Introduction
As a metabolic disorder, diabetes has been defined
as an independent risk factor for various types of cardiovascular
disease. It has been recognized that endothelial cell dysfunction
and apoptosis represent the beginning of diabetes-associated
vascular disease (1,2). The maintenance of the balance between
the proliferation, apoptosis and necrosis of endothelial cells is
multi-factorial and involves the interaction between vascular
endothelial growth factor (VEGF) and tumor necrosis factor-like
cytokine 1A (TL1A).
VEGF, considered as the most important cytokine in
promoting endothelial cell growth, exhibits a crucial role in
maintaining the stability of vascular endothelial structure and
function by stimulating endothelial cell proliferation and
regeneration while restraining apoptosis. Elevated VEGF levels are
observed as a cardiac response under conditions of
ischemia-hypoxia, while impairments in VEGF expression and activity
have been identified in patients with diabetes (3–5). It
has been reported that patients without diabetes exhibited
increased VEGF expression in response to myocardial ischemia and
infarction but that diabetic patients exhibited reduced VEGF levels
in the myocardium and in chronic wounds (6,7). The
downregulation of VEGF expressed in tissues, aside from the kidney
and the retina, contributes to the decrease in ischemia-induced
collateral vessel formation in patients with diabetes, resulting in
necrosis and irreversible damage (3). Thus, VEGF is a key component in the
mechanism that regulates endothelial cell proliferation and
neovascularization.
TL1A is a unique endogenous inhibitor of
angiogenesis (8,9) that is predominantly produced by
endothelial cells and that performs its physiological functions by
binding to its receptors, DR3 and DcR3 (10). As a multifunctional member of the
tumor necrosis factor super-family, TL1A participates in numerous
physiological processes, such as the immune response (11), and in pathologies such as
inflammatory bowel disease (12)
and atherosclerosis (13,14). It has been widely accepted that
TL1A exerts an inhibitory effect on endothelial cell proliferation
during different growth stages by promoting apoptosis or growth
arrest (15). Previous experiments
revealed that senescent human umbilical vein endothelial cells
(HUVECs) displayed enhanced TL1A expression (16). Additionally, TL1A inhibits tumor
angiogenesis and tumor growth, and these effects are consistent
with the observation that TL1A expression is significantly
decreased in various types of cancer, such as ovarian (17) and breast cancer (18), as well as in wound tissue (19). Thus, TL1A functions as a suppressor
of neovascularization and endothelial cell proliferation.
As important components of the mechanisms regulating
neovascularization and cell proliferation, the balance between TL1A
and VEGF ensures the stability of the established vasculature. The
present study investigated TL1A and VEGF expression in high
glucose-induced apoptotic HUVECs, with the aim of elucidating the
mechanism by which TL1A and VEGF act on HUVECs. The experiments
aimed to provide a novel strategy to protect endothelial cells from
high glucose-induced apoptosis.
Materials and methods
Chemicals and reagents
Anti-glyceraldehyde 3-phosphate dehydrogenase
(GAPDH) mouse monoclonal antibody (mAb; HRP-60004) and the
horseradish peroxidase (HRP)-conjugated Affinipure goat anti-mouse
(SA00001-1) and anti-rabbit (SA00001-2) secondary immunoglobulin
(Ig)G (H+L) antibodies were purchased from Proteintech Group
(Wuhan, China). Anti-phospho-phosphoinositide 3-kinase (PI3K) p85
(Tyr458)/p55 (Tyr199) rabbit polyclonal antibody (pAb; 4228), the
anti-PI3K p85 rabbit pAb (4257), the anti-phospho-Akt (Ser473)
rabbit mAb (4060), the anti-Akt rabbit mAb (4691), the
anti-phospho-endothelial nitric oxide synthase (eNOS) (Ser1177)
rabbit pAb (9571), and the anti-eNOS rabbit pAb (9572) were
purchased from Cell Signaling Technologies Inc. (Beverly, MA, USA).
Anti-VEGF mouse pAb (ab46154) and anti-TL1A rabbit pAb (ab21272)
were purchased from Abcam (Cambridge, UK). Endothelial cell medium
(ECM) was purchased from ScienCell Research Laboratories (San
Diego, CA, USA).
Cell culture
The HUVECs used in this study were purchased from
ScienCell Research Laboratories. The cells were cultured in ECM at
37°C in a humidified incubator containing 5% CO2. Cultured
endothelial cells of 3–5 passages were used for the following
experiments. HUVECs were cultured in conditioned medium with
various concentrations of glucose (5.5, 15 and 33 mM/l) for various
time periods (0, 24 and 48 h). Furthermore, HUVECs were transfected
with TL1A siRNA, negative control siRNA or VEGF DNA. Following
transfection for 12 h, cells were cultured in conditioned medium
with 5.5 mM/l NG or 33 mM/l HG for 0, 24 or 48 h.
