Introduction
Prostate cancer (PCa) is the most common male
malignancy, and one of the leading causes of cancer-associated
mortality among men in Western countries (1). Extensive studies have been conducted
in an effort to understand the genetic mechanisms underlying PCa
initiation and progression. Using a systems biology approach,
Tomlins et al (2) reported
that the androgen response gene transmembrane protease, serine 2
(TMPRSS2) was fused to the E-twenty-six (ETS) family gene
v-ETS avian erythroblastosis virus E26 oncogene homolog (ERG) and
ETS variant 1 (ETV1) in certain patients with PCa. Among the
various types of fusion genes, the TMPRSS2:ERG fusion is the
most common, and has been identified in 40–70% of samples from
patients with PCa (3).
Functional studies of TMPRSS2:ERG have
suggested that it is involved in cellular proliferation,
differentiation and invasion (4–7),
while its association with additional cellular functions, and in
particular, apoptosis, remains unknown. Shao et al (8) reported that celastrol, a nuclear
factor-κB inhibitor, can suppress TMPRSS2:ERG-expressing PCa
cell growth by inducing apoptosis, implying a possible
anti-apoptotic function for the TMPRSS2:ERG fusion gene.
As an effective anticancer drug, cisplatin is widely
used in the treatment of many types of solid tumors, including
ovarian, testicular, bladder, cervical, head and neck, and
small-cell lung cancer (9,10). Cisplatin is able to interact with
DNA, and results in the formation of interstrand and/or intrastrand
cross-links (11), with such
severe DNA damage considered a major factor in triggering cancer
cell apoptosis. However, cisplatin treatment of PCa has been a
subject of debate, as while earlier studies suggested a lack of
activity, more recent studies using palliation and prostate
specific antigen (PSA) endpoints have suggested a greater degree of
clinical benefit (12). Therefore,
it is of interest to understand why PCa cells have a low response
to cisplatin-induced apoptosis, and whether the TMPRSS2:ERG
fusion gene may serve a role in this response.
In the present study, the role of TMPRSS2:ERG
in cell death, specifically, apoptosis induced by cisplatin was
investigated. The TMPRSS2:ERG fusion gene was demonstrated
to serve an important role in the regulation of apoptosis induced
by cisplatin, and activating transcription factor 5 (ATF5)
was suggested to be involved in this process.
Materials and methods
Cell culture and treatment
Human embryonic kidney 293 (HEK293) cells and the
prostate cancer cell lines DU145 and VCaP cells were purchased from
the American Type Culture Collection (Manassas, VA, USA) and the
cells were grown in Dulbecco's modified Eagle's medium (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), supplemented
with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific,
Inc.), in a humidified atmosphere containing 5% CO2 at
37°C. Cells were plated in 6-well plates and treated at the
indicated times or doses with cisplatin (Sigma-Aldrich, St. Louis,
MO, USA).
DNA constructs and transfection
cDNA corresponding to the most common truncated ERG
(ΔERG), the TMPRSS2:ERG fusion gene (T1/E5), was constructed
using pEGP-C1 (Clontech Laboratories, Inc., Mountainview, CA, USA)
with PstI and BglII sites to generate a green
fluorescent protein (GFP)-ΔERG over-expressing vector. Kanamycin
resistant colonies were selected and verified by sequencing. Cells
were transfected with FugeneHD (Roche Diagnostics, Basel,
Switzerland) following the manufacturer's instructions. In brief, 5
µl FugeneHD was diluted with 50 µl Opti-MEM (Gibco;
Thermo Fisher Scientific, Inc.), mixed and incubated at room
temperature for 5 min. Subsequently, 1.5 µg plasmid DNA was
added to the diluted FugeneHD and mixed, followed by incubation for
20 min at room temperature, following which the FugeneHD-DNA
mixture was added to the cells.
To establish the stable cell line, the HEK293 cells
were transfected as described above. Following selection with 0.8
mg/ml G418 (Gibco, Thermo Fisher Scientific, Inc.) for 14 days,
resistant clones were tested for the expression of ΔERG. The
selected stable cell line was designated as Δ293, and the cells
were cultured further in the medium supplemented with 300
µg/ml G418 for maintenance. Cells transfected with the
GFP-empty vector were designated as E293 and were used as a
control.
Scratch assay
Cells were seeded at 1×105 cells/well in
triplicate in collagen IV coated 6-well plates (Corning
Incorporated, Corning, NY, USA). After 24 h, a scratch through the
central axis of the plate was gently made using a pipette tip. The
migration of Δ293 and E293 cells into the scratched region was
assessed quantitatively at 12 h following the introduction of the
scratch in the monolayer.
Cell proliferation assay
Cells were seeded at 5×103 cells/well
into a 96-well plate. Cell proliferation was examined using a Cell
Counting Kit-8 (Dojindo Molecular Technologies, Inc., Kumamoto,
Japan) following the manufacturer's instructions. Absorbance at 450
nm was measured in quintuplicate using a Tecan Infinite M200 plate
reader (Tecan Trading AG, Männedorf, Switzerland) at the indicated
time points.
ΔERG knock down
Knockdown of ΔERG expression was achieved by the
transfection of ΔERG short interfering RNA (siRNA). The ΔERG siRNA
was sequenced by Shanghai GenePharma Co., Ltd., (Shanghai, China),
the sequences were as follows: Sense, 5′-CCA GAC GUC AAC AUC UUG
UTT-3′ and antisense, 5′-ACA AGA UGU UGA CGU CUG GTT-3′, which were
synthesized by. A nonspecific control strand (UUC UCC GAA CGU GUC
ACG UTT) was used as the negative control (NC).
The siRNA was transfected into the cells using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.)
following the manufacturer's instructions.
Immunoblot analysis
Cells were harvested at the indicated times and
washed three times in phosphate-buffered saline (PBS) prior to
lysis. Protein concentrations were measured using a Bicinchoninic
Acid Protein Assay Reagent kit (Pierce Biotechnology, Inc.,
Rockford, IL, USA). Equal amounts of the protein (20 µg)
were subjected to 12 or 15% sodium dodecyl sulfate polyacrylamide
gel electrophoresis (Bio-Rad Laboratories, Inc., Hercules, CA,
USA), then transferred to polyvinylidene fluoride membranes (EMD
Millipore, Billerica, MA, USA), which were incubated first with the
primary antibody at 4°C overnight, followed incubation with IR
Dye-conjugated secondary antibodies for 1 h at room temperature.
