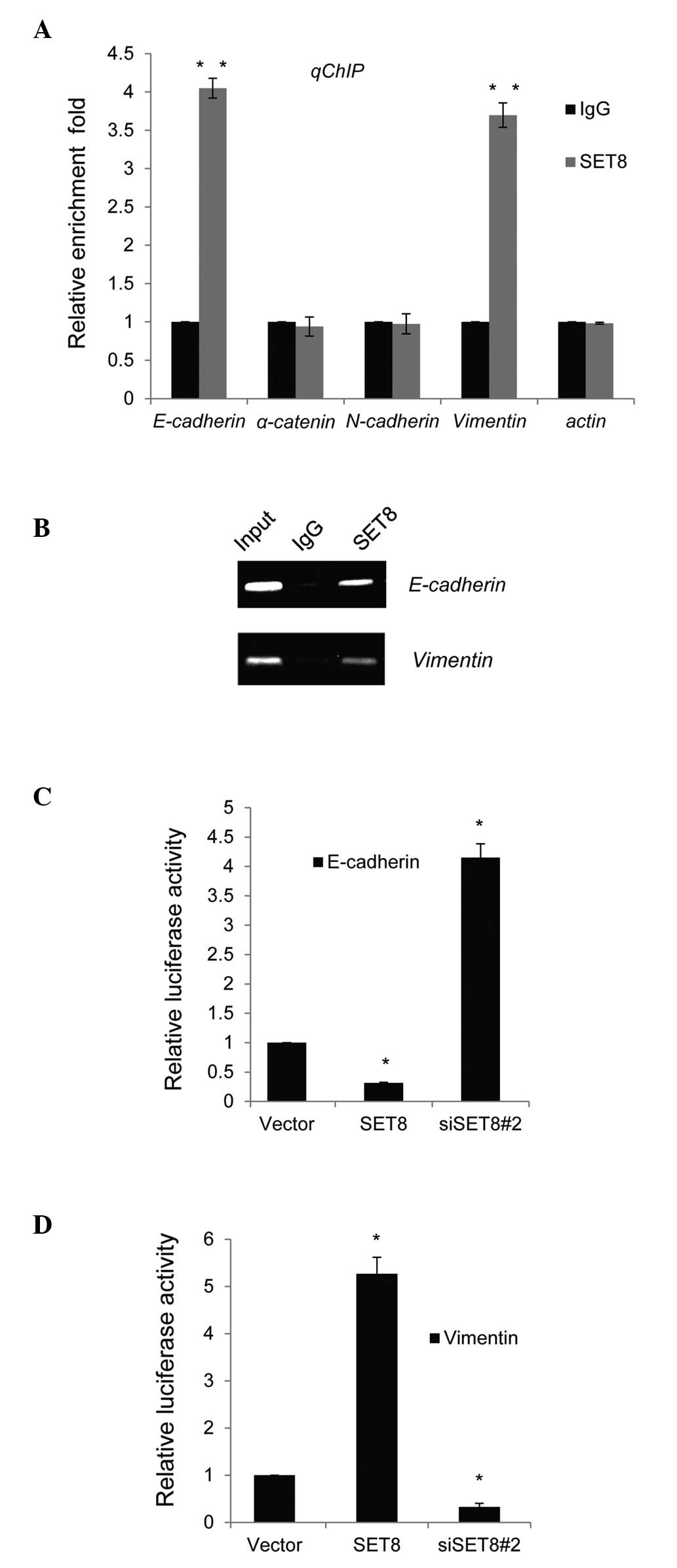
SET8 directly regulates E-cadherin and
vimentin transcription
To further assess the molecular mechanism of the
regulatory roles of SET8 in transcription, qChIP assays were
performed in PC-3 cells. Several key genes of molecular pathways
involved in the EMT and metastasis, namely E-cadherin, α-catenin,
N-cadherin vimentin and actin, were selected for detection of
possible binding to SET8 with their gene promoter regions. The
binding of SET8 to the promoter sequences of E-cadherin and
vimentin was obviously higher than that to normal IgG (Fig. 3A). Similar results were obtained in
ChIP PCR assays in PC-3 cells, as bright bands were obtained in the
anti-SET8 groups when primers for the E-cadherin or vimentin
promoter were used, while no bands were obtained with normal IgG
(Fig. 3B).
