Introduction
As hypoxic pulmonary arterial hypertension (PAH)
progresses, the pulmonary vascular resistance and pulmonary
arterial pressure increase as a result of pulmonary vessel
remodeling (PVR), vasoconstriction, and thrombosis in situ
(1,2). The imbalance between cell
proliferation and cell apoptosis in pulmonary artery smooth muscle
cells (PASMCs) contributes to medial pulmonary vascular
hypertrophy, a major pathophysiological change during PVR (3–6). The
inhibition of apoptosis and the promotion of cell growth of PASMCs
may lead to their overgrowth and result in medial hypertrophy of
pulmonary vessels. This may lead to a decrease in the inner-lumen
diameter and thus induce increased resistance of pulmonary
arteries, which may then elevate the arterial pressure (7).
Transforming growth factor-β (TGF-β) triggers
numerous cellular responses through various receptors and
intracellular transduction pathways (8–10).
The members of the TGF-β family are multifunctional proteins that
are important mediators in pulmonary fibrosis and vascular
remodeling (11–13). The three mammalian isoforms of TGF
include TGF-β1, TGF-β2 and TGF-β3, and are involved in cell
proliferation, differentiation, migration and apoptosis regulation
(14). Previous studies have
indicated that abnormalities of the TGF-β signaling pathway are
linked to the pathogenesis of PAH (12,15,16),
and that TGF-β1 protects against apoptosis of pulmonary artery
endothelial cells (17,18). However, the mechanism responsible
for the survival of PASMCs and the involvement of TGF-β1 in this
mechanism remain unclear.
The phosphatidylinositol 3-kinase/protein kinase B
(PI3K/Akt) signaling pathway is an important pro-survival pathway.
The hyperactivation of Akt may lead to the inhibition of apoptosis
in various cell types (19).
Growth factors may promote cell proliferation and antagonize cell
apoptosis by activating the PI3K/Akt pathway (20). Previous studies have identified
that Akt plays an inhibitory role in cell apoptosis by reducing the
expression of certain pro-apoptotic proteins (21–23).
Additionally, the PI3K/Akt pathway is important in the progression
of PAH (24). Therefore, the
PI3K/Akt pathway may participate in the growth and survival of
PASMCs in response to TGF-β1 signaling.
As there are currently no effective treatment
methods for PAH, more research is required in order to explore the
molecular mechanisms underlying the progression of PAH, which may
help to identify novel treatment methods to interfere with the
development of the disease. The present study hypothesized that
TGF-β1 protects against cell apoptosis in PASMCs via the PI3K/Akt
signaling pathway, resulting in the overgrowth of PASMCs and the
medial thickening of the pulmonary artery. The results from the
current study demonstrated that TGF-β1 inhibits the apoptotic
change induced by serum deprivation, and that the inhibitory
effects of TGF-β1 on cell apoptosis are mediated by the PI3K/Akt
signaling pathway. These findings indicate that TGF-β1 and its
downstream effectors may be potential targets to treat pulmonary
artery hypertension.
Materials and methods
Materials
Recombinant human TGF-β1, which was dissolved in
deionized water, was obtained from PeproTech, Inc. (Rocky Hill, NJ,
USA). Antibodies against Akt (polyclonal; rabbit anti-rat;
dilution, 1:1,000; cat. no. 9272), phosphorylated-Akt (monoclonal;
rabbit anti-rat; dilution, 1:1,000; cat. no. 4060), and β-actin
(monoclonal; rabbit anti-rat; dilution, 1:1,000; cat. no. 8457)
were purchased from Cell Signaling Technology, Inc. (Beverly, MA,
USA). Anti-α-actin (mouse monoclonal immunoglobulin M; dilution,
1:100; sc-58670) was purchased from Santa Cruz Biotechnology Inc.
(Dallas, TX, USA). LY294002 and assay kits used to examine the
release of lactate dehydrogenase (LDH), caspase-3 and caspase-9
were purchased from Beyotime Institute of Biotechnology (Haimen,
China). Methanol, chloroform, dimethyl sulfoxide, fetal bovine
serum (FBS), phosphate buffered saline containing 0.1% Tween-20
(PBS-T) and bovine serum albumin were purchased from Thermo Fisher
Scientific, Inc. (Waltham, MA, USA).
Experimental animals
A total of 6 adult Wistar rats (age, 6 weeks; mean
weight, 200 g) were obtained from the Experimental Animal Center of
Harbin Medical University (Harbin, China). The experimental
procedures applied in this study were approved by the Institutional
Animal Care and Use Committee of Harbin Medical University. The
rats were conditioned at a controlled ambient temperature of 22±2°C
with 50±10% relative humidity and a 12 h light-dark cycle (lights
on at 8:00 am). All rats were provided with standard chow and water
ad libitum.
Cell culture
Rats were sacrificed at 6 weeks of age by cervical
dislocation. The outer diameter of pulmonary arteries (distal,
200–500 µm) was dissected from the lungs of adult Wistar
rats under an optical microscope (SZ61; Olympus Corporation, Tokyo,
Japan). The extracted segments were cut open, then the adventitia
and endothelium of pulmonary arteries were stripped mechanically.