The primary antibodies used were as follows: Rabbit polyclonal
anti-ERG (1:500; Santa Cruz Biotechnology, Inc., Dallas, TX, USA;
cat. no. sc-376293); rabbit polyclonal anti-ATF5 (1:500; Santa Cruz
Biotechnology, Inc.; sc-377168); rabbit monoclonal
anti-cleaved-caspase-3 (1:1,000; Cell Signaling Technology, Inc.,
Danvers, MA, USA; cat. no. 9661); rabbit polyclonal anti-poly ADP
ribose polymerase (PARP-1; 1:1,000; Cell Signaling Technology,
Inc.; cat. no. 9542); mouse monoclonal anti-β-actin (1:1,000; Cell
Signaling Technology, Inc.; cat. no. 3700); rabbit monoclonal
anti-H3 (1:1,000; Cell Signaling Technology, Inc.; cat. no. 4499);
and mouse monoclonal anti-γH2AX (1:3,000; EMD Millipore; cat. no.
05-636). Alexa Fluor 488-conjugated goat anti-mouse (1:5,000; cat.
no. A-11001) and goat anti-rabbit (1:5,000; cat. no. A11008) IgG
were obtained from Thermo Fisher Scientific, Inc.. The results were
observed using an Odyssey Infrared Imaging System (Li-Cor, Inc.,
Lincoln, NB, USA).
Flow cytometry
Cells were harvested at 48 h post cisplatin
treatment. Cells were centrifuged at 300 x g for 5 min at 4°C and
washed with PBS three times, then resuspended in 1X Binding Buffer.
Subsequently, Annexin V-phycoerythrin and 7-aminoactinomycin D (EMD
Millipore) were added, and cells were incubated at room temperature
for 5 min in the dark, following which the apoptotic cells were
analyzed by flow cytometry (FC500 MCL; Beckman Coulter, Inc., Brea,
CA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA from E293 and Δ293 cells was extracted
using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.), and 5
µg of total RNA was reverse-transcribed to cDNA using
SuperscriptIII Reverse Transcriptase (Invitrogen; Thermo Fisher
Scientific, Inc.). Prior to this, the RNA was treated with DNase I
(Sangon Biotech Co., Ltd., Shanghai, China) to remove DNA
contamination. The 20 µl PCR reaction was set up using SYBR
Green Premix Ex Taq kit (Takara Bio, Inc., Otsu, Japan) on a
96-well plate, and conducted using a ABI Prism Sequence Detection
System 7500 (Applied Biosystems, Inc.; Thermo Fisher Scientific,
Inc.) with the following PCR conditions: 95°C for 1 min; 95°C for
10 sec, 60°C for 1 min, repeated for 40 cycles. The primers used
were as follows: Sense, 5′-GAG TGG GCG GTG AAA GAA TA-3′ and
antisense, 5′-AGA AGG ATG TCG GCG TTG TA-3′ for ERG; sense, 5′-TGC
GTG ACA TAA GGA GAA-3′ and anti-sense, 5′-AAG GAA GGC TGG AAG
AGT-3′ for β-actin; sense, 5′-TGG AAA GCG TAG ACA AGG AGAT-3′ and
antisense, 5′-AAG GCT CTA GGT GGT CAT TCAG-3′ for BCL-XL; sense,
5′-CCA TCT ACT GCC GCA ACG AG-3′ and antisense, 5′-CTC AGA GCC
GCCGAC TTG TT-3′ for ATF5; sense, 5′-GGA TTG TGG CCT TCT TTGA-3′
and antisense, 5′-GTG CCG GTT CAG GTA CTCA-3′ for BCL-2; sense,
5′-GGA CGG CGG TGA TGG ACG-3′ and antisense, 5′-GAA GCA AAA GGG CCC
CTGT-3′ for BAX; and sense, 5′-CCG CCA CTA CCA CCA CTT GA-3′ and
antisense, 5′-GGG AAC GCA GCG AAC CGA AT-3′ for BIM. Data analysis
was performed using the 2−ΔΔCq method for relative
quantification (13), and all
samples were normalized to β-actin, which served as an endogenous
control.
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed using the EZ ChIP kit
(EMD Millipore) according to the manufacturer's instructions.
Briefly, cross-linked chromatin was sonicated and
immunoprecipitated with the indicated antibody. Three independent
ChIP analyses were performed to prepare the DNA for future
analysis. Immunoprecipitated DNA was purified and quantified by
RT-qPCR as described above. The primers were as follows: Sense,
5′-AAA AGA CGG ACA TCC ACC TCG-3′ and antisense, 5′-CAA GGG ATG GAG
AAA AGG ACG-3′ for ATF5.
Statistical analysis
The data are presented as the mean ± standard
deviation from a minimum of three independent experiments. The
differences between groups were assessed using an unpaired
two-tailed Student's t-test or one-way analysis of variance.
P<0.05 was considered to indicate a statistically significant
difference.
Results
ΔERG inhibits cisplatin-induced apoptosis
in PCa cell lines
The present study evaluated the toxicity of
cisplatin on DU145 and VcaP cells. It was observed that these two
types of cell were relatively resistant to the toxic effect of
cisplatin as only at concentrations of 50 µM or higher was
significant cell death observed (data not shown). To examine the
function of fusion genes in apoptosis, GFP-tagged
TMPRSS2:ERG (T1/E5) (∆ERG) was transiently expressed in
DU145 cells, which do not express this fusion gene. The cells were
then treated with 50 µM cisplatin for 48 h, and the
apoptotic cell population was analyzed by flow cytometry. As shown
in Fig. 1A, the ∆ERG protein was
expressed in the transfected DU145 cells as indicated by western
blotting. Following cisplatin treatment, the apoptotic cell ratio
in the control DU145 cells was 30.59%. However, this ratio was
reduced to 14.67% in ∆ERG trans-fected DU145 cells (P<0.01;
Fig. 1B). To further investigate
the function of ∆ERG in apoptosis, VCaP cells, which harbor the
TMPRSS2:ERG fusion gene, were transfected with siRNAs to
knockdown the expression of the fusion gene (Fig. 1C). Consistent with the idea that
the fusion protein inhibits apop-tosis, the apoptotic cell ratio
increased from 13.59% in control cells to 21.35% in
siRNA-transfected cells (P<0.05; Fig. 1D).