Western blot analysis
Western blot analysis was performed as follows:
Total proteins were extracted from cells cultured in a 6-well plate
(3×105 cells/well) in triplicate using radioimmunoprecipitation
assay buffer (Invitrogen; Thermo Fisher Scientific Inc., Waltham,
MA, USA). After centrifugation at 16,100 × g at 4°C, the
supernatant was collected, and the protein concentration was
quantified using a bicinchoninic acid protein assay kit (Pierce,
Rockford, IL, USA). Then, 30 µg of each total protein
extract was separated via 8% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis gels (Amresco, LLC, Solon, OH, USA) and
transferred to a polyvinylidene difluoride membrane (Roche
Diagnostics, Indianapolis, IN, USA). The membranes were blocked in
5% milk or 5% bovine serum albumin (Invitrogen; Thermo Fisher
Scientific, Inc.), depending on the level of background, and then
probed with the following primary antibodies overnight at 4°C:
Mouse anti-VEGF (1:500) pAb and rabbit
anti-phospho-phosphoinositide 3-kinase (PI3K) p85 (Tyr458)/p55
(Tyr199) pAb, anti-PI3K p85, anti-phospho-Akt (Ser473), anti-Akt,
anti-phospho-endothelial nitric oxide synthase (eNOS) (Ser1177),
anti-eNOS (all 1:1,000) and anti-TL1A (1:500) and mouse anti-GAPDH
(1:10,000). The following day, the membranes were washed three
times with 1X Tris-buffered saline with Tween-20 and incubated in
HRP-conjugated Affinipure goat anti-mouse (1:1,000) and anti-rabbit
(1:2,000) secondary IgG (H+L) antibodies secondary antibodies for 1
h at room temperature. Protein signals were visualized using
enhanced chemiluminescence (Millipore, Boston, MA, USA), and the
gray analysis of bands were relatively quantified using Quantity
One software (version 4.62; Bio-Rad Laboratories, Hercules, CA,
USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from HUVECs using TRIzol
reagent (Sigma-Aldrich, St. Louis, MO, USA) according to the
manufacturer's instructions, and the RNA concentrations were
measured using a NanoDrop 2000 (Thermo Fisher Scientific Inc.). RNA
was reverse transcribed as follows: 4µl sample RNA (0.5
µg/µl), 1 µl random primer (25 µM) and
1 µl RNase-free dH2O (both Bio-Rad Laboratories, Inc.) were
mixed together in a 0.2 ml Eppendorf tube, incubated at 70°C for 10
min and immediately cooled on ice for ≥2 min. Following
centrifugation at 3,000 × g for several seconds, 0.25 µl
RTase M-MLV, 2 µl M-MLV buffer (5X), 0.5 µl dNTPs
(2.5 mM each), 0.25 µl RNase inhibitor (40 u/µl; all
Bio-Rad Laboratories, Inc.) and 1 µl RNase-free dH2O was
added and incubated at 30°C for 10 min, followed by 1 h at 42°C and
15 min at 70°C prior to cooling on ice. qPCR for VEGF and TL1A was
performed using a qPCR kit (SYBR Green; Roche, Ltd., Basel,
Switzerland). The primer sequences used were as follows: Forward:
5′-TCTCTACCCCAGGTCAGACG-3′ and reverse: 5′-TCCCCAAACTCCTGGTCAGA-3′
for VEGF; forward: 5′-TGTGCAGTTCCAGGCTCTAAAA-3′ and reverse:
5′-CCGTCTGCTCTAAGAGGTGCAT-3′ for TL1A; and forward:
5′-GGTGGTCTCCTCTGACTTCAACA-3′ and reverse: 5′-GTT GCT GTA GCC AAA
TTC GTT GT-3′ for GAPDH (Invitrogen; Thermo Fisher Scientific
Inc.). The reverse transcription conditions were as follows: 95°C
for 30 sec for pre-incubation, followed by 45 cycles of 50°C for 40
sec and 72°C for 40 sec. Transcript amplification and detection
were performed using a LightCycler480 thermal cycler (Roche
Diagnostics GmbH, Manheim, Germany) as follows: Pre-incubation for
5 min at 95°C, amplification via 45 cycles of 10 sec at 95°C, 10
sec at 60°C and 20 sec at 72°C, and a final cooling step of 30 sec
at 40°C (ramp rate of 4.4°C/s). The relative quantity of each gene
was obtained using the ΔΔCq method (20).
Gene silencing
RNA interference was performed using TL1A small
interfering (si)RNA and control siRNA (RiboBio, Guangzhou, China).
Cell cultures at 40–60% confluence were transfected with siRNA at a
final concentration of 50 nM using Lipofectamine 2000 (Invitrogen;
Thermo Fisher Scientific Inc.) according to the manufacturer's
instructions. After transfection (12 h), HUVECs were used for
experimentation.
VEGF overexpression
The cells were seeded in 6-well plates (3×105
cells/well) and were incubated for 24 h prior to transfection. VEGF
DNA was transfected into the HUVECs at a final concentration of 3
µg/µl using Lipofectamine 2000. Following
transfection for 4–6 h, the transfection medium was replaced with
ECM and culturing was continued for 24 h prior to further
treatment.
Apoptosis assay via flow cytometry
(FCM)
Apoptosis was measured using an Annexin
V-fluorescein isothiocyanate (FITC) Apoptosis Assay kit (Nanjing
KeyGen Biotech. Co. Ltd., Nanjing, China) according to the
manufacturer's instructions. After transfection with TL1A siRNA or
VEGF DNA, the HUVECs incubated in high glucose(33 mM/l) for various
time periods (0, 24 or 48 h) were harvested and stained with
Annexin V-FITC and propidium iodide (PI; 1:100 BD Biosciences;
Franklin Lakes, NJ, USA) for 15 min at room temperature in the
dark. Following incubation, flow cytometry (FACS Calbur; BD
Biosciences) was performed to detect the apoptosis of HUVECs. The
axes were set as Annexin V on the horizontal axis and PI on the
vertical axis, in order to determine the baseline of the axes
according to the blank control and distinguish normal cells from
apoptotic cells. Four-quadrant were used; the left lower quadrant
(Annexin V−-PI−) represented normal cells,
the left upper quadrant (Annexin V−-PI+)
represents fragmented or injured cells, the right lower quadrant
(Annexin V+-PI−) represented viable apoptotic
cells, the right upper quadrant(Annexin
V+-PI+) represented non-viable apoptotic or
non-viable non-apoptotic cells. Both viable apoptotic and
non-viable apoptotic cells or non-viable non-apoptotic cells were
used to calculate cell apoptosis.
Statistical analysis
All experiments were performed at least three times
and all data are expressed as the mean ± standard deviation.
Statistical analysis of the data was performed via one-way analysis
of variance using SPSS 13.0 software (SPSS, Inc., Chicago, IL,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
TL1A expression is induced in HUVECs in
response to high glucose
To determine whether TL1A is induced in response to
high glucose, HUVECs were treated with different concentrations of
glucose (5.5, 15 or 33 mM) for different periods (0, 24 or 48 h)
and the changes in TL1A expression were determined via RT-qPCR and
western blot analysis. As shown in Fig. 1A and B, TL1A expression at the mRNA
and protein levels was induced by high glucose, resulting in a
significant increase compared with the controls at 24 and 48 h.