To assess whether SET8 directly targeted the
promoter regions of E-cadherin and vimentin, luciferase reporter
assays were performed in PC-3 cells. E-cadherin or vimentin
promoter-driven luciferase reporter vectors were transfected into
PC-3 cells with simultaneous SET8 overexpression or depletion. The
results indicated that SET8 overexpression or knockdown resulted in
repressed or enhanced E-cadherin reporter activity, respectively
(Fig. 3C). By contrast,
overexpression or silencing of SET8 led to a significant activation
or repression, respectively, of the vimentin reporter vector
(Fig. 3D).
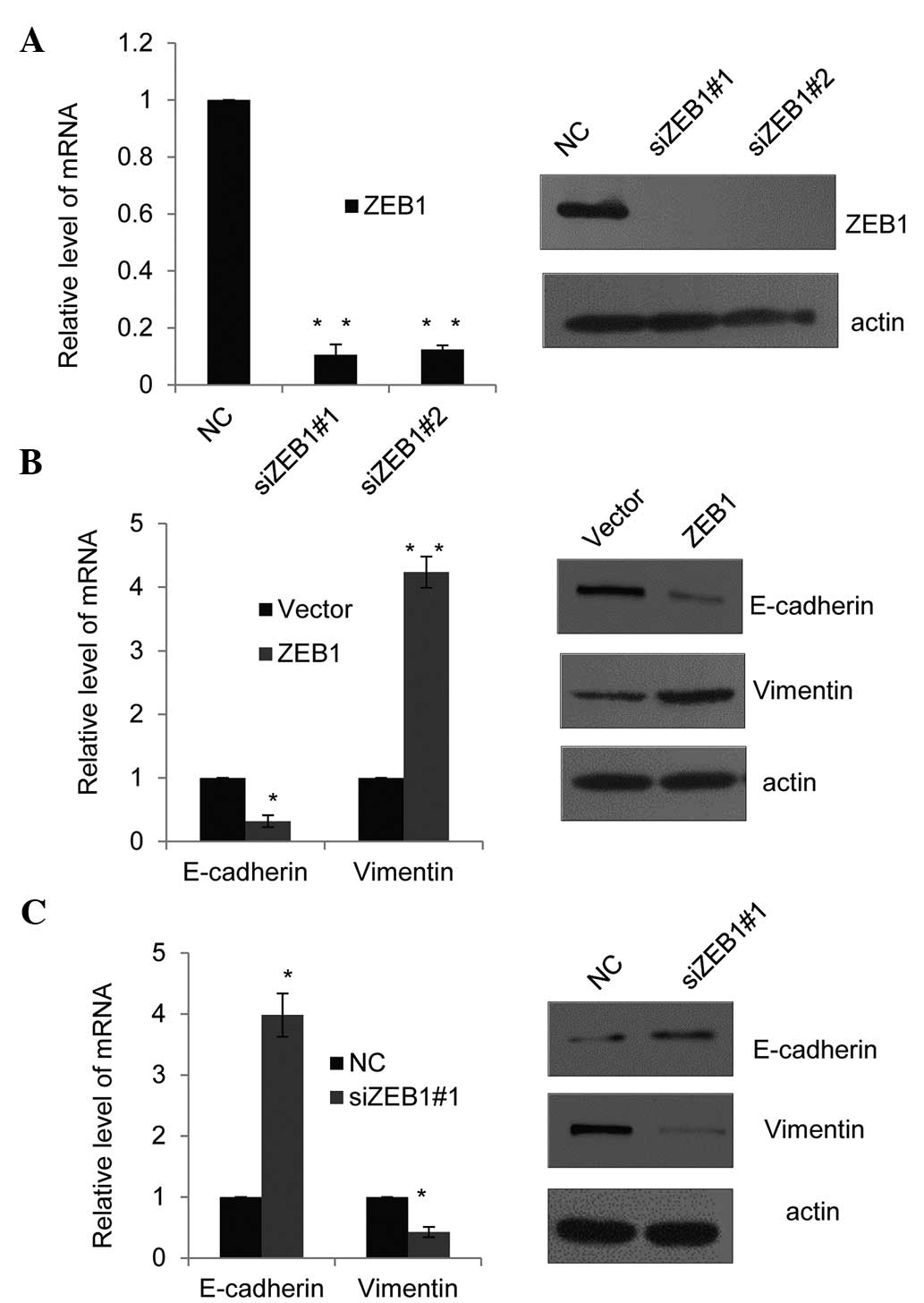
SET8 cooperates with ZEB1 to affect the
expression of E-cadherin and vimentin
Since the recruitment of ZEB1 to the promoter of
E-cadherin had been previously reported (22), the present study assessed whether
SET8 and ZEB1 have similar or joint effects on E-cadherin
expression patterns, and whether they jointly affect vimentin
expression. First, the efficiency of two different ZEB1-specific
siRNAs was assessed using RT-qPCR and western blot analysis in PC-3
cells (Fig. 4A). ZEB1 siRNA#2 was
revealed to slightly more efficiently silence ZEB1 than ZEB1
siRNA#1 and was therefore selected for use in the subsequent
experiment. Consistent with SET8 promoter occupancy (Fig. 3A and B) and the effects of SET8 on
the expression of the indicated epithelial or mesenchymal markers
(Fig. 1C and D). In
ZEB1-overexpressing PC-3 cells, the mRNA (Fig. 4B, left panel) and protein
expression (Fig. 4B, right panel)
of E-cadherin was decreased, while in ZEB1-silenced PC-3 cells,