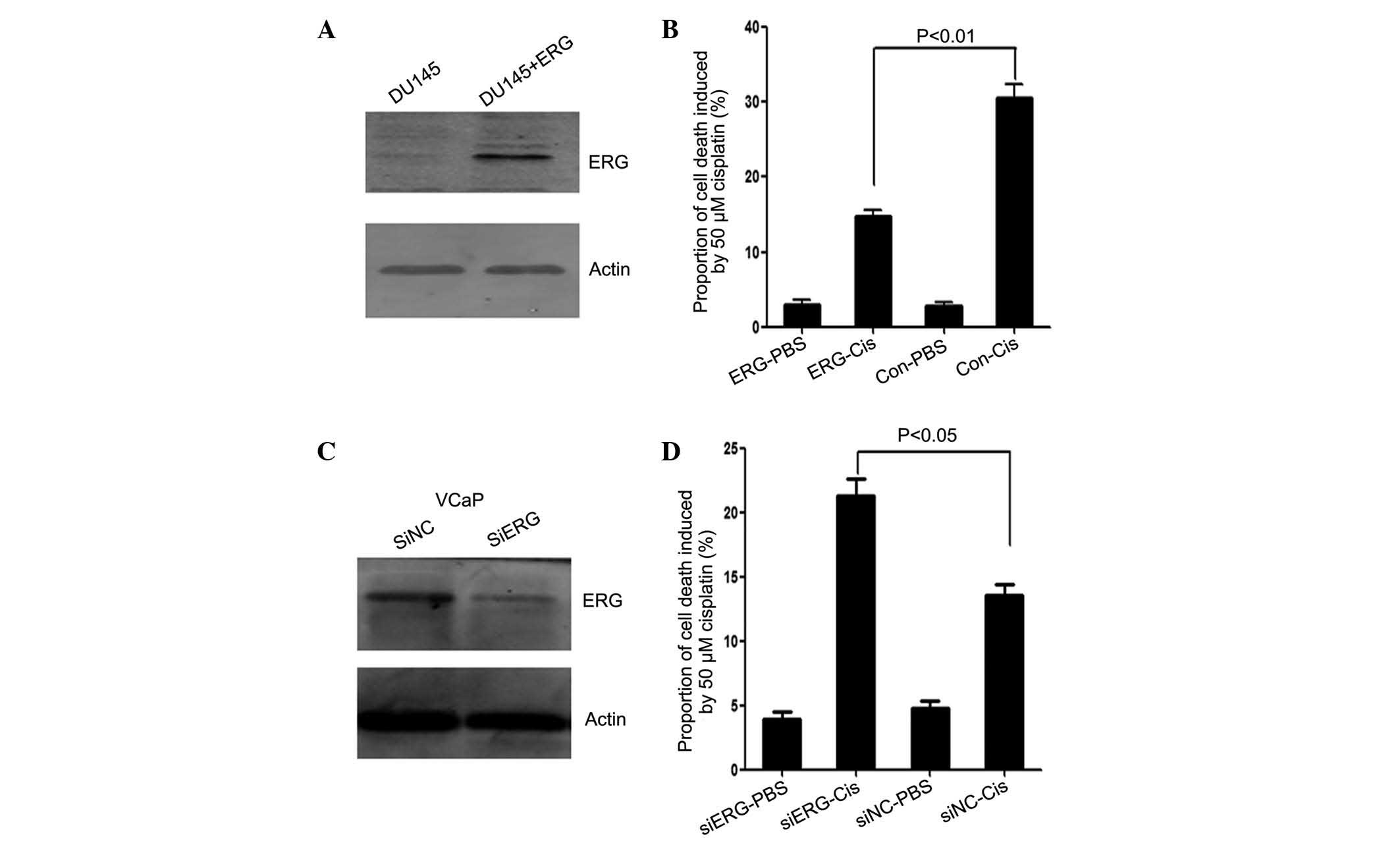 | Figure 1Effects of ΔERG on cisplatin-induced
apoptosis in DU145 and VCaP cells. At 24 h following transfection,
cells were treated with 50 µM cisplatin for 48 h, and
apoptosis was detected by 7-aminoactinomycin D and annexin-V
phycoerythrin double staining. (A) Western blotting of ΔERG protein
expression in DU145 cells transfected with GFP-vector or GFP-ΔERG.
(B) Quantitative results of cisplatin-induced apoptosis, as
measured by flow cytometry analysis in DU145 cells. (C) Western
blotting of ΔERG protein expression in VCaP cells transfected with
control or specific siRNA directed against ΔERG. (D) Quantitative
results of cisplatin-induced apoptosis as measured by flow
cytometry analysis in VCaP cells. ΔERG, truncated v-E-twenty-six
avian erythro-blastosis virus E26 oncogene homolog; GFP, green
fluorescent protein; siRNA, short interfering RNA; PBS,
phosphate-buffered saline; Cis, cisplatin; siNC, si-negative
control; Con, control. |
Expression of ΔERG renders HEK293 cells
resistant to cisplatin-induced apoptosis
The toxic effects of cisplatin on HEK293 cells were
also examined. It was demonstrated that compared with PCa cells,
HEK293 cells were more sensitive to the toxic effect of cisplatin,
as at a concentration of 1 µM marked cell death was observed
and at 100 µM, almost all the cells had undergone cell death
(data not shown). To determine whether ΔERG only has the
anti-apoptosis function in PCa cells but not in other types of
cell, a stable cell line expressing the ΔERG gene was established
using HEK 293 cells (Δ293 cells), with cells transfected with the
empty plasmid serving as controls (E293 cells). GFP fluorescence
demonstrated that the majority of the ΔERG fusion protein was
localized within the nucleus, and western blot analysis using a
commercial anti-ERG antibody confirmed the expression of ΔERG
(Fig. 2A). The effect of ΔERG on
cell growth was investigated and the results indicated that the
expression of the ΔERG gene in HEK293 cells did not affect cell
growth (data not shown). To further examine whether ΔERG does
indeed serve a role in protecting cells from cisplatin-induced
apoptosis, Δ293 cells were treated with 50 µm cisplatin for
48 h. The cell viability of E293 cells was 10% with cisplatin
treatment, while in Δ293 cells, the viability was ~46%, suggesting
that ΔERG can protect Δ293 cells from cell death induced by
cisplatin treatment (Fig. 2D).
Further analysis revealed that the apoptotic cell ratio in E293
cells was 67.26%, while in Δ293 cells it was 25.89% (P<0.01;
Fig. 2E). To verify that apoptosis
had indeed occurred, a number of apoptosis-associated proteins,
including caspase 3 and PARP-1, were examined by western blotting
following cisplatin treatment. As shown in Fig. 2F, caspase 3 and PARP-1 cleavage was
apparent in E293 cells following cisplatin treatment, however, was
less abundant in Δ293 cells under the same conditions. To further
demonstrate that ΔERG was responsible for the protective effect,
ΔERG was knocked down in Δ293 cells using specific siRNA (Fig. 2G), which indicated that loss of
ΔERG can restore the sensitivity of Δ293 cells to cisplatin
treatment (Fig. 2H). Taken
together, these data demonstrate that ∆ERG is able to protect cells
from apoptosis induced by cisplatin.