PASMCs were dispersed according to previously established methods
(25,26). Cells were cultured in Dulbecco's
modified Eagle's medium (DMEM) containing 20% fetal bovine serum in
an atmosphere containing 5% CO2 at 37°C, in a humidified
incubator. Subsequently, anti-α-actin was used to determine the
purity of the primary cultured PASMCs. The cells of passages 2–3
were used in all the experiments. Serum deprivation was used in
order to induce apoptosis in PASMCs, thus the cells were incubated
in DMEM without any serum for 24 h. Cells were divided into three
groups: Control, SD, and SD + TGF-β1. Cells cultured in complete
medium (DMEM + 10% FBS) were used as the control group. Cells
cultured in DMEM without FBS were used as the model of serum
deprivation, and cells in the SD + TGF-β1 group were serum-deprived
and cultured with 10 ng TGF-β1 for 24 h.
Quantitative polymerase chain reaction
(qPCR)
In order to determine the mRNA expression levels of
B-cell lymphoma 2 (Bcl-2) and Bcl-2-associated X (Bax) in PASMCs
obtained from Wistar rats, qPCR was performed. The PASMCs were
divided into three groups (Control, SD and SD + TGF-β1). LY294002
(10 µm) was used to block the PI3K/Akt signaling pathway.
After 24 h following treatment, the RNA from 1×106 cells
from each group was extracted using TRIzol reagent (Invitrogen;
Thermo Fisher Scientific, Inc.). Then, reverse transcription was
performed to obtain cDNA using a PrimeScript RT Reagent kit
(RR037A) from Takara Biotechnology Co., Ltd. (Dalian, China).
Reagents used from the kit included 2 µl 5X PrimeScript
buffer, 0.5 µl PrimeScript RT Enzyme Mix I, 0.5 µl
Oligo dT Primer (50 µM), 0.5 µl Random 6-mers (100
µM), 500 ng RNA and 10 µl RNase Free dH2O.
The reverse transcription thermocycling conditions were as follows:
37°C For 15 min and 85°C for 5 sec, then stored 4°C. An Applied
Biosystems 7300 Fast Real-Time PCR system (Thermo Fisher
Scientific, Inc.) was used to perform all our qPCR experiments and
a BLAST program (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to
confirm the specificity of the primers. The total reaction volume
was 20 µl containing: 1x SYBR Premix Ex Taq II (RP820A;
Takara Biotechnology Co., Ltd.), 10 µM forward and reverse
primers, 0.4 µl ROX reference dye (Takara Biotechnology Co.,
Ltd.), and 2 µl of cDNA. The qPCR conditions were as
follows: 95°C for 30 sec 40 cycles of 95°C for 5 sec, and 60°C for
31 sec, followed by routine melting curve analysis. The sequences
of the primers were as follows: Bcl-2 forward,
5′-CGGGAGAACAGGGTATGA-3′ and reverse, 5′-CAGGCTGGAAGGAGAAGAT-3′
(149 bp); Bax forward, 5′-ATCCACCAAGAAGCTGAG-3′ and reverse,
5′-GTAGAAGAGGGCAACCAC-3′ (184 bp); and β-actin
forward:5′-AGGCCCCTCTGAACCCTAAG-3′ and reverse,
5′-CCAGAGGCATACAGGGACAAC-3′ (118 bp). Relative quantification of
target gene expression was calculated using the 2−ΔΔCq
method (27). The ratio of
Bcl-2/Bax was then calculated.
Western blot analysis
The cells were cultured in 6 well plates, and
1×106 cells were added to each well. Cells were divided
into three groups: Control, SD, and SD + TGF-β1. Their growth was
arrested for 24 h prior to adding 10 ng TGF-β1 under serum
deprivation conditions (SD + TGF-β1 group). LY294002 (10 µm)
was used to block the PI3K/Akt signaling pathway. Cells cultured in
complete medium were the control group. The cells were then washed
three times with ice-cold phosphate-buffered saline, 24 h after the
treatment was applied. Subsequently, the cells were treated with
200 µl lysis buffer, containing 50 mM Tris (pH 7.4), 150 mM
NaCl, 1% Triton X-100, 1 mM EDTA and 2 mM PMSF, and then incubated
for 30 min on ice. The lysates were sonicated for 1 min and
centrifuged at 17,000 × g for 15 min at 4°C. The protein
concentrations in the supernatant were determined using the Bio-Rad
Protein Assay kit (Bio-Rad Laboratories, Inc., Berkeley, CA, USA).
The western blot protocol followed to determine the protein
expression of the samples was similar to that reported in a
previous study (28). Briefly, 50
µg protein was electrophoresed on SDS polyacrylamide gels,
and then transferred onto polyvinylidene diflfluoride membranes
(Merck Millipore, Darmstadt, Germany). Then, 5% bovine serum
albumen was used to block the membrane for 1 h at room temperature.
The membrane was then incubated with phosphorylated-Akt and Akt
primary antibodies at 4°C overnight. After washing for 35 min with
PBS-T, the membrane was incubated with an alkaline
phosphatase-conjugated secondary antibody (monoclonal; goat
anti-rabbit; dilution, 1:5,000; #7074; Cell Signaling Technology,
Danvers, MA, USA) for 1 h at room temperature. After washing for 35
min with PBS-T, immunoreactivity was detected using an enhanced
chemiluminescence western blotting detection kit (Amersham
Biosciences, Piscataway, NJ, USA) and exposed to X-ray film.