E-cadherin was increased at the mRNA and protein level (Fig. 4C). Furthermore, ZEB1 overexpression
enhanced vimentin mRNA and protein expression, while in
ZEB1-depleted PC-3 cells, vimentin was downregulated (Fig. 4B and C). These results supported
the notion that SET8 and ZEB1 are required for trans-repression of
E-cadherin and trans-activation of vimentin. It appeared that SET8
functions in a dual mode in ZEB1-regulated gene expression.
SET8-induced H4K20me1 has a dual function
in ZEB1-regulated gene expression
qChIP assays using anti-H4K20me1, anti-H4K20me2 and
anti-H4K20me3 in PC-3 cells indicated that H4K20 was mono-, di- and
trimethylated in E-cadherin and vimentin promoters (Fig. 5A). To explore whether the
monomethylation of H420 was catalyzed by SET8 in the promoter of
E-cadherin and vimentin, SET8 was silenced in PC-3 cells and the
methylation status of H4K20 in E-cadherin and vimentin promoters
was assessed by qChIP. The results indicated that knockdown of SET8
was associated with a sharp reduction of monomethylation of H4K20,
while only a slight decrease in di- and trimethylation of H4K20 was
observed in the E-cadherin (Fig.
5B) and vimentin promoters (Fig.
5C). Of note, ZEB1 knockdown also led to a marked decrease of
H4K20me1 in the E-cadherin (Fig.
5B) and vimentin (Fig. 5C)
promoters. These results further supported the hypothesis that SET8
is recruited to the E-cadherin and vimentin promoters by ZEB1 to
monomethylate H4K20. Collectively, these experiments indicated that
SET8 is recruited to the E-cadherin promoter by ZEB1 to repress its
transcription and to the vimentin promoter to activate its
transcription through its H4K20 monomethylation activity.