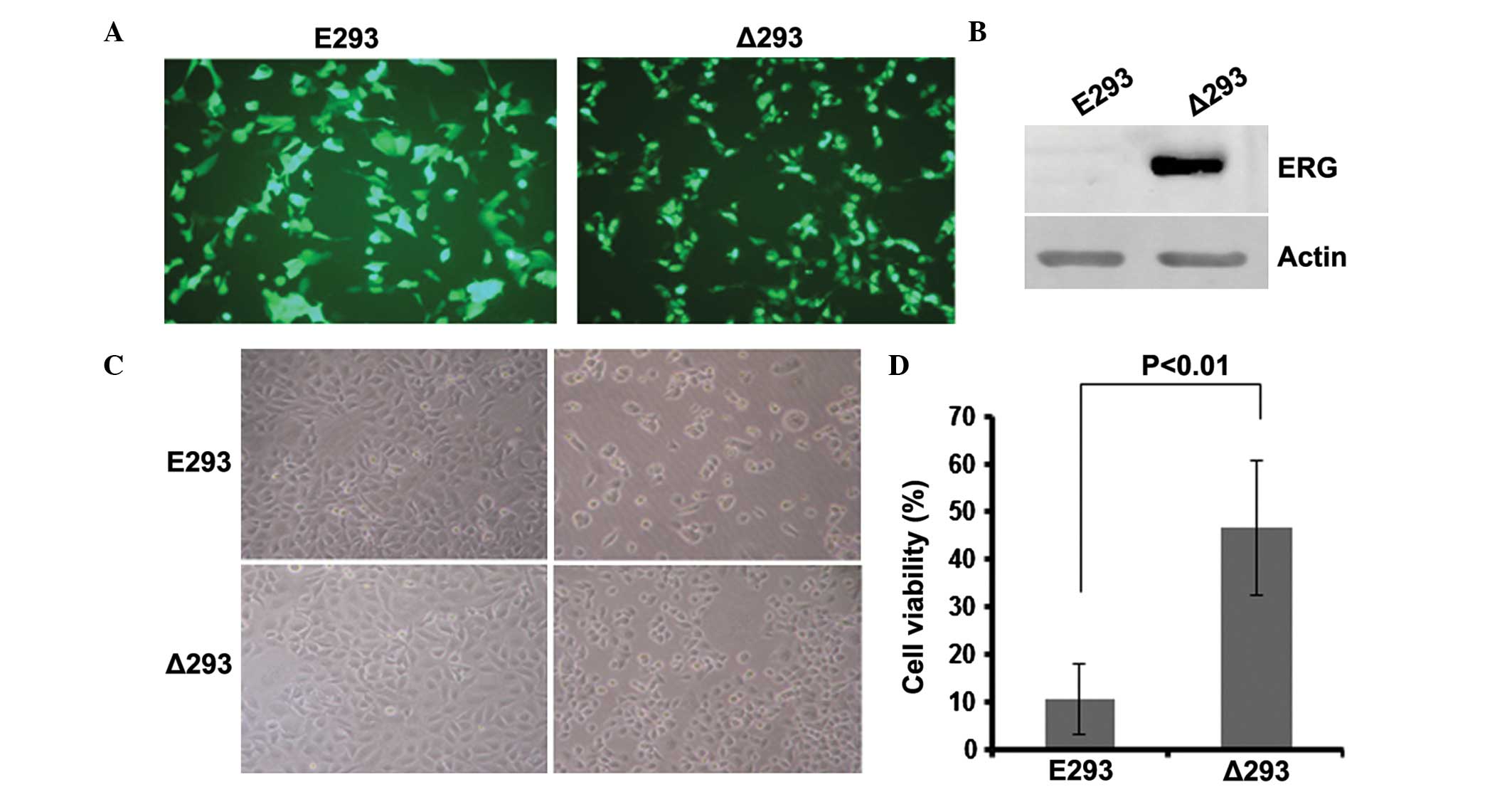 | Figure 2Effects of ΔERG overexpression on
cisplatin-induced apoptosis in HEK293 cells. (A) Fluorescent images
of the expression of ΔERG in E293 or Δ293 cells (magnification,
×200). (B) Western blotting for ΔERG expression. (C) Representative
phase contrast microscopy images of E293 and Δ293 cell density and
morphological alterations following 50 µM cisplatin
treatment for 48 h (magnification, ×200). (D) Cell viability as
measured with a Cell Counting Kit-8 kit. ΔERG, truncated
v-E-twenty-six avian erythroblastosis virus E26 oncogene homolog;
E293, p-enhanced green fluorescent vector-transfected HEK293 cells;
Δ293, ΔERG-transfected HEK293 cells. Effects of ΔERG overexpression
on cisplatin-induced apoptosis in HEK293 cells. (E) Quantitative
results of flow cytometry analyses for cell apoptosis using
7-aminoactinomycin D and annexin-V phycoerythrin double staining.
Cells were treated with 50 µM cisplatin for 48 h. (F)
Western blotting for cleaved caspase-3 and cleaved PARP-1. E293 and
Δ293 cells were treated with 50 µM cisplatin for 48 h, total
cell lysates were prepared and 10 µg aliquots of total
proteins were subjected to 15% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis. (G) Western blotting
was used to measure ΔERG protein expression in Δ293 cells
transfected with control or specific siRNA against ΔERG. (H)
Quantitative results of flow cytometric analyses for cell
apoptosis. ΔERG, truncated v-E-twenty-six avian erythroblastosis
virus E26 oncogene homolog; E293, p-enhanced green fluorescent
vector-transfected HEK293 cells; Δ293, ΔERG-transfected HEK293
cells; PARP1, poly ADP-ribose polymerase-1; siRNA, short
interfering RNA; PBS, phosphate-buffered saline; siNC, si-negative
control. |
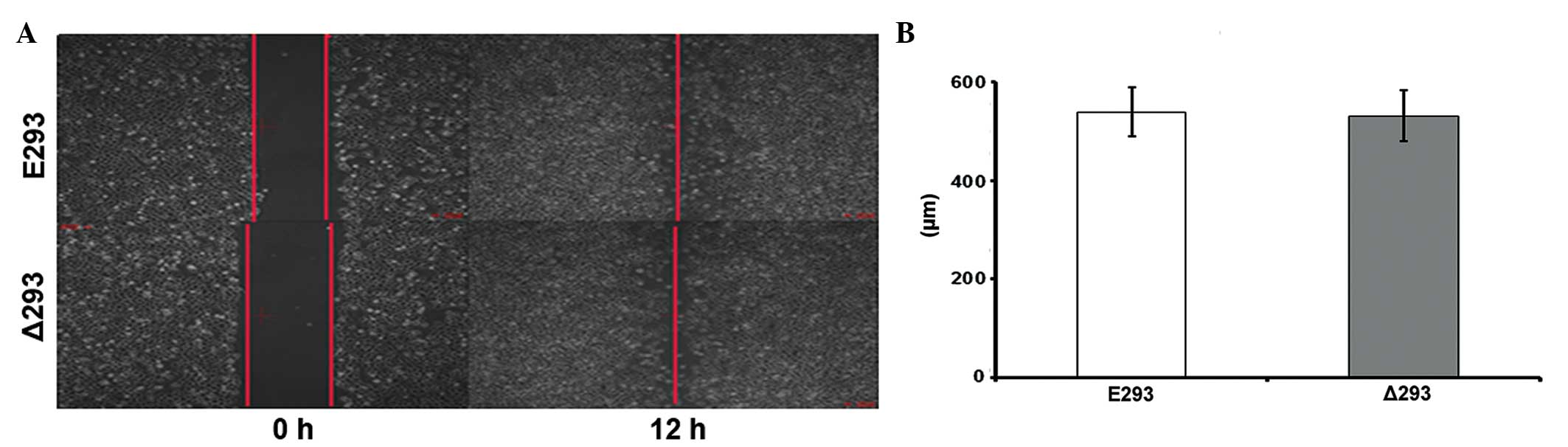
It has been previously reported that a major
function of TMPRSS2:ERG is to promote PCa cell migration and
invasion (1,14–18).