MTT assay
The MTT assay was performed according to the method
published by Ma et al (26), in order to determine the cell
viability. Briefly, 1×104 cells were added to each well
in 96-well culture plates and prepared using the same method as
used in the western blot analysis. After incubation at 37°C for 24
h following treatment, the cells were incubated for 4 h in 0.5% MTT
(Beyotime Institute of Biotechnology). The supernatant was then
removed, and 150 µl dimethyl sulfoxide was added to each
well. The plates were then agitated on a plate shaker for 10 min at
room temperature. A spectrophotometer (Epoch 2; BioTek Instruments,
Inc., Winooski, VT, USA) was used to read the absorbance at 490 nm.
The measured absorbance value was used to represent the number of
living cells.
LDH assay
The expression levels of LDH were determined using a
Cytotoxicity Detection kit (Beyotime Institute of Biotechnology).
The experiments were carried out as previous studies (26). Briefly, 100 µl culture
medium and an equal volume of LDH substrate solution was added to
the culture medium for 30 min at room temperature. The reaction was
stopped by adding 0.1 M NaOH to the mixture, and a
spectrophotometer (Epoch 2; BioTek Instruments, Inc.) was used to
detect the absorbance at 440 nm.
Caspase-3 and caspase-9 activity
assay
The cleavage of two chromogenic caspase substrates
was determined, including the caspase-3 substrate Ac-DEVD-pNA (also
known as N-acetyl-Asp-Glu-Val-Asp p-nitroanilide) and the caspace-9
substrate Ac-LEHD-pNA (also known as N-acetyl-Leu-Glu-His-Asp
p-nitroanilide). The experimental procedures followed the
manufacturer's protocols and a previously published method
(29). Briefly, 50 µg total
protein was added to 50 µl reaction buffer (Beyotime