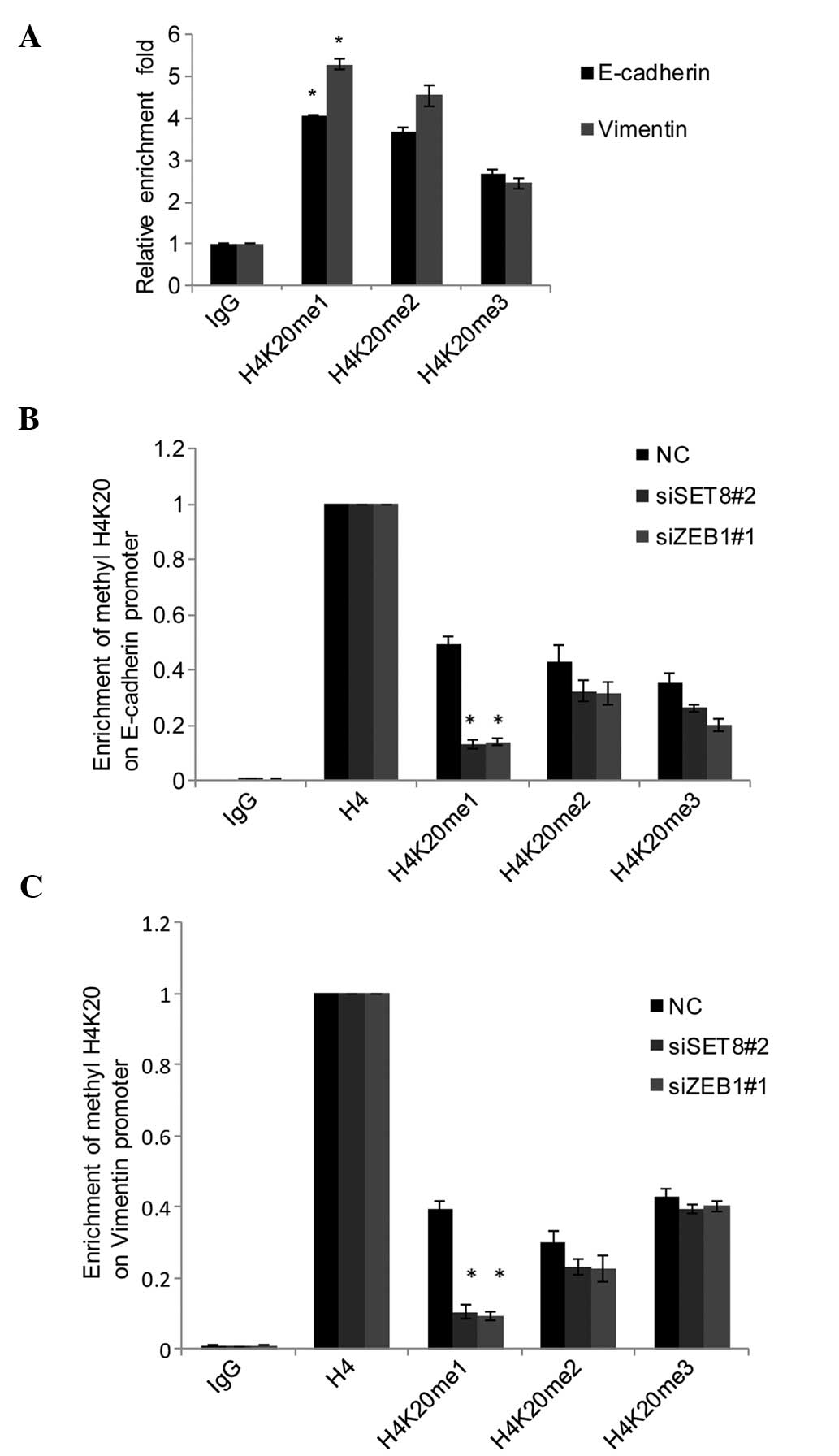 | Figure 5SET8-mediated H4K20me1 has a dual
function in ZEB1-regulated gene expression. (A) qChIP assays of the
promoter of E-cadherin and vimentin were performed with
anti-H4K20me1, anti-H4K20me2 and anti-H4K20me3 with normal IgG used
for the negative control. (B) SET8 was silenced using siRNA in PC-3
cells, and qChIP of the promoter of E-cadherin and vimentin was
performed with antibodies against H4K20me1, H4K20me2 and H4K20me3
with normalization to H4 as the control. (C) In ZEB1-depleted PC-3
cells, qChIP assays of the promoter of E-cadherin and vimentin were
performed with mono-, di-, and trimethylated H4K20-specific
antibodies, and anti-H4 set as 100%. Values are expressed as the
mean ± standard deviation of three experiments.