Therefore, the effect of ΔERG on cell migration was investigated
using a scratch assay. Of note, no significant difference was
observed in the scratch assay results between the Δ293 and E293
cells (Fig. 3), suggesting that
overex-pressing ΔERG did not affect cell motility, at least in
HEK293 cells.
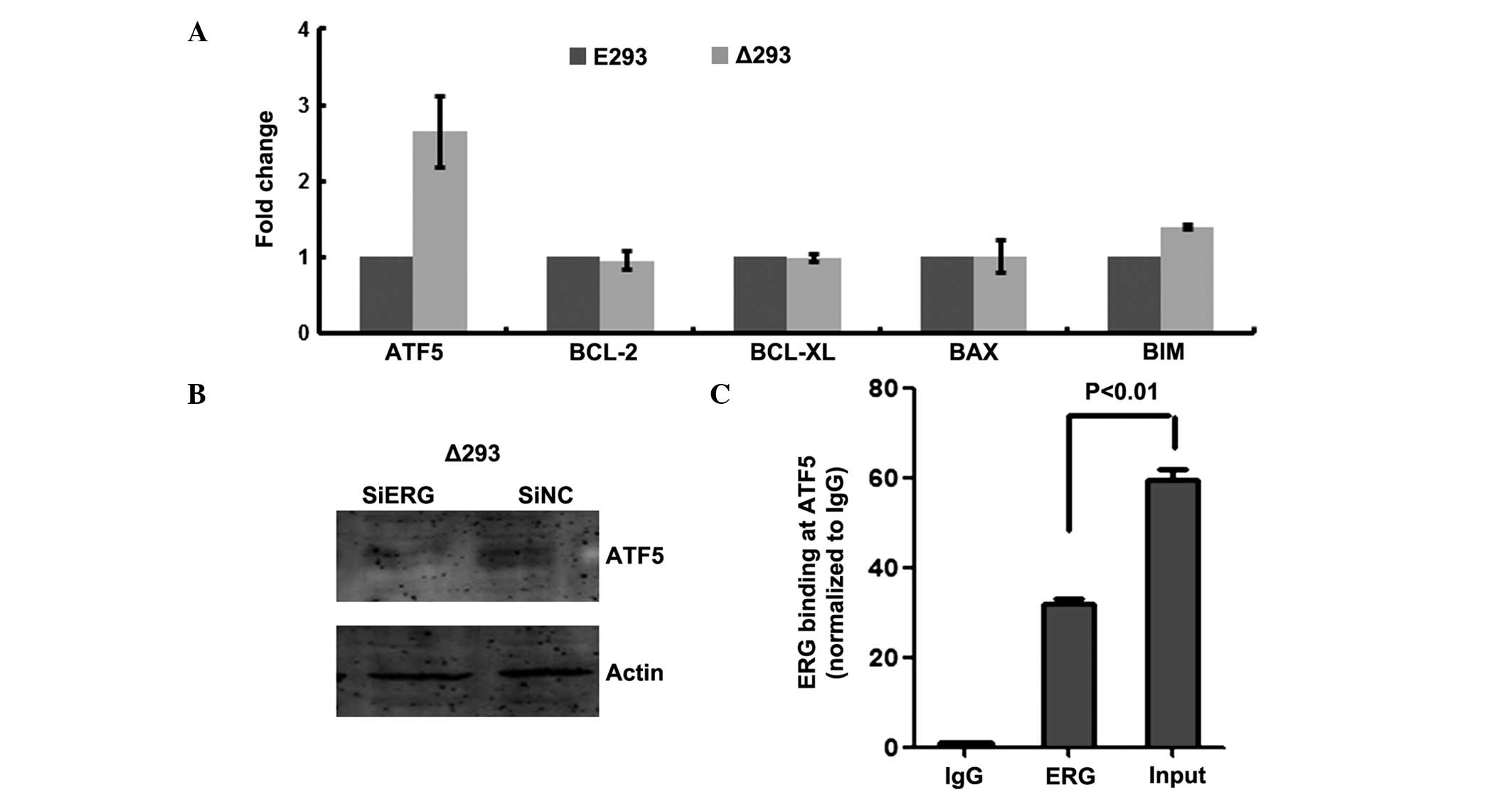
ΔERG upregulates expression of the
anti-apoptotic gene ATF5
Considering the potential anti-apoptotic function of
ΔERG, and given that the protein functions as a transcription
factor, whether it was able to influence the expression of key
regulators in apoptosis, such as members of the BCL-2 family, was
of interest. Therefore, RT-qPCR was conducted to examine the
expression of the BCL-2 family members BCL-2, BCL-XL, BIM and BAX
in the presence and absence of ΔERG. Notably, none of these genes
showed differential expression between E293 and Δ293 cells
(Fig. 4A). However, ATF5 (also
known as ATFx), a member of the basic zipper protein ATF/cAMP
response element-binding protein family which can suppress
apoptosis (19), was observed to
be up-regulated in Δ293 cells (Fig.