Institute of Biotechnology), which contained 10 µl
Ac-DEVD-pNA (2 mM) or 10 µl Ac-LEHD-pNA (2 mM), and the
samples were incubated at 37°C for 2 h. The absorbance of yellow
pNA cleaved from their corresponding precursors was measured using
a spectrophotometer (Epoch 2; BioTek Instruments, Inc.) at 405 nm.
The absorbance was used to represent the activity of
caspase-3/9.
Statistical analysis
Statistical analysis was performed using SPSS
version 15.0 for Windows (SPSS, Inc., Chicago, IL, USA).
Experiments were performed in triplicate. All values were
represented as the mean ± standard error of mean. One-way analysis
of variance and t-test analysis (two-tailed) were used to determine
the statistical significance of differences between the means of
different groups. P<0.05 indicated a statistically significant
difference.
Results
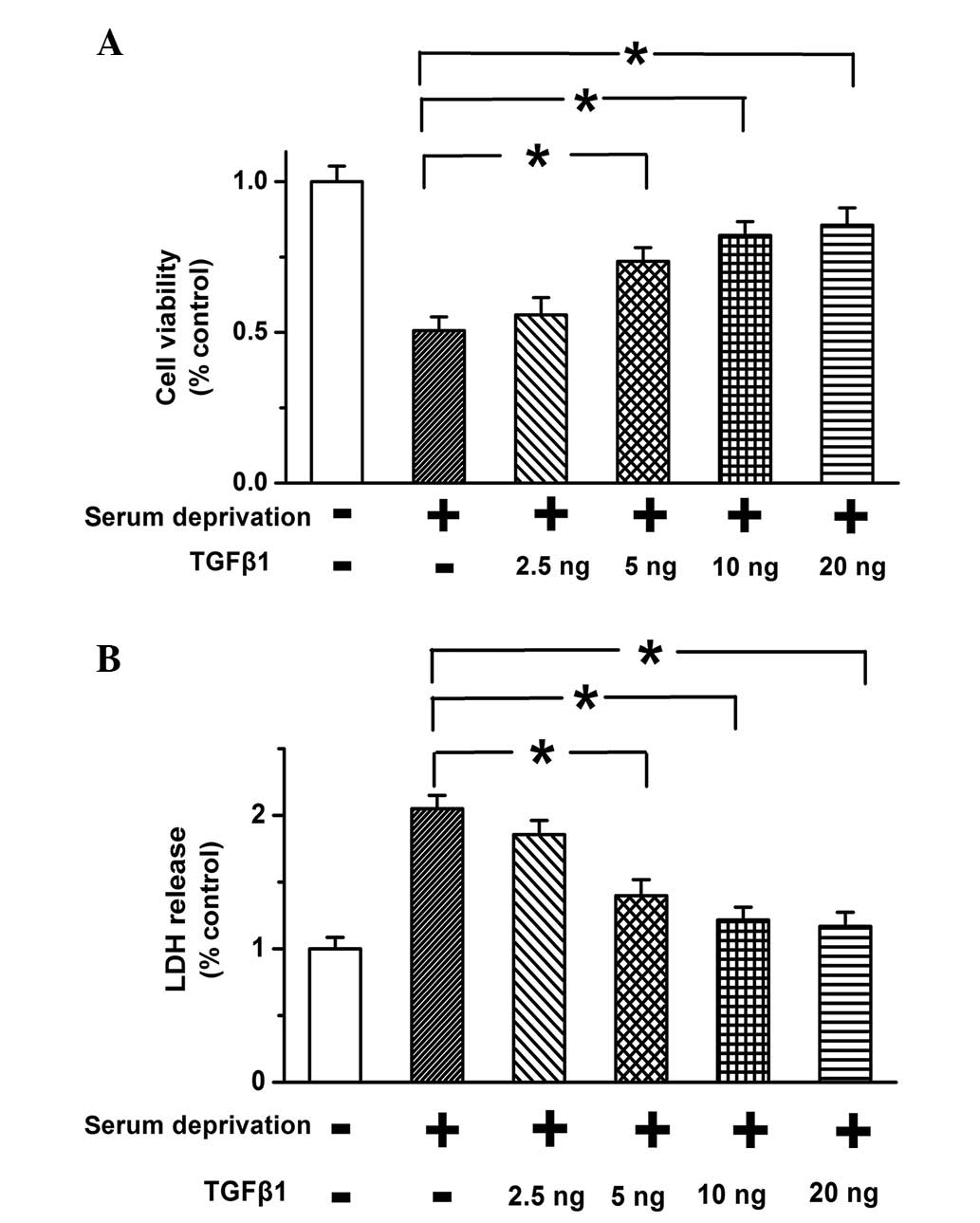
TGF-β1 promotes the survival of starved
PASMCs in a dose-dependent manner
MTT assay was applied to determine the effects of
TGF-β1 (10 ng) on cell viability of PASMCs. Serum-deprivation was
used to induce cell apoptosis. As shown in Fig. 1A, the decrease in cell viability
due to serum deprivation was reduced following TGF-β1 treatment in
a dose-dependent manner. TGF-β1 significantly improved cell
viability when used at a concentration of ≥10 ng. The effects of
TGF-β1 on cell death were also examined, and serum deprivation was
found to result in increased release of LDH, which was reversed by
the addition of 10 ng TGF-β1 (Fig.
1B; P<0.05). The results suggest that TGF-β1 improves cell
viability and inhibits cell death in a dose-dependent manner in
PASMCs.
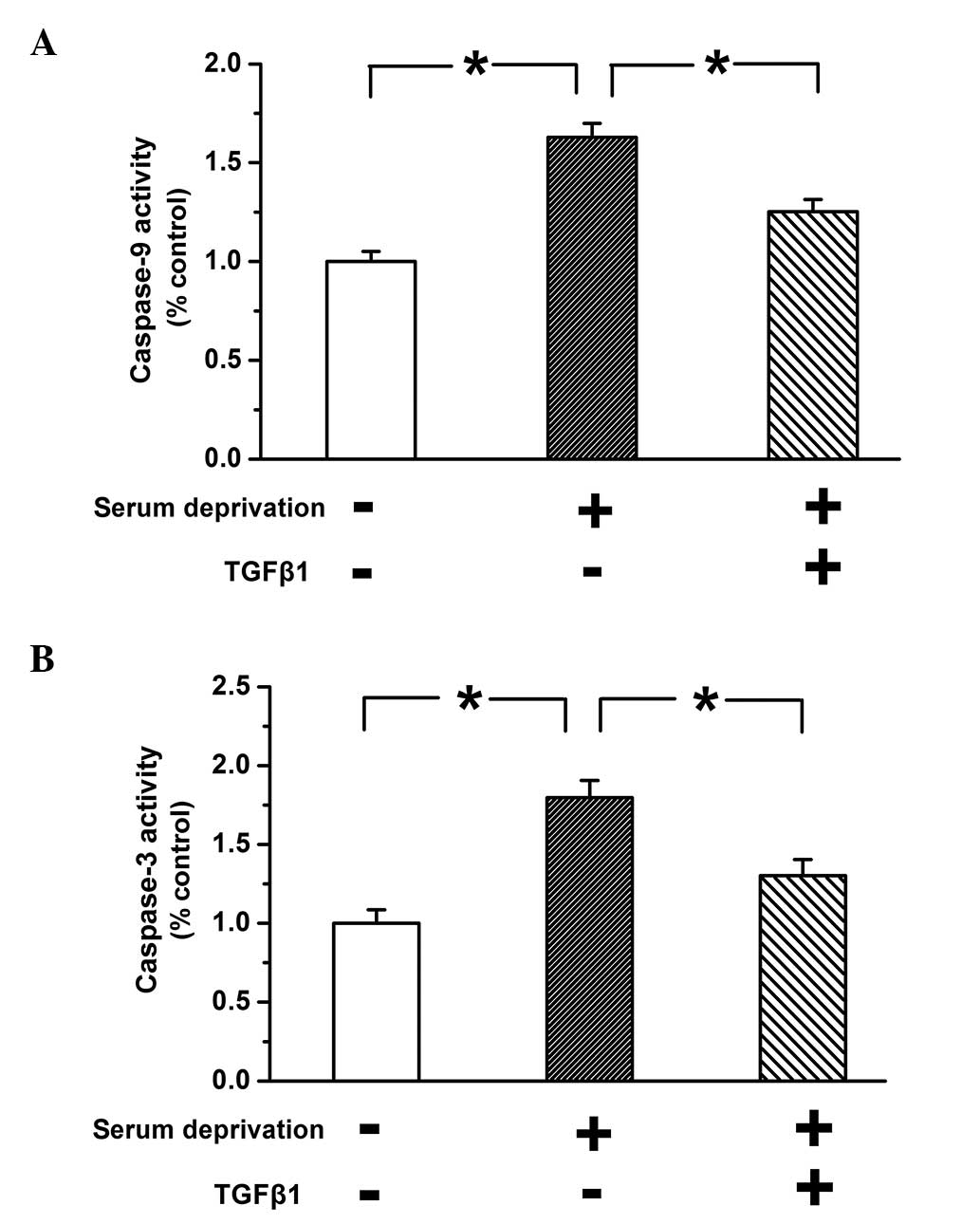
TGF-β1 inhibits the apoptosis induced by
serum deprivation in PASMCs