These results demonstrated that high glucose induces TL1A
expression in HUVECs in a concentration-dependent manner.
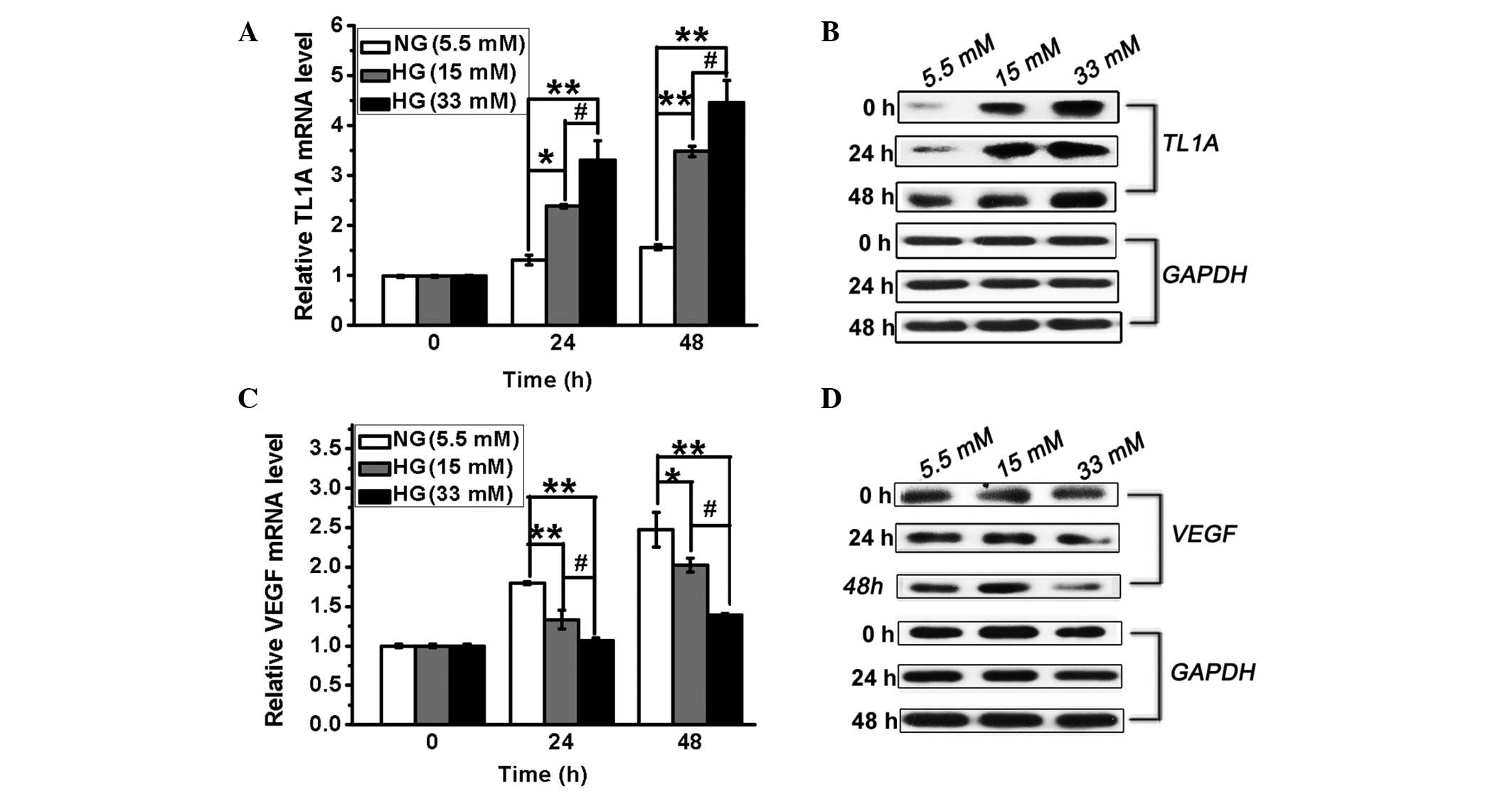 | Figure 1High glucose levels result in
upregulation of TL1A expression and downregulation of VEGF
expression in HUVECs. HUVECs were cultured in conditioned medium
with different concentrations of glucose (5.5, 15 and 33 mM/l).
TL1A mRNA and protein levels were analyzed by (A) RT-qPCR and (B)
western blot analysis at different time intervals, respectively.
VEGF mRNA and protein levels were analyzed by (C) RT-qPCR and (D)
western blot analysis at different time intervals, respectively.
GAPDH served as a control. NG, normal glucose. HG, high glucose;
HUVEC, human umbilical vein endothelial cell; TL1A, tumor necrosis
factor-like cytokine 1A; VEGF, vascular endothelial growth factor;
RT-qPCR, reverse transcription-quantitative polymerase chain
reaction; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
*P<0.05, **P<0.01 vs. NG. #P<0.05 vs. HG. |
VEGF expression is reduced in HUVECs in
response to high glucose
Different VEGF-regulating mechanisms resulted in
distinct levels of VEGF expression between retinal and cardiac
tissue in response to high glucose (3). A previous study has shown that VEGF
expression was increased by 2-fold in the retina and in glomeruli
in diabetes (3). To evaluate VEGF
expression in HUVECs under hyperglycemic conditions, the HUVECs
were treated as described above and the expression of VEGF was
measured by RT-qPCR and western blot analysis. As shown in Fig. 1C and D, VEGF expression was
decreased in the presence of high glucose compared with normal
glucose. Thus, VEGF expression is inhibited in HUVECs cultured in
the presence of high glucose.