4A). To further examine the possibility that ATF5 expression
was regulated by ΔERG, the protein levels of ATF5 were examined by
western blotting in Δ293 cells, transfected with either control
siRNA or with siRNA designed to knockdown ΔERG. As shown in
Fig. 4B, the ATF5 protein levels
were reduced when ΔERG was knocked down, suggesting that ΔERG
serves a role in the regulation of ATF5 expression. As ERG is a
transcription factor, it was then investigated whether ΔERG is able
to bind directly to the ATF5 gene to regulate ATF5
expression. To investigate this, a ChIP assay was performed in Δ293
cells using a specific anti-ERG antibody, which indicated that ΔERG
was able to bind to the ATF5 promoter region in Δ293 cells
(Fig. 4C).
Expression of ΔERG protects cells from
cisplatin-induced DNA damage
As a classical DNA damage agent, cisplatin is
thought to induce apoptosis, at least in part, through the
generation of DNA damage. Therefore, the effect of ΔERG expression
on cisplatin-induced DNA damage was assessed. E293 and Δ293 cells
were treated with cisplatin (50 µM) for 24 h, and the level
of γH2AX, a sensitive indicator for DNA damage, was examined by
western blotting. As shown in Fig.
5, cisplatin treatment increased the level of γH2AX in the Δ293
and E293 cells. However, the increase was much smaller in the Δ293
cells (lanes 3, 4 and 5) compared with the E293 cells (lanes 8 and
9), indicating that the expression of ΔERG may partially protect
cells from cisplatin-induced DNA damage.
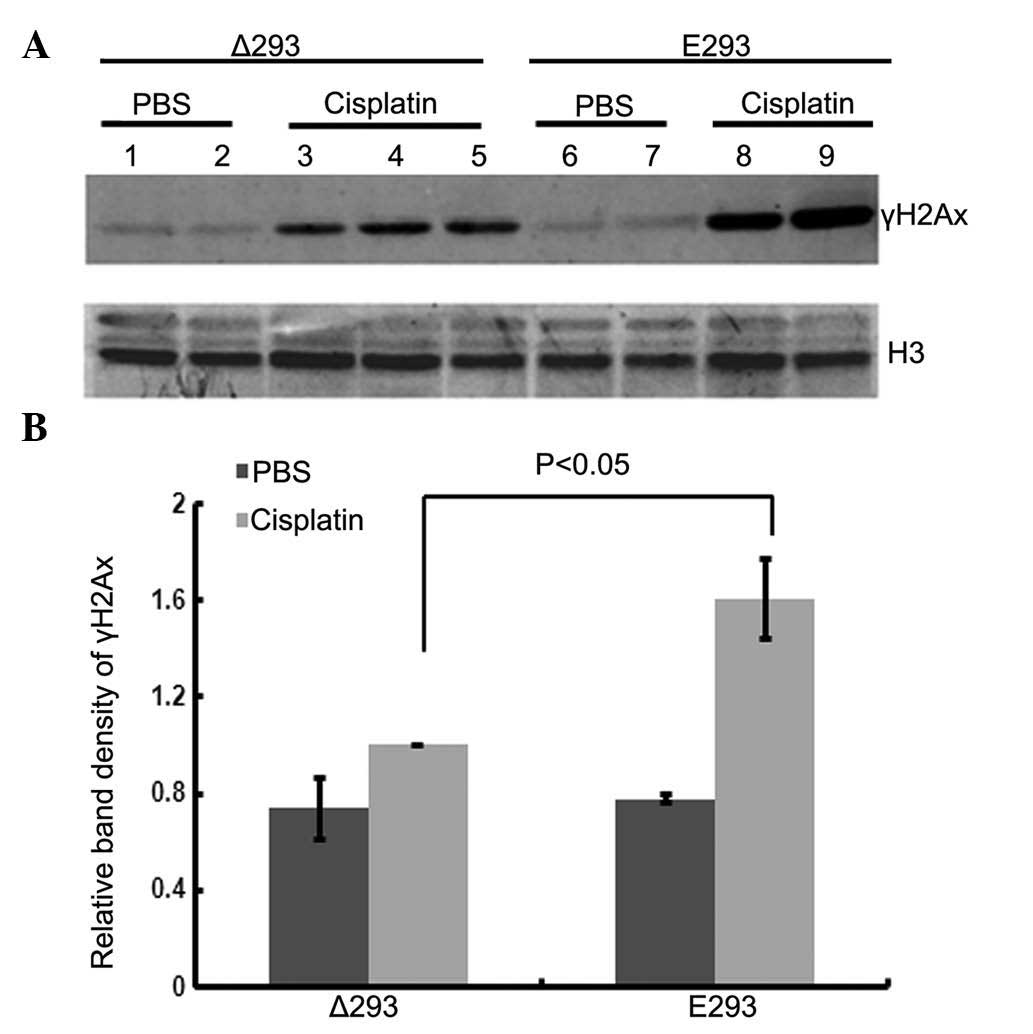 | Figure 5ΔERG overexpression reduces
cisplatin-induced DNA damage in Δ293 cells. (A) Western blotting
detection of γH2AX protein in E293 and Δ293 cells following 50
µM cisplatin treatment for 24 h. Lanes 1 and 2, Δ293 cells
treated with PBS; lanes 3, 4 and 5, Δ293 cells treated with 50
µM cispl-atin; lanes 6 and 7, E293 cells treated with PBS;
and lanes 8 and 9, E293 cells treated with 50 µM cisplatin.
(B) Densitometry analysis of western blotting results. The relative
band densities of γH2AX were normalized to H3, and the value of the
bands from Δ293 cells treated with cisplatin was arbitrarily set as
1. The values were derived from at least three independent
experiments. P<0.05 vs. E293 is indicated. ΔERG, truncated
v-E-twenty-six avian eryth-roblastosis virus E26 oncogene homolog;
Δ293, ΔERG-transfected HEK293 cells; E293, p-enhanced green
fluorescent vector-transfected HEK293 cells; PBS,
phosphate-buffered saline; H3, histone H3. |
Discussion
The potential role of TMPRSS2:ERG fusion in
prostate tumorigenesis and tumor progression has been under
intensive investigation. Enhancing cell invasion and metastasis has
been regarded as the major function of this protein, and although
overexpression of ERG was observed to induce a neoplastic phenotype
in prostate cells in certain studies (5,6), it
was thought that the protein may require the cooperation of
additional genes, such as phosphatase and tensin homolog or members
of the phosphatidylinositol 3-kinase pathway (14,15),
to result in transformation. However, its association with
apoptosis/cell death, a major mechanism for the majority of cancer
chemotherapeutic drugs, was relatively unexplored prior to the
current study. Platinum chemotherapy drugs, such as cisplatin and
carboplatin, have moderate single-agent activity in PCa (12). However, when combined with a taxane
and estramustine, high response rates were observed in several
clinical trials (20,21). Considering the high prevalence of
TMPRSS2:ERG fusion in patients with PCa, its potential role
in the relatively low response rate of PCa to cisplatin was
explored.