Caspase-3 and caspase-9 are synthesized by the
precursor proteins procaspase-3 and procaspase-9, respectively, in
response to apoptotic stimuli; subsequently, they are then
activated, triggering cell apoptosis (30). Therefore, the activity of caspase-3
and caspase-9 was examined to determine whether TGF-β1 inhibited
the apoptosis of PASMCs. As shown in Fig. 2, the activity of caspase-3 and
caspase-9 was significantly greater in untreated serum-deprived
cells compared with the control group (P<0.05). This effect was
reversed by treatment with TGF-β1 (10 ng), which indicates that
TGF-β1 inhibits apoptosis induced by serum deprivation.
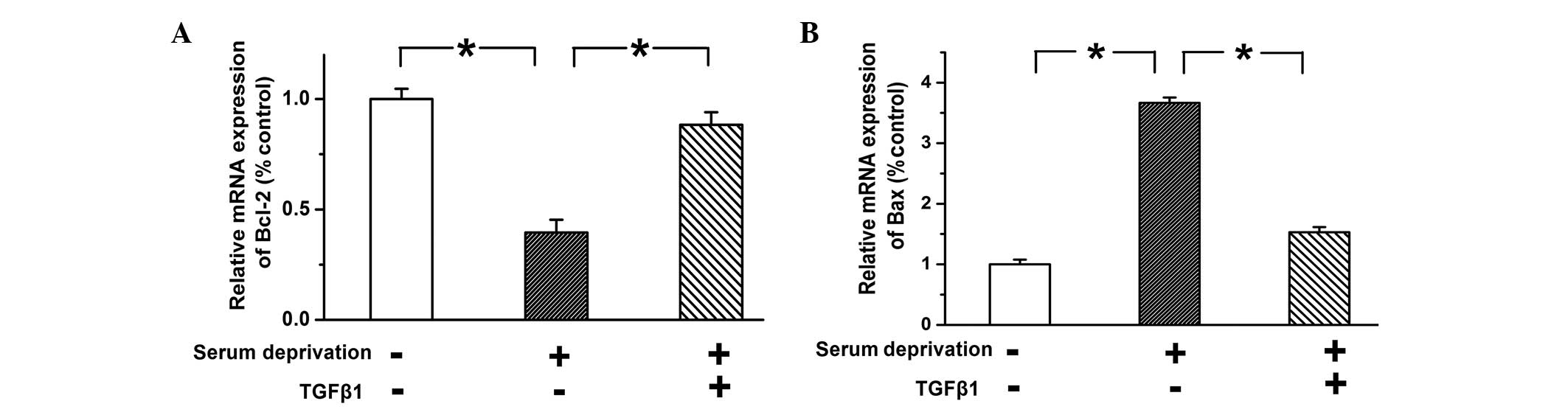
TGF-β1 regulates the expression of
mitochondrial membrane proteins to inhibit apoptosis in PASMCs
Bcl-2 and Bax are two important apoptosis-associated
proteins, located on the outer membrane of mitochondria. Bcl-2 is
an anti-apoptotic protein and Bax is a pro-apoptotic protein. They
participate in maintaining mitochondrial integrity and regulating
mitochondrial-dependent apoptosis (31). In the present study, TGF-β1 was
found to inhibit the activation of caspase-9, a key molecular in
mitochondrial-dependent apoptosis, and thus the protein expression
levels of Bcl-2 and Bax were examined. The mRNA expression levels
of Bcl-2 and Bax were examined using qPCR. The expression of Bcl-2
was reduced and the expression of Bax was increased in
serum-deprived PASMCs compared with the control cells, while
treatment with TGF-β1 (10 ng) reversed these trends (Fig. 3; n=3; P<0.05). These results
indicated that TGF-β1 inhibits apoptosis by upregulating the
expression of Bcl-2 and downregulating the expression of Bax, thus
increasing the ratio of Bcl-2/Bax.