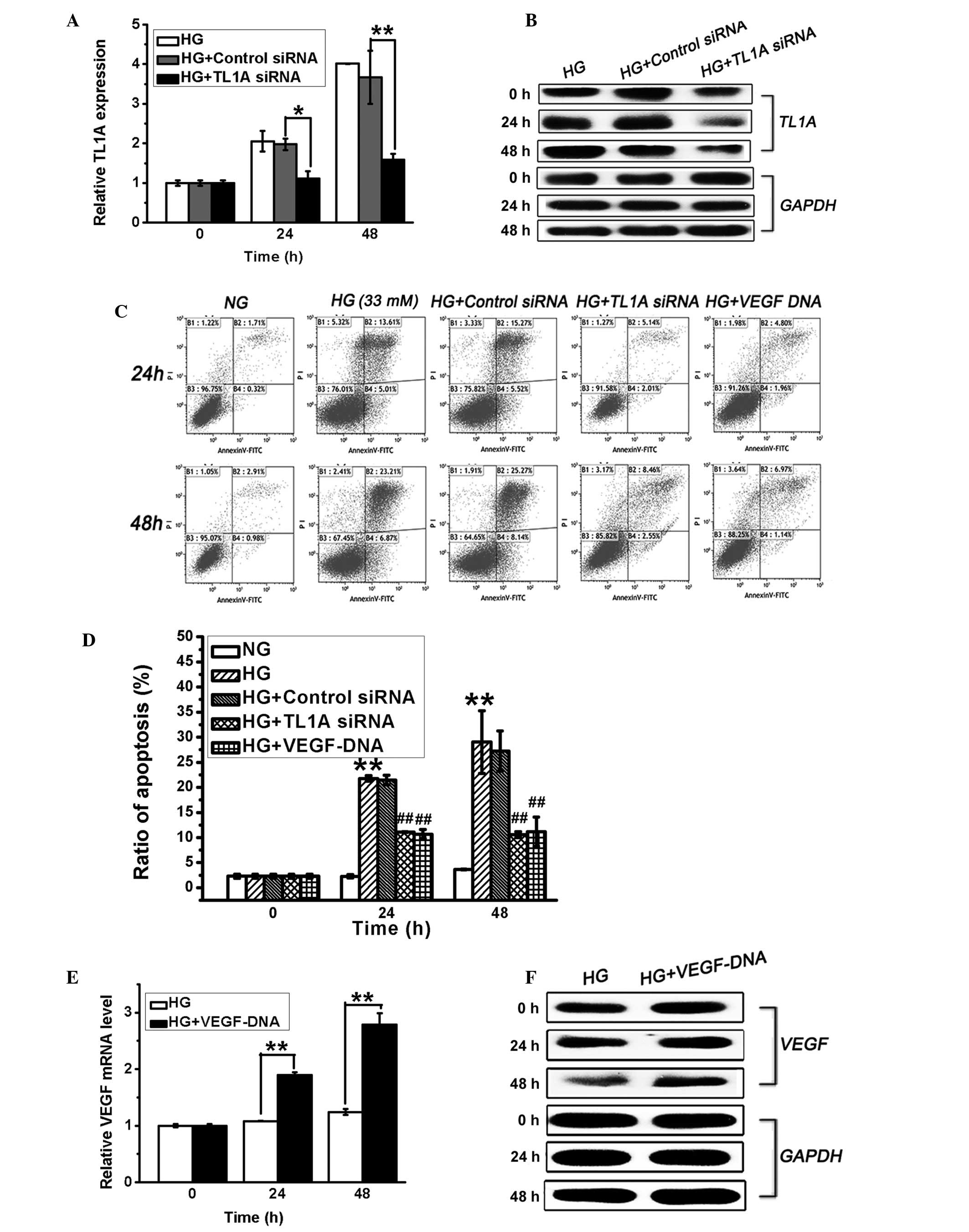
Increase in TL1A expression and the
decrease in VEGF expression promotes HUVEC apoptosis in response to
high glucose
TL1A and VEGF expression levels in HUVECs displayed
opposing changes in response to the high glucose stimulus, and it
has previously been demonstrated that high glucose levels induce
endothelial cell apoptosis (21).
To determine the association between the changes in TL1A and VEGF
expression and the induction of HUVEC apoptosis in hyperglycemia,
HUVECs were transfected with VEGF DNA (3 µg/µl) or
TL1A siRNA (50 nM). Apoptosis of these cells was then evaluated via
flow cytometry. As shown in Fig. 2A
and B, TL1A siRNA effectively suppressed high glucose-induced
TL1A expression at the mRNA and protein levels after stimulation in
high glucose for 24 and 48 h. Furthermore, the silencing of high
glucose-induced TL1A expression significantly attenuated high
glucose-induced cell apoptosis (Fig.
2C and D). Furthermore, the transfection of VEGF DNA, which
increased the VEGF levels (Fig. 2E and
F) in HUVECs, also protected against high glucose-induced cell
apoptosis (Fig. 2C and D). The
transfection of HUVECs with negative control siRNA exerted no
effect on TL1A expression or cell apoptosis (Fig. 2). These findings demonstrated that
VEGF is protective against high glucose-induced HUVEC apoptosis and
that high glucose-induced TL1A expression promotes HUVEC
apoptosis.