*P<0.05 vs. NC or IgG. IgG, immunoglobulin G; qChIP,
quantitative chromatin immunoprecipitation; siSET8, small
interfering RNA specific for SET8; NC, negative control;
H4K20me1/2/3, mono/di/trimethylated histone H4K20; ZEB1, zinc
finger E-box-binding homeobox 1. |
Discussion
Cancer-cell invasion and metastasis are multistep
processes comprising alterations in cell adhesion as well as
transformation of cells to phenotypes with enhanced invasive and
migratory potential (23).
E-cadherin generally functions as a tumor suppressor gene and
maintains the polarity as well as structural integrity of
epithelial cell. Loss or dysfunction of E-cadherin is closely
associated with tumorigenesis, invasion and metastasis in numerous
cancer types.
The present study demonstrated that depletion of
SET8 inhibits PC-3-cell migration and invasion in vitro;
furthermore, silencing of SET8 in non-transformed PZ-HPV-7 prostate
epithelial cells was shown to disrupt their epithelial-type
morphology. Silencing of SET8 in PC-3 cells led to decreases in the
expression of the epithelial markers E-cadherin and α-catenin, and
increased the expression of the mesenchymal markers N-cadherin and
vimentin implying that SET8 promotes EMT and enhances the invasive
capacity of PC-3 PCa cells. Furthermore, SET8 was shown to exert a
dual transcriptional regulatory function comprising the repression
of E-cadherin expression and the activation of vimentin expression.
A co-IP assay indicated that SET8 mediates the EMT by physically
binding to the EMT-inducing transcription factor ZEB1.
The important role of SET8 as a histone
H4K20-specific methyltransferase was further proved by its binding
toanti-H4K20me1 in the promoter regions of E-cadherin and vimentin.
Furthermore, ZEB1 knockdown also led to a marked decrease of
H4K20me1 in the E-cadherin and vimentin promoters. Although SET8
directly targeted the promoters of E-cadherin and vimentin, binding
with ZEB1 resulted in a more marked change on the these promoters,
which indicated that ZEB1 may enhance the enzyme activity of SET8.
The present study revealed a role for SET8 in facilitating EMT and
the invasive potential of PCa cells, suggesting that SET8
represents a potential therapeutic target for treating or
preventing metastasis of PCa.
In conclusion, the present study revealed that SET8
is a positive effector of the EMT and PCa metastasis, by directly
repressing the transcription of E-cadherin and activating that of
vimentin via methylating H4K20me1; furthermore, it physically binds
to ZEB1. ZEB1 is a transcription factor, the function of which is
to directly bind DNA, and since the physical association of ZEB1
with SET8 was determined in the present study, it was reasonable to
hypothesize that ZEB1 cooperates with SET8 to enhance prostate
cancer cell metastasis. SET8 represents a promising therapeutic
target for reversing or preventing EMT and tumor metastasis, which
are major events in PCa progression and aggravation.
However, it remains elusive whether SET8 has any
additional molecular targets involved in PCa-cell proliferation and
invasion. It is known that neoplastic progression is attributable
to a large number of genes; therefore, it is necessary to further
explore the potential role of SET8 in the genesis and progression
of PCa.
Acknowledgments
This study was supported by grants from the Zhejiang
Provincial Foundation of National Science (no. LY13H160030) and the
Scientific and Technological Developing Scheme of Hangzhou (no.
20130633B33).
References
|
1
|
Beltran H, Beer TM, Saad F, Sternberg C
and Tagawa ST: New therapies for castration-resistant prostate
cancer: Efficacy and safety. Eur Urol. 60:279–290. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Martin GS: Cell signaling and cancer.
Cancer Cell. 4:167–174. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kase S, Sugio K, Yamazaki K, Okamoto T,
Yano T and Sugimachi K: Expression of E-cadherin and beta-catenin
in human non-small cell lung cancer and the clinical significance.