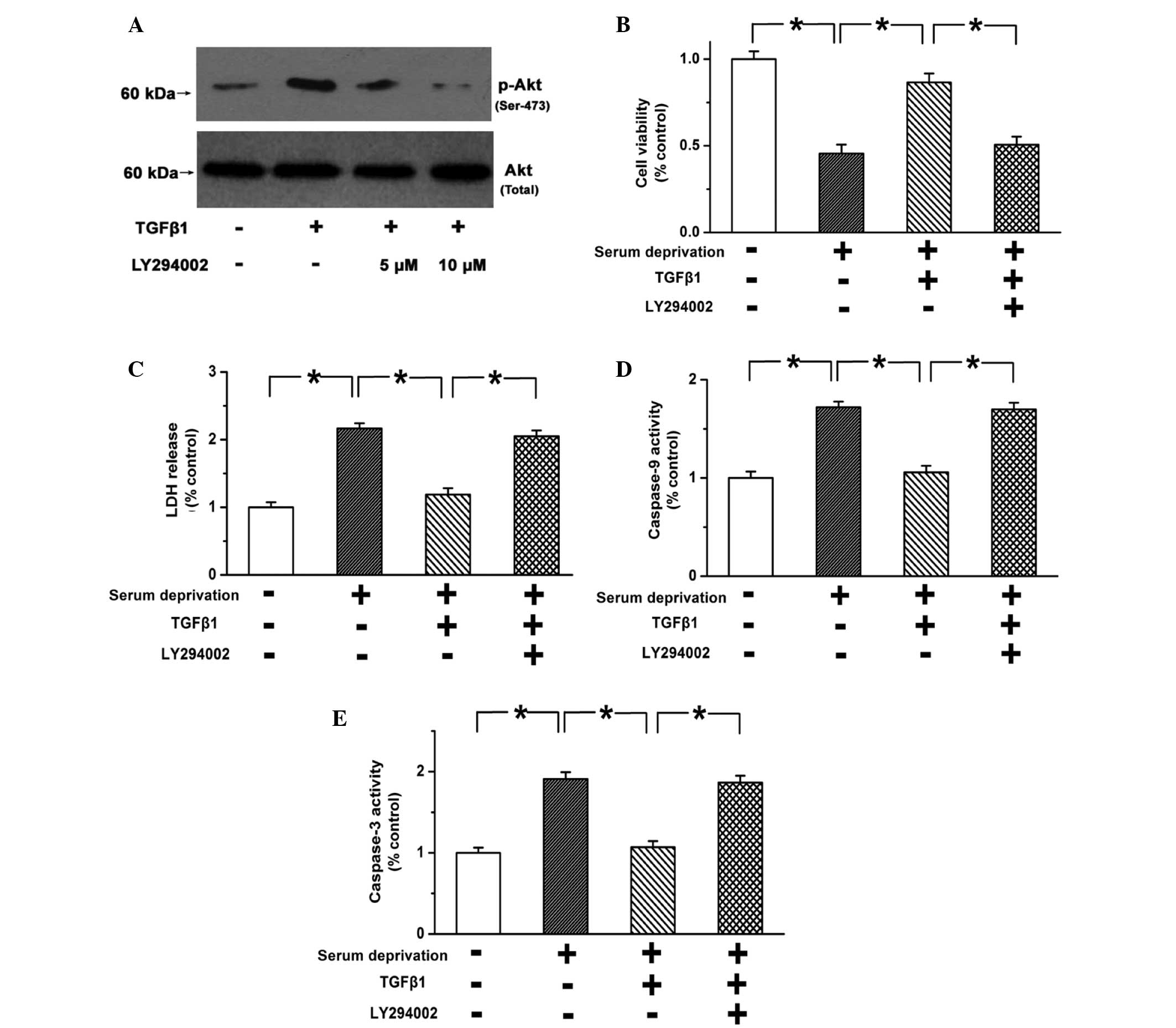
TGF-β1 activates the PI3K/Akt pathway in
PASMCs, but the inhibitory effect of TGF-β1 on cell apoptosis is
abolished following limitation of the PI3K/Akt pathway
The PI3K/Akt pathway is one of the most important
survival pathways, and has been reported to always be activated in
PASMCs during PAH (32). TGF-β1
(10 ng) was found to significantly promote the phosphorylation of
Akt compared with the control group (Fig. 4; P<0.05). LY294002 (10
µM) effectively blocked the activation of the PI3K/Akt
pathway (Fig. 4A; n=3; P<0.05).
In addition, the pro-survival effects of TGF-β1 on PASMCs were
weakened following the blocking of the PI3K/Akt pathway (Fig. 4B and C; n=3; P<0.05). Caspase-3
and caspase-9 activity was not inhibited by TGF-β1 when the
PI3K/Akt pathway was blocked (Fig. 4D
and E; n=3; P<0.05). These results indicate that TGF-β1
inhibits the apoptosis of PASMCs via the PI3K/Akt pathway.
Discussion
The medial hypertrophy of pulmonary arterial vessels
during the progression of PAH is an important pathophysiological
change. Previous studies have identified that overgrowth of PASMCs
contributes to the hypertrophy of pulmonary vascular media
(4–6). The present study provides novel
evidence indicating that TGF-β1 inhibits the apoptosis of PASMCs
through the activation of the PI3K/Akt signaling pathway.