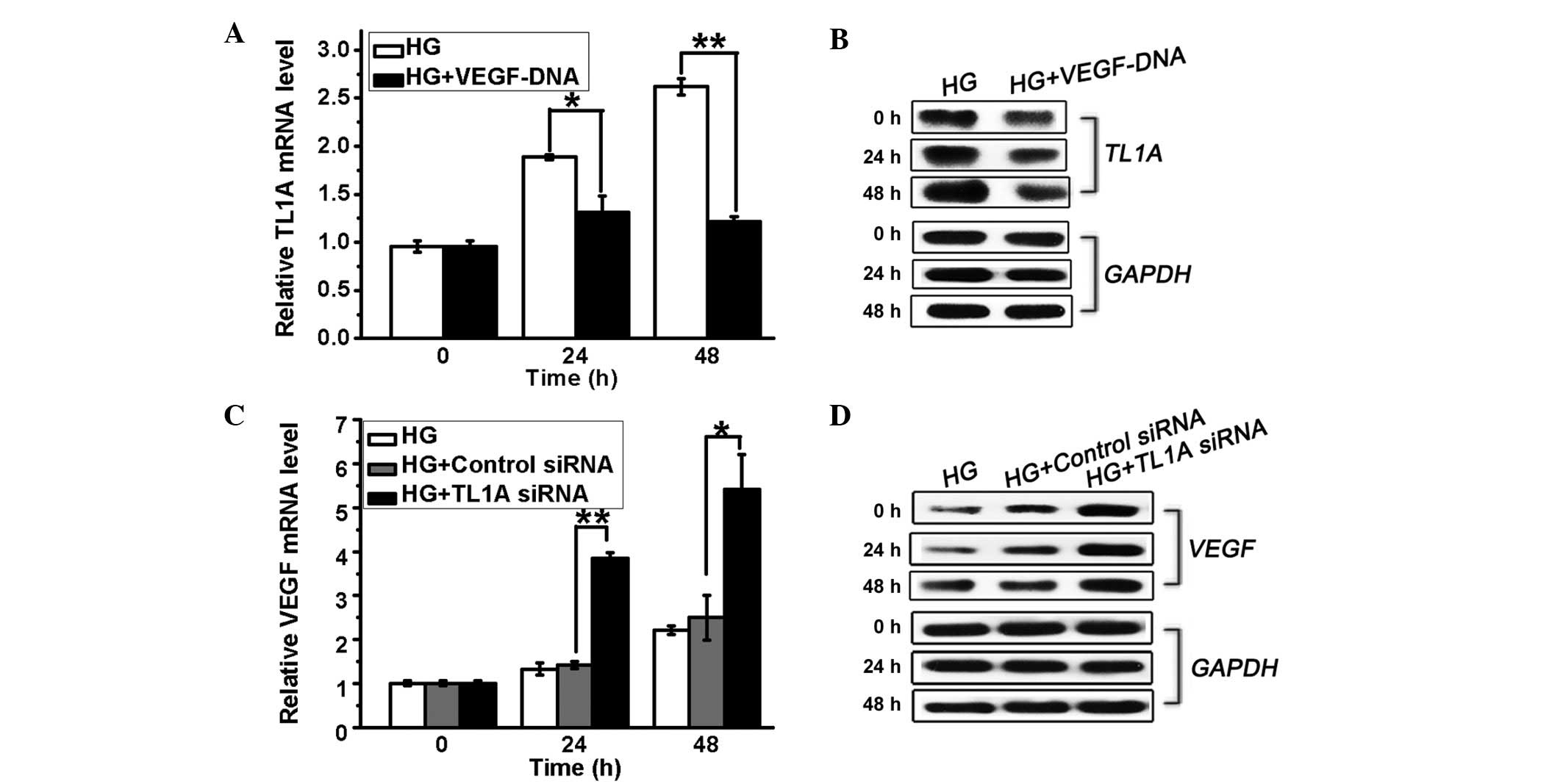
TL1A and VEGF are mutually inhibited in
hyperglycemia
A previous study indicated that VEGF expressed in
cancer cells is responsible for the downregulation of TL1A in
ovarian cancer (17). In order to
investigate the correlation between TL1A and VEGF expression in
response to high glucose, HUVECs were transfected with TL1A siRNA
or VEGF DNA and cultured in high glucose. It was demonstrated that
high glucose-induced TL1A expression was inhibited by VEGF
overexpression (Fig. 3A and B). By
contrast, the HUVECs transfected with TL1A siRNA (50 nM) displayed
increased VEGF expression at the mRNA and protein levels compared
with the control cells (Fig. 3C and
D). Thus, high glucose-induced TL1A expression further inhibits
VEGF expression, whereas the high glucose-induced downregulation of
VEGF production results in a weakened inhibitory effect of VEGF on
TL1A accumulation, resulting in elevated TL1A expression and
reduced VEGF production in HUVECs in response to high glucose.
Enhanced TL1A expression and reduced VEGF
production accelerate HUVEC apoptosis by attenuating the
PI3K/Akt/eNOS signaling pathway
The PI3K/Akt/eNOS signaling pathway has been
demonstrated to act as an important component of the mechanism that
protects the cardiovasculature from endothelial cell injury. The
attenuation of the PI3K/Akt/eNOS signaling pathway is associated
with high glucose-induced HUVEC apoptosis (21). It was hypothesized that high
glucose induces TL1A expression and reduces VEGF expression,
thereby increasing HUVEC apoptosis by attenuating the PI3K/Akt/eNOS
pathway. To test this hypothesis, HUVECs were transfected with TL1A
siRNA or VEGF DNA and were cultured in high glucose for different
periods. Then, PI3K/Akt/eNOS expression was assessed via western
blot analysis. As shown in Fig.
4A, the phosphorylation of PI3K, Akt and eNOS was enhanced in
cells transfected with TL1A siRNA compared with those transfected
with negative control siRNA. Identical results were obtained for
the HUVECs transfected with VEGF DNA to those for the HUVEC
transfected with TL1A siRNA. Based on the above results, it was
concluded that VEGF protects against high glucose-induced HUVEC
apoptosis by activating the PI3K/Akt/eNOS pathway but that TL1A
promotes HUVEC apoptosis in response to high glucose by attenuating
the PI3K/Akt/eNOS pathway.