Indeed, PCa cells were observed to be relatively
resistant to the cytotoxic effects of cisplatin. For example, only
at doses of 50 µM or above were significant levels of cell
death induced by cisplatin in either DU145 or VCaP cells (data not
shown). By contrast, significant cell death was not induced in
HEK293 cells by cisplatin at concentrations as low as 1 µM,
and high levels of cell death was observed at 100 µM (data
not shown). Therefore, 50 µM was selected as the test
concentration for all subsequent experiments. The response of the
PCa cell lines to cisplatin treatment was investigated in DU145
cells, which do not express the fusion gene and VCaP cells, which
harbor the fusion gene. This indicated that cisplatin is able to
induce up to 30% apoptosis in DU145 cells, while the drug induces
~13% apoptosis in VCaP cells (Fig.
1). However, when ΔERG was expressed in DU145 cells, the
apoptotic cell ratio was reduced to ~15%. In comparison, knocking
down the endogenous fusion gene expressed in the VCaP cell line led
to an increased apoptotic cell ratio. Thus, this indicates that
ΔERG may influence the response of these cells to cisplatin. To
discover whether or not the anti-apoptotic effect of ΔERG was
limited to PCa cells, ΔERG was introduced into the HEK293 cells.
Notably, no significant effect of ΔERG on HEK293 cell growth or
migration was observed (data not shown and Fig. 3). These observations are in
contradiction to the report by Tian et al (7) in which it was shown that the
overexpression of TMPRSS2:ERG in PCa cells markedly
increased invasion. A possible explanation may be that different
types of cells were used, with HEK293 cells originating from the
kidney rather than the prostate, and thus may have a different
genetic background. Alternatively, an effect of ΔERG on HEK293
cells was observed with respect to apoptosis. Although cisplatin
induced apoptosis in ~70% of the HEK293 cells, which only expressed
the GFP-tag, the apoptotic cell ratio was around 30% in the Δ293
cells that expressed the ΔERG (Fig.
2). An additional piece of evidence for the anti-apoptotic
function of ΔERG comes from the observation that knocking down ΔERG
in Δ293 cells rendered the cells sensitive to cisplatin (Fig. 2). Taken together, these data
indicate an anti-apoptotic effect of ΔERG. Therefore, future
treatment planning for patients with PCa may be enhanced by
assessing the status of the TMPRSS2 gene fusion.
Once the anti-apoptotic function of the fusion gene
had been determined in the current study, the underlying molecular
mechanism was further probed. The BCL-2 family of proteins include
both anti-apoptotic (BCL-2, BCL-XL and MCL-1) and apoptotic (BAK,
BAX, BID and BIM) members (22).
The regulation and balance of the BCL-2 family proteins in a
particular cell results in either the inhibition or induction of
the apoptotic signaling pathways (23). Therefore, the expression of several
BCL-2 family members was examined. Notably, none of the BCL-2
family members examined exhibited differential expression between
the E293 and Δ293 cells (Fig. 4),
suggesting that they may not be involved in the anti-apoptotic
activity of ΔERG. However, it was observed that the expression of
ATF5, a transcriptional activator with anti-apoptotic activity, was
increased in Δ293 cells. It has been reported that when ATF5 was
stably expressed in an IL-3-dependent cell line, apoptosis was
suppressed through cytokine deprivation (24). Additionally, overexpression of ATF5
suppressed apoptosis induced by serum withdrawal in HeLa cells
(25). By contrast, inhibition of
endogenous ATF5 activity by the introduction of a dominant-negative
form of ATF5 led to apop-tosis in asynchronously growing cells
(24,25). To determine whether ΔERG was
involved in the regulation of ATF5 expression, ΔERG was knocked
down in Δ293 cells, and the protein levels of ATF5 were examined by
western blotting. As shown in Fig.
4, knock down ΔERG resulted in a reduction in ATF5 expression.
Subsequently, ChIP analysis was performed, and the results
indicated that ΔERG is able to bind directly to the promoter region
of ATF5 (Fig. 4). Based on these
observations, ΔERG is proposed to potentially upregulate the
expression of ATF5, which in turn exerts anti-apoptotic activities
through the activation of downstream signaling pathways.
Finally, since cisplatin-induced DNA damage is
considered to be a major factor in triggering apoptosis, it was
examined whether ΔERG is able to influence cisplatin-induced DNA
damage. γH2AX is regarded to be a sensitive indicator for DNA
damage (26). The results of the
present study indicated that expression of the γH2AX protein was
significantly reduced in Δ293 cells, suggesting that overexpression
of ΔERG may protect cells from cisplatin-induced DNA damage
(Fig. 5). The role of
TMPRSS2:ERG in the DNA damage process remains controversial.
TMPRSS2:ERG has been previously shown to interact with the
enzyme PARP1 and the catalytic subunit of DNA protein kinase in a
DNA-dependent manner, and overexpression of similar fusion genes
induces DNA double-strand breaks (27). In addition, Han et al
(28) observed that overexpression
of TMPRSS2:ERG increases PARP1 activity and clonogenic
survival (28). Haffner et
al (29) reported that
androgen signaling promotes co-recruitment of the androgen receptor
and topoi-somerase II beta (TOP2B) to sites of TMPRSS2:ERG
genomic breakpoints, triggering recombinogenic TOP2B-mediated DNA
double-strand breaks (DSBs). Furthermore, androgen stimulation
resulted in de novo production of TMPRSS2:ERG fusion
transcripts in a process that required TOP2B and components of the
DSB repair machinery (29). Thus,
how ΔERG may interfere with the DNA damage process remains clear,
and the potential link between ΔERG and DNA damage is of great
interest for future studies.
In summary, the current study indicated that the
ΔERG protein is able to inhibit cisplatin-induced DNA damage and
cell death, and that the ATF5 gene may participate in such
events.
Acknowledgments
The current study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81172692,
81302398, 81502752 and 81373036), Department of Science and
Technology, Zhejiang Province (grant no. 2013C14016), the Hangzhou
City S&T Committee (grant no. 20130633B50) and the Zhejiang
Provincial Natural Science Foundation (grant no. Q16H220001).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics for Hispanics/Latinos, 2012. CA Cancer J Clin.
62:283–298. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tomlins SA, Rhodes DR, Perner S,
Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J,
Kuefer R, et al: Recurrent fusion of TMPRSS2 and ETS transcription
factor genes in prostate cancer. Science. 310:644–648. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Falzarano SM and Magi-Galluzzi C: ERG
protein expression as a biomarker of prostate cancer. Biomark Med.