In normal tissues, cell apoptosis is strictly
controlled and there is a balance between apoptosis and
proliferation. However in pathological conditions, this balance is
often disturbed, leading to the overgrowth of cells and the
progression of various diseases. PAH is characterized by sustained
vasoconstriction, thickening of the pulmonary artery walls and
vascular remodeling (2,33,34).
Medial wall thickening usually results from the overgrowth of
PASMCs, a major medial component of pulmonary vascular vessels.
Previous studies have indicated that the increase of cell
proliferation and the decrease of cell apoptosis may lead to
overgrowth of PASMCs (14,35). This subsequently triggers medial
hypertrophy, arterial remodeling and vascular lumen narrowing
(14,35). In addition, apoptosis is regarded
to play a key role during vascular remodeling (36,37).
Therefore, it is necessary to determine the molecular pathway that
mediates the inhibitory effects of hypoxia on PASMCs apoptosis, as
the findings may provide a novel therapeutic target for future
treatments.
Hypoxia is a major trigger of PAH; however, the
precise underlying mechanisms are not fully understood. Previous
studies have suggested that TGF-β1 is activated by hypoxia
(3). Furthermore, there is growing
evidence that abnormalities in the TGF-β1 signaling pathway may be
linked to the pathogenesis of PAH (12,15,16).
Previous studies have indicated that TGF-β1 may participate in the
regulation of the development of hypoxic PAH; however, the role of
TGF-β1 in the survival of PASMCs remains unclear. The current study
determined that TGF-β1 promotes the survival of PASMCs in a
dose-dependent manner in starved PASMCs, and TGF-β1 inhibits the
apoptosis by regulating the expression of mitochondrial membrane
proteins. However, the protective effects of TGF-β1 were markedly
weakened subsequent to the blocking of the PI3K/Akt pathway.
Therefore, it is likely that TGF-β1 mitigates PASMCs apoptosis and
thus promotes pulmonary arterial medial hypertrophy via the
PI3K/Akt pathway.
In conclusion, the present study indicates that
TGF-β1 protects PASMCs from apoptosis by activating the PI3K/Akt
signaling pathway, which leads to medial change of pulmonary
vessels during the progression of PAH. Notably, the current study
determined that the PI3K/Akt pathway mediates the
apoptosis-inhibition effect of TGF-β1 in the survival of PASMCs and
thus offers a novel treatment target for PAH.
Acknowledgments
This study was supported by a grant from the Youth
Science Foundation of Heilongjiang Province (no. QC05C44).
References
|
1
|
Chan SY and Loscalzo J: Pathogenic
mechanisms of pulmonary arterial hypertension. J Mol Cell Cardiol.
44:14–30. 2008. View Article : Google Scholar :
|
|
2
|
Humbert M, Sitbon O and Simonneau G:
Treatment of pulmonary arterial hypertension. N Engl J Med.
351:1425–1436. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Archer S and Rich S: Primary pulmonary
hypertension: A vascular biology and translational research 'Work
in progress'. Circulation. 102:2781–2791. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
De Caestecker M and Meyrick B: Bone
morphogenetic proteins, genetics and the pathophysiology of primary
pulmonary hypertension. Respir Res. 2:193–197. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stenmark KR and Mecham RP: Cellular and
molecular mechanisms of pulmonary vascular remodeling. Annu Rev
Physiol. 59:89–144. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Voelkel NF and Tuder RM: Cellular and
molecular biology of vascular smooth muscle cells in pulmonary
hypertension. Pulm Pharmacol Ther. 10:231–241. 1997. View Article : Google Scholar
|
|
7
|
McMurtry MS, Bonnet S, Wu X, Dyck JR,
Haromy A, Hashimoto K and Michelakis ED: Dichloroacetate prevents
and reverses pulmonary hypertension by inducing pulmonary artery
smooth muscle cell apoptosis. Circ Res. 95:830–840. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Derynck R, Zhang Y and Feng XH: Smads:
Transcriptional activators of TGF-beta responses. Cell. 95:737–740.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Agrotis A, Kalinina N and Bobik A:
Transforming growth factor-beta, cell signaling and cardiovascular
disorders. Curr Vasc Pharmacol. 3:55–61. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bartram U and Speer CP: The role of
transforming growth factor beta in lung development and disease.
Chest. 125:754–765. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu YD, Hua J, Mui A, O'Connor R,
Grotendorst G and Khalil N: Release of biologically active
TGF-beta1 by alveolar epithelial cells results in pulmonary
fibrosis. Am J Physiol Lung Cell Mol Physiol. 285:L527–L539. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rabinovitch M: The mouse through the
looking glass: A new door into the pathophysiology of pulmonary
hypertension. Circ Res. 94:1001–1004. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Arcot SS, Lipke DW, Gillespie MN and Olson
JW: Alterations of growth factor transcripts in rat lungs during
development of monocrotaline-induced pulmonary hypertension.