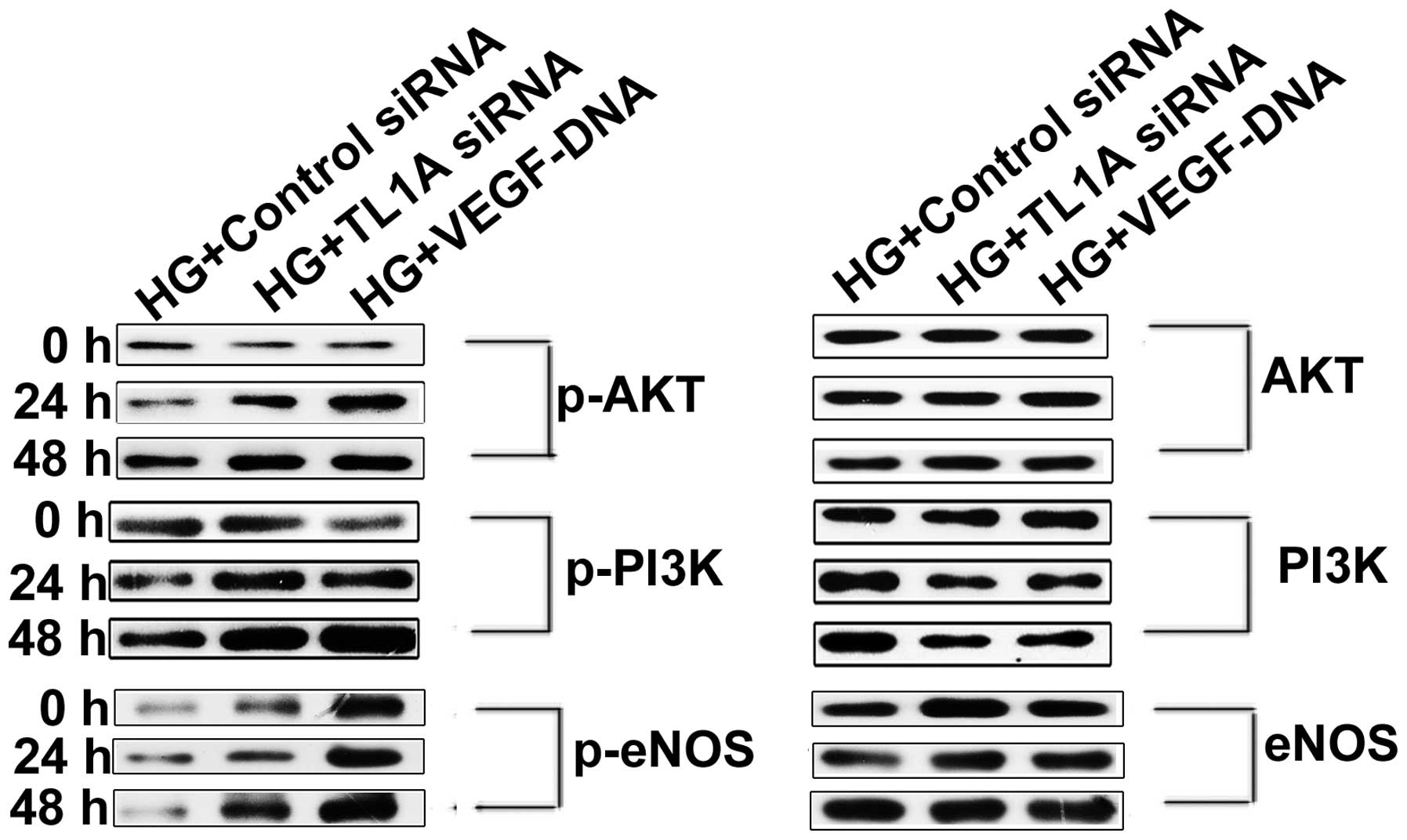 | Figure 4Elevated expression of TL1A and
reduced expression of VEGF in response to high glucose attenuated
the PI3K-Akt-eNOS signaling pathway. HUVECs were transfected with
TL1A siRNA, negative control siRNA and VEGF DNA separately. After
transfection for 12 h, cells were cultured in conditioned medium
with high glucose (33 mM/l) for indicated time points. Western blot
analysis was used to determine the quantity of PI3K, Akt, eNOS and
their phosphorylated forms. HG, high glucose; siRNA, small
interfering RNA; TL1A, tumor necrosis factor-like cytokine 1A;
VEGF, vascular endothelial growth factor; HUVECs, human umbilical
vein endothelial cells; p-, phosphorylated; PI3K, phosphoinositide
3-kinase; eNOS, endothelial nitric oxide synthase. |
Discussion
A large body of evidence suggests that endothelial
cell injury and apoptosis are likely responsible for
diabetes-related cardiovascular complications. The balance between
VEGF and TL1A, a pair of endogenous angiogenesis effectors that
perform opposing functions, is critical for maintaining vascular
integrity and homeostasis (22).
The disruption of the balance between VEGF and TL1A promotes the
onset of various types of disease. This study provided a
comprehensive analysis of the correlation between high
glucose-induced HUVEC apoptosis and the imbalance of VEGF and TL1A
expression, and preliminary results demonstrating the mutual
inhibitory effects between VEGF and TL1A under high glucose
conditions were obtained.
It has been widely accepted that apoptosis and
senescence are considered as important processes that contribute to
vascular dysfunction and pathology. Previous studies have
demonstrated that VEGF may aid in the survival and proliferation of
endothelial cells and in the protection of cells from apoptosis by
inducing the expression of anti-apoptotic and antioxidant proteins
(23,24). Furthermore, adequate VEGF levels
are required for the regeneration and repair of endothelial cells
(18). This concept was
concomitant with our findings that high glucose induced the
inhibition of VEGF expression, resulting in increased cell
apoptosis compared with normal glucose levels. Alternatively, the
overexpression of VEGF largely ameliorated high glucose-induced
HUVEC apoptosis. By contrast, TL1A has been demonstrated to arrest
the growth of quiescent endothelial cells and to induce the
apoptosis of proliferating endothelial cells (25). Mück et al (26) reported that the overexpression of
TL1A triggers premature senescence in HUVECs and that the silencing
of TL1A partially alleviated HUVEC senescence. In the present
study, the exposure of HUVECs to high glucose induced an increase
in the apoptosis rate and the TL1A levels compared with the
control. Further investigation revealed that the apoptosis of
TL1A-silenced HUVECs was significantly reduced compared with that
of the control cells. Based on these findings, it was proposed that
decreased anti-apoptotic VEGF expression and pro-apoptotic TL1A
accumulation in hyperglycemia is detrimental to survival and
proliferation, finally leading to the high glucose-induced
apoptosis in endothelial cells.
The above observations indicated that the disrupted
balance between VEGF and TL1A results in increased HUVEC apoptosis.