Clin Cancer Res. 6:4789–4796. 2000.
|
|
4
|
Hay ED: An overview of
epithelio-mesenchymal transformation. Acta Anat (Basel). 154:8–20.
1995. View Article : Google Scholar
|
|
5
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cano A, Perez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hajra KM, Chen DY and Fearon ER: The SLUG
zinc-finger protein represses E-cadherin in breast cancer. Cancer
Res. 62:1613–1618. 2002.PubMed/NCBI
|
|
8
|
Smit MA and Peeper DS: Zeb1 is required
for TrkB-induced epithelial-mesenchymal transition, anoikis
resistance and metastasis. Oncogene. 30:3735–3744. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Comijn J, Berx G, Vermassen P, Verschueren
K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D and van Roy
F: The two-handed E box binding zinc finger protein SIP1
downregulates E-cadherin and induces invasion. Mol Cell.
7:1267–1278. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang J, Mani SA, Donaher JL, Ramaswamy S,
Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A and
Weinberg RA: Twist, a master regulator of morphogenesis, plays an
essential role in tumor metastasis. Cell. 117:927–939. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Singh S, Sadacharan S, Su S, Belldegrun A,
Persad S and Singh G: Overexpression of vimentin: Role in the
invasive phenotype in an androgen-independent model of prostate
cancer. Cancer Res. 63:2306–2311. 2003.PubMed/NCBI
|
|
12
|
Zhao Y, Yan Q, Long X, Chen X and Wang Y:
Vimentin affects the mobility and invasiveness of prostate cancer
cells. Cell Biochem Funct. 26:571–577. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kouzarides T: Chromatin modifications and
their function. Cell. 128:693–705. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shilatifard A: Chromatin modifications by
methylation and ubiquitination: Implications in the regulation of
gene expression. Ann Rev Biochem. 75:243–269. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Esteller M: Cancer epigenomics: DNA
methylomes and histone-modification maps. Nat Rev Genet. 8:286–298.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gronbaek K, Treppendahl M, Asmar F and
Guldberg P: Epigenetic Changes in cancer as potential targets for
prophylaxis and maintenance therapy. Basic Clin Pharmacol Toxicol.
103:389–396. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Couture JF, Collazo E, Brunzelle JS and
Trievel RC: Structural and functional analysis of SET8, a histone
H4 Lys-20 methyltransferase. Genes Dev. 19:1455–1465. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fang J, Feng Q, Ketel CS, Wang H, Cao R,
Xia L, Erdjument-Bromage H, Tempst P, Simon JA and Zhang Y:
Purification and functional characterization of SET8, a nucleosomal
histone H4-lysine 20-specific methyltransferase. Curr Biol.
12:1086–1099. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang F, Sun L, Li Q, Han X, Lei L, Zhang H
and Shang Y: SET8 promotes epithelial-mesenchymal transition and
confers TWIST dual transcriptional activities. EMBO J. 31:110–123.
2012. View Article : Google Scholar :
|
|
20
|
Li Z, Nie F, Wang S and Li L: Histone H4
Lys 20 monomethylation by histone methylase SET8 mediates Wnt
target gene activation. Proc Natl Acad Sci USA. 108:3116–3123.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Talasz H, Lindner HH, Sarg B and Helliger
W: Histone H4-lysine 20 monomethylation is increased in promoter
and coding regions of active genes and correlates with
hyperacetylation. J Biol Chem. 280:38814–38822. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wong TS, Gao W and Chan JY: Transcription
regulation of E-cadherin by zinc finger E-box binding homeobox
proteins in solid tumors. Biomed Res Int. 2014:9215642014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Park SI, Zhang J, Phillips KA, Araujo JC,
Najjar AM, Volgin AY, Gelovani JG, Kim SJ, Wang Z and Gallick GE:
Targeting SRC family kinases inhibits growth and lymph node
metastases of prostate cancer in an orthotopic nude mouse model.
Cancer Res. 68:3323–3333. 2008. View Article : Google Scholar : PubMed/NCBI
|