Biochem Pharmacol. 46:1086–1091. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Morrell NW, Yang X, Upton PD, Jourdan KB,
Morgan N, Sheares KK and Trembath RC: Altered growth responses of
pulmonary artery smooth muscle cells from patients with primary
pulmonary hypertension to transforming growth factor-beta(1) and
bone morphogenetic proteins. Circulation. 104:790–795. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lu Q: Transforming growth factor-beta1
protects against pulmonary artery endothelial cell apoptosis via
ALK5. Am J Physiol Lung Cell Mol Physiol. 295:L123–L133. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mauro M, Kim J, Costello C and Laurence J:
Role of transforming growth factor beta1 in microvascular
endothelial cell apoptosis associated with thrombotic
thrombocytopenic purpura and hemolytic-uremic syndrome. Am J
Hematol. 66:12–22. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Datta SR, Brunet A and Greenberg ME:
Cellular survival: A play in three Akts. Genes Dev. 13:2905–2927.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Downward J: Mechanisms and consequences of
activation of protein kinase B/Akt. Curr Opin Cell Biol.
10:262–267. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo
P, Hu LS, Anderson MJ, Arden KC, Blenis J and Greenberg ME: Akt
promotes cell survival by phosphorylating and inhibiting a Forkhead
transcription factor. Cell. 96:857–868. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Y, Song YH, Mohler J and Delafontaine
P: ANG II induces apoptosis of human vascular smooth muscle via
extrinsic pathway involving inhibition of Akt phosphorylation and
increased FasL expression. Am J Physiol Heart Circ Physiol.
290:H2116–H2123. 2006. View Article : Google Scholar
|
|
23
|
Wang XQ, Sun P and Paller AS: Inhibition
of integrin-linked kinase/protein kinase B/Akt signaling: Mechanism
for ganglioside-induced apoptosis. J Biol Chem. 276:44504–44511.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Garat CV, Fankell D, Erickson PF, Reusch
JEB, Bauer NN, McMurtry IF and Klemm DJ: Platelet-derived growth
factor BB induces nuclear export and proteasomal degradation of
CREB via phosphatidylinositol 3-kinase/Akt signaling in pulmonary
artery smooth muscle cells. Mol Cell Biol. 26:4934–4948. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang L, Ma J, Li Y, Guo L, Ran Y, Liu S,
Jiang C and Zhu D: 15-Hydroxyeicosatetraenoic acid (15-HETE)
protects pulmonary artery smooth muscle cells against apoptosis via
HSP90. Life Sci. 87:223–231. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ma J, Liang S, Wang Z, Zhang L, Jiang J,
Zheng J, Yu L, Zheng X, Wang R and Zhu D: ROCK pathway participates
in the processes that 15-hydroxyeicosatetraenoic acid (15-HETE)
mediated the pulmonary vascular remodeling induced by hypoxia in
rat. J Cell Physiol. 222:82–94. 2010. View Article : Google Scholar
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
28
|
Ma J, Zhang L, Han W, Shen T, Ma C, Liu Y,
Nie X, Liu M, Ran Y and Zhu D: Activation of JNK/c-Jun is required
for the proliferation, survival, and angiogenesis induced by EET in
pulmonary artery endothelial cells. J Lipid Res. 53:1093–105. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang Z, Tang X, Li Y, Leu C, Guo L, Zheng
X and Zhu D: 20-Hydroxyeicosatetraenoic acid inhibits the apoptotic
responses in pulmonary artery smooth muscle cells. Eur J Pharmacol.
588:9–17. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kobayashi T, Masumoto J, Tada T, Nomiyama
T, Hongo K and Nakayama J: Prognostic significance of the
immunohistochemical staining of cleaved caspase-3, an activated
form of caspase-3, in gliomas. Clin Cancer Res. 13:3868–3874. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Adams JM and Cory S: The Bcl-2 protein
family: Arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li L, Xu M, Li X, Lv C, Zhang X, Yu H,
Zhang M, Fu Y, Meng H and Zhou J: Platelet-derived growth factor-B
(PDGF-B) induced by hypoxia promotes the survival of pulmonary
arterial endothelial cells through the PI3K/Akt/Stat3 pathway. Cell
Physiol Biochem. 35:441–451. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mandegar M, Fung YC, Huang W, Remillard
CV, Rubin LJ and Yuan JX: Cellular and molecular mechanisms of
pulmonary vascular remodeling: Role in the development of pulmonary
hypertension. Microvasc Res. 68:75–103. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pidgeon GP, Tamosiuniene R, Chen G,
Leonard I, Belton O, Bradford A and Fitzgerald DJ: Intravascular
thrombosis after hypoxia-induced pulmonary hypertension: Regulation
by cyclooxygenase-2. Circulation. 110:2701–2707. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang S, Fantozzi I, Tigno DD, Yi ES,
Platoshyn O, Thistlethwaite PA, Kriett JM, Yung G, Rubin LJ and
Yuan JX: Bone morphogenetic proteins induce apoptosis in human
pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol
Physiol. 285:L740–L754. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gibbons GH and Dzau VJ: The emerging
concept of vascular remodeling. N Engl J Med. 330:1431–1438. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Uhal BD: Apoptosis in lung fibrosis and
repair. Chest. 122(Suppl): 293S–298S. 2002. View Article : Google Scholar : PubMed/NCBI
|