It has been widely accepted that maintaining the balance between
TL1A and VEGF expression depends on various mechanisms, including
their mutual effects. Deng et al (17) reported that HUVECs cultured in
OVCAR-3 conditioned medium containing various concentrations of
recombinant human VEGF exhibited significantly downregulated TL1A
levels. Consistently, based on the present study, HUVECs exposed to
high glucose in which VEGF was overexpressed via transfection with
VEGF DNA also exhibited reduced TL1A levels, whereas VEGF
expression was increased following the inhibition of TL1A
expression. In addition, TL1A regulates the relative levels of two
different isoforms of the VEGF receptor 1, membrane-bound (mFlt1)
and soluble (sFlt1) VEGF receptor 1; and the VEGF receptor 1 is
capable of inducing pro-angiogenic mFlt1 degradation and
anti-angiogenic sFlt1 accumulation (27). Thus, TL1A and VEGF exert mutual
inhibitory effects under high glucose conditions, and these effects
may ensure the maintenance of the balance between VEGF and TL1A
expression. It was speculated that high glucose-induced TL1A
expression may be partially responsible for the reduction in VEGF
expression. Furthermore, cell survival may be enhanced by
increasing VEGF expression via the alleviation of high
glucose-induced TL1A expression, thereby attenuating the inhibition
of VEGF.
A previous study demonstrated that high
glucose-induced apoptosis of human vascular endothelial cells is
mediated by the activation of reactive oxygen species
(ROS)-dependent c-Jun N-terminal protein kinase (JNK) and caspase-3
(28), and is prevented by the
PI3K/Akt/eNOS pathway (21). It
was demonstrated that the high glucose-induced increase in TL1A
expression and reduction in VEGF expression promotes HUVEC
apoptosis. Thus, to investigate whether the pro-apoptotic effect of
enhanced TL1A expression and reduced VEGF expression is associated
with the apoptotic and survival pathways described above, the
activation of the PI3K/Akt/eNOS pathway was detected. HUVECs under
high glucose stimulation displayed decreased VEGF expression and
increased TL1A production accompanied by attenuated PI3K/Akt/eNOS
pathway activation. Further investigation revealed that the
PI3K/Akt/eNOS pathway was activated following the silencing of TL1A
expression or the overexpression of VEGF; this activation was
concomitant with decreased HUVEC apoptosis. The above results
indicate that VEGF may protect against high glucose-induced
endothelial cell apoptosis by activating the PI3K/Akt/eNOS pathway
and that the attenuation of the PI3K/Akt/eNOS pathway may have
pathological relevance to TL1A-induced apoptosis.
In conclusion, this study demonstrated that
decreased VEGF levels and increased TL1A expression promote the
apoptosis of HUVECs in response to high glucose conditions.
Moreover, the activities of VEGF and TL1A are mutually inhibited.
As a result, to prevent the occurrence and development of high
glucose-induced endothelial cell injury and apoptosis, methods
targeted to restore the balance between TL1A and VEGF expression by
suppressing TL1A expression and enhancing VEGF production may be
effective. Although the present observations strongly support the
concept that TL1A is important in high glucose-induced cell
apoptosis and although TL1A has been demonstrated to exert its
pro-apoptotic effects in endothelial cells under high glucose
conditions via multiple DR3-related signaling pathways, such as the
activation of ROS-dependent JNK and caspase-3 (29), the relevance of this downstream
pathway to pro-apoptosis pathways, such as the nuclear factor-κB
pathway remains to be fully elucidated. Further studies to
determine the mechanisms underlying the maintenance of the balance
between the anti-apoptotic effector VEGF and the pro-apoptotic
effector TL1A are required.
Acknowledgments
This study was supported by the Science and
Technology Plan Project of Guangdong Province (grant no.
2010B080701043). The authors would like to thank Mr. Wendong Fan
et al for their technical help and Miss Ying Chen for
assistance with writing.
References
|
1
|
Minamino T, Miyauchi H, Yoshida T, Ishida
Y, Yoshida H and Komuro I: Endothelial cell senescence in human
atherosclerosis: Role of telomere in endothelial dysfunction.
Circulation. 105:1541–1544. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen J and Goligorsky MS: Premature
senescence of endothelial cells: Methusaleh's dilemma. Am J Physiol
Heart Circ Physiol. 290:H1729–H1739. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chou E, Suzuma I, Way KJ, Opland D,
Clermont AC, Naruse K, Suzuma K, Bowling NL, Vlahos CJ, Aiello LP
and King GL: Decreased cardiac expression of vascular endothelial
growth factor and its receptors in insulin-resistant and diabetic
states: A possible explanation for impaired collateral formation in
cardiac tissue. Circulation. 105:373–379. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Waltenberger J, Lange J and Kranz A:
Vascular endothelial growth factor-A-induced chemotaxis of
monocytes is attenuated in patients with diabetes mellitus: A
potential predictor for the individual capacity to develop
collaterals. Circulation. 102:185–190. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yoon YS, Uchida S, Masuo O, Cejna M, Park
JS, Gwon HC, Kirchmair R, Bahlman F, Walter D, Curry C, et al:
Progressive attenuation of myocardial vascular endothelial growth
factor expression is a seminal event in diabetic cardiomyopathy:
Restoration of microvascular homeostasis and recovery of cardiac
function in diabetic cardiomyopathy after replenishment of local
vascular endothelial growth factor. Circulation. 111:2073–2085.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abaci A, Oğuzhan A, Kahraman S, Eryol NK,
Unal S, Arinç H and Ergin A: Effect of diabetes mellitus on
formation of coronary collateral vessels. Circulation.
99:2239–2242. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rivard A, Silver M, Chen D, Kearney M,
Magner M, Annex B, Peters K and Isner JM: Rescue of
diabetes-related impairment of angiogenesis by intramuscular gene
therapy with adeno-VEGF. Am J Pathol. 154:355–363. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhai YF, Ni J, Jiang GW, Lu J, Xing L,
Lincoln C, Carter KC, Janat F, Kozak D, Xu S, et al: VEGI, a novel
cytokine of the tumor necrosis factor family, is an angiogenesis
inhibitor that suppresses the growth of colon carcinomas in vivo.