7:851–865. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Helgeson BE, Tomlins SA, Shah N, Laxman B,
Cao Q, Prensner JR, Cao X, Singla N, Montie JE, Varambally S, et
al: Characterization of TMPRSS2:ETV5 and SLC45A3:ETV5 gene fusions
in prostate cancer. Cancer Res. 68:73–80. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tomlins SA, Laxman B, Varambally S, Cao X,
Yu J, Helgeson BE, Cao Q, Prensner JR, Rubin MA, Shah RB, et al:
Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia.
10:177–188. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Klezovitch O, Risk M, Coleman I, Lucas JM,
Null M, True LD, Nelson PS and Vasioukhin V: A causal role for ERG
in neoplastic transformation of prostate epithelium. Proc Natl Acad
Sci USA. 105:2105–2110. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tian TV, Tomavo N, Huot L, Flourens A,
Bonnelye E, Flajollet S, Hot D, Leroy X, de Launoit Y and
Duterque-Coquillaud M: Identification of novel TMPRSS2:ERG
mechanisms in prostate cancer metastasis: Involvement of MMP9 and
PLXNA2. Oncogene. 33:2204–2214. 2014. View Article : Google Scholar
|
|
8
|
Shao L, Zhou Z, Cai Y, Castro P, Dakhov O,
Shi P, Bai Y, Ji H, Shen W and Wang J: Celastrol suppresses tumor
cell growth through targeting an AR-ERG-NF-kB pathway in
TMPRSS2/ERG fusion gene expressing prostate cancer. PLoS One.
8:e583912013. View Article : Google Scholar
|
|
9
|
Loehrer PJ and Einhorn LH: Drugs five
years later. Cisplatin Ann Intern Med. 100:704–713. 1984.
View Article : Google Scholar
|
|
10
|
Prestayko AW, D'Aoust JC, Issell BF and
Crooke ST: Cisplatin (cis-diamminedichloroplatinum II). Cancer
Treat Rev. 6:17–39. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Eastman A: The formation, isolation and
characterization of DNA adducts produced by anticancer platinum
complexes. Pharmacol Ther. 34:155–166. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Oh WK, Tay MH and Huang J: Is there a role
for platinum chemotherapy in the treatment of patients with
hormone-refractory prostate cancer? Cancer. 109:477–486. 2007.
View Article : Google Scholar
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
14
|
Carver BS, Tran J, Gopalan A, Chen Z,
Shaikh S, Carracedo A, Alimonti A, Nardella C, Varmeh S, Scardino
PT, et al: Aberrant ERG expression cooperates with loss of PTEN to
promote cancer progression in the prostate. Nat Genet. 41:619–624.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
King JC, Xu J, Wongvipat J, Hieronymus H,
Carver BS, Leung DH, Taylor BS, Sander C, Cardiff RD, Couto SS, et
al: Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation
in prostate oncogenesis. Nat Genet. 41:524–526. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Esgueva R, Perner S, J LaFargue C, Scheble
V, Stephan C, Lein M, Fritzsche FR, Dietel M, Kristiansen G and
Rubin MA: Prevalence of TMPRSS2-ERG and SLC45A3-ERG gene fusions in
a large prostatectomy cohort. Mod Pathol. 23:539–546. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Galluzzi L, Aaronson SA, Abrams J, Alnemri
ES, Andrews DW, Baehrecke EH, Bazan NG, Blagosklonny MV, Blomgren
K, Borner C, et al: Guidelines for the use and interpretation of
assays for monitoring cell death in higher eukaryotes. Cell Death
Differ. 16:1093–1107. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Galluzzi L, Vitale I, Abrams JM, Alnemri
ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry
WS, Fulda S, et al: Molecular definitions of cell death
subroutines: Recommendations of the nomenclature committee on cell
death 2012. Cell Death Differ. 19:107–120. 2012. View Article : Google Scholar :
|
|
19
|
Hai TW, Liu F, Coukos WJ and Green MR:
Transcription factor ATF cDNA clones: An extensive family of
leucine zipper proteins able to selectively form DNA-binding
heterodimers. Genes Dev. 3:2083–2090. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Papandreou CN, Daliani DD, Thall PF, Tu
SM, Wang X, Reyes A, Troncoso P and Logothetis CJ: Results of a
phase II study with doxorubicin, etoposide, and cisplatin in
patients with fully char-acterized small-cell carcinoma of the
prostate. J Clin Oncol. 20:3072–3080. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Berry W, Friedland D, Fleagle J, Jackson
D, Ilegbodu D, Boehm KA and Asmar L: A phase II study of weekly
paclitaxel/estramustine/carboplatin in hormone-refractory prostate
cancer. Clin Genitourin Cancer. 5:131–137. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chipuk JE, Moldoveanu T, Llambi F, Parsons
MJ and Green DR: The BCL-2 family reunion. Mol Cell. 37:299–310.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Harris MH and Thompson CB: The role of the
Bcl-2 family in the regulation of outer mitochondrial membrane
permeability. Cell Death Differ. 7:1182–1191. 2000. View Article : Google Scholar
|
|
24
|
Persengiev SP and Green MR: The role of
ATF/CREB family members in cell growth, survival and apoptosis.
Apoptosis. 8:225–228. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Persengiev SP, Devireddy LR and Green MR:
Inhibition of apoptosis by ATFx: A novel role for a member of the
ATF/CREB family of mammalian bZIP transcription factors. Genes Dev.
16:1806–1814. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhou C, Li Z, Diao H, Yu Y, Zhu W, Dai Y,
Chen FF and Yang J: DNA damage evaluated by gammaH2AX foci
formation by a selective group of chemical/physical stressors.
Mutat Res. 604:8–18. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brenner JC, Ateeq B, Li Y, Yocum AK, Cao
Q, Asangani IA, Patel S, Wang X, Liang H, Yu J, et al: Mechanistic
rationale for inhibition of poly (ADP-ribose) polymerase in ETS
gene fusion-positive prostate cancer. Cancer Cell. 19:664–678.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Han S, Brenner JC, Sabolch A, Jackson W,
Speers C, Wilder-Romans K, Knudsen KE, Lawrence TS, Chinnaiyan AM
and Feng FY: Targeted radiosensitization of ETS fusion-positive
prostate cancer through PARP1 inhibition. Neoplasia. 15:1207–1217.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Haffner MC, Aryee MJ, Toubaji A, Esopi DM,
Albadine R, Gurel B, Isaacs WB, Bova GS, Liu W, Xu J, et al:
Androgen-induced TOP2B-mediated double-strand breaks and prostate
cancer gene rearrangements. Nat Genet. 42:668–675. 2010. View Article : Google Scholar : PubMed/NCBI
|