Faseb J. 13:181–189. 1999.PubMed/NCBI
|
|
9
|
Chew LJ, Pan H, Yu J, Tian S, Huang WQ,
Zhang JY, Pang S and Li LY: A novel secreted splice variant of
vascular endothelial cell growth inhibitor. FASEB J. 16:742–744.
2002.PubMed/NCBI
|
|
10
|
Migone TS, Zhang J, Luo X, Zhuang L, Chen
C, Hu B, Hong JS, Perry JW, Chen SF, Zhou JX, et al: TL1A is a
TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell
costimulator. Immunity. 16:479–492. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Meylan F, Davidson TS, Kahle E, Kinder M,
Acharya K, Jankovic D, Bundoc V, Hodges M, Shevach EM, Keane-Myers
A, et al: The TNF-family receptor DR3 is essential for diverse T
cell-mediated inflammatory diseases. Immunity. 29:79–89. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bamias G, Martin C III, Marini M, Hoang S,
Mishina M, Ross WG, Sachedina MA, Friel CM, Mize J, Bickston SJ, et
al: Expression, localization and functional activity of TL1A, a
novel Th1-polarizing cytokine in inflammatory bowel disease. J
Immunol. 171:4868–4874. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kang YJ, Kim WJ, Bae HU, Kim DI, Park YB,
Park JE, Kwon BS and Lee WH: Involvement of TL1A and DR3 in
induction of pro-inflammatory cytokines and matrix
metalloproteinase-9 in atherogenesis. Cytokine. 29:229–235. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim WJ, Kang YJ, Suk K, Park JE, Kwon BS
and Lee WH: Comparative analysis of the expression patterns of
various TNFSF/TNFRSF in atherosclerotic plaques. Immunol Invest.
37:359–373. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fang L, Adkins B, Deyev V and Podack ER:
Essential role of TNF receptor superfamily 25 (TNFRSF25) in the
development of allergic lung inflammation. J Exp Med.
205:1037–1048. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hampel B, Fortschegger K, Ressler S, Chang
MW, Unterluggauer H, Breitwieser A, Sommergruber W, Fitzky B,
Lepperdinger G, Jansen-Dürr P, et al: Increased expression of
extracellular proteins as a hallmark of human endothelial cell in
vitro senescence. Exp Gerontol. 41:474–481. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Deng WM, Gu X, Lu Y, Gu C, Zheng Y, Zhang
Z, Chen L, Yao Z and Li LY: Down-modulation of TNFSF15 in ovarian
cancer by VEGF and MCP-1 is a pre-requisite for tumor
neovascularization. Angiogenesis. 15:71–85. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Parr C, Gan CH, Watkins G and Jiang WG:
Reduced vascular endothelial growth inhibitor (VEGI) expression is
associated with poor prognosis in breast cancer patients.
Angiogenesis. 9:73–81. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Conway KP, Price P, Harding KG and Jiang
WG: The role of vascular endothelial growth inhibitor in wound
healing. Int Wound J. 4:55–64. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Ho FM, Lin WW, Chen BC, Chao CM, Yang CR,
Lin LY, Lai CC, Liu SH and Liau CS: High glucose-induced apoptosis
in human vascular endothelial cells is mediated through NF-kappa B
and c-Jun NH2-terminal kinase pathway and prevented by
PI3K/Akt/eNOS pathway. Cell Signal. 18:391–399. 2006. View Article : Google Scholar
|
|
22
|
Folkman J, Watson K, Ingber D and Hanahan
D: Induction of angiogenesis during the transition from hyperplasia
to neoplasia. Nature. 339:58–61. 1989. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang F, Tang Z, Hou X, Lennartsson J, Li
Y, Koch AW, Scotney P, Lee C, Arjunan P, Dong L, et al: VEGF-B is
dispensable for blood vessel growth but critical for their survival
and VEGF-B targeting inhibits pathological angiogenesis. P Natl
Acad Sci USA. 106:6152–6157. 2009. View Article : Google Scholar
|
|
24
|
Abid MR, Schoots IG, Spokes KC, Wu SQ,
Mawhinney C and Aird WC: Vascular endothelial growth
factor-mediated induction of manganese superoxide dismutase occurs
through redox-dependent regulation of forkhead and
IkappaB/NF-kappaB. J Biol Chem. 279:44030–44038. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yu JY, Tian S, Metheny-Barlow L, Chew LJ,
Hayes AJ, Pan H, Yu GL and Li LY: Modulation of endothelial cell
growth arrest and apoptosis by vascular endothelial growth
inhibitor. Circ Res. 89:1161–1167. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mück C, Herndler-Brandstetter D, Micutkova
L, Grubeck-Loebenstein B and Jansen-Dürr P: Two functionally
distinct isoforms of TL1A (TNFSF15) generated by differential
ectodomain shedding. J Gerontol A Biol Sci Med Sci. 65:1165–1180.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qi JW, Qin TT, Xu LX, Zhang K, Yang GL, Li
J, Xiao HY, Zhang ZS and Li LY: TNFSF15 inhibits vasculogenesis by
regulating relative levels of membrane-bound and soluble isoforms
of VEGF receptor 1. P Natl Acad Sci USA. 110:13863–13868. 2013.
View Article : Google Scholar
|
|
28
|
Ho FM, Liu SH, Liau CS, Huang PJ and
Lin-Shiau SY: High glucose-induced apoptosis in human endothelial
cells is mediated by sequential activations of c-Jun NH2-terminal
kinase and caspase-3. Circulation. 101:2618–2624. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yue TL, Ni J, Romanic AM, et al: TL1, a
novel tumor necrosis factor-like cytokine, induces apoptosis in
endothelial cells-Involvement of activation of stress protein
kinases (stress-activated protein kinase and p38 mitogen-activated
protein kinase) and caspase-3-like protease. J Biol Chem.
274:1479–1486. 1999. View Article : Google Scholar : PubMed/NCBI
|