Introduction
Autophagy refers to any lysosomal degradation
pathway and is essential for survival, differentiation, development
and homeostasis (1). During
autophagy, an autophagosome, a double-membraned vesicle
sequestering cytoplasmic materials, and a lysosome, fuse together
to form an autolysosome (2). The
two principal physiological roles of autophagy are to maintain
synthesis of macromolecules and ATP, and to eliminate defective or
abnormal proteins and organelles. These functions allow cells to
survive various metabolic stresses, such as nutritional and growth
factor depletion, or hypoxia, and be protected from infection,
neurodegenerative diseases, genomic instabilities or tumor
initiation (3). Various
pathological conditions are associated with defects in autophagy,
as it serves an important role in homeostasis (1). Autophagy has been considered a tumor
suppressor pathway (4–6) following the establishment of an
association between autophagy and cancer in 1999, when the
autophagy-related gene (ATG) beclin-1 was identified as a candidate
tumor suppressor (7). Numerous
genetic links have been identified between defects in autophagy and
cancer. Tumor suppressor genes involved in the upstream inhibition
of mammalian target of rapamycin (mTOR) signaling, including
phosphatase and tensin homolog (4), tuberous sclerosis 1, and tuberous
sclerosis 2 (5), stimulated
autophagy. By contrast, TOR-activating oncogene products, such as
Ras, phosphoinositide 3-kinase (PI3K) and protein kinase B (Akt),
inhibit autophagy (6).
Furthermore, a previous study hypothesized that chemoradiotherapy
induces the accumulation of autophagosomes in various cancer cell
lines and eliminates cancer cells by induction of
caspase-independent autophagic and caspase-dependent cell death
(8). However, autophagy may
promote survival of cancer cells during chemotherapy and serve a
role in chemoresistance (9).
Therefore, inhibiting autophagy by targeting certain
autophagy-related (ATG) genes may accelerate, rather than prevent
cell death (10).
Previous studies in the field of hematology have
primarily focused on elucidating the role of autophagy in chronic
myeloid, chronic lymphoid and acute promyelocytic leukemias, and
autophagy was associated with resistance to tyrosine kinase
inhibitors, histone deacetylase inhibitors and hypomethylating
agents (11–14). However, to the best of our
knowledge, no previous study has investigated the possible
association between autophagy and chemoresistance to cytosine
arabinoside (1-β-d-arabinofuranosylcytosine; Ara-C) in acute
myeloid leukemia (AML). Ara-C is a nucleoside analog used to induce
complete remission or for post-remission therapy in AML (15). Ara-C acts as an anti-metabolite and
induces cell death by competing with pyrimidine for incorporation
into replicative DNA, which inhibits DNA polymerase activity and
leads to chain elongation termination. Due to the important role of
Ara-C in AML treatment, acquired resistance to Ara-C is one of the
multiple factors leading to AML persistence or relapse. Therefore,
novel therapeutic strategies are required to overcome the Ara-C
resistance in myeloid leukemia cells, which is hypothesized to
develop through various mechanisms. In the present study, the role
of autophagy in myeloid leukemic cell lines was assessed according
to Ara-C sensitivity, and whether inhibiting autophagy would
overcome Ara-C resistance was investigated.
Materials and methods
Reagents and antibodies
Ara-C was purchased from Sigma-Aldrich (St. Louis,
MO, USA), and was dissolved in distilled water and stored as a 100
mM stock solution. Hydroxychloroquine (HCQ) was purchased from
Myung In Pharmaceutical Company, Ltd. (Seoul, South Korea) and
dissolved in dimethyl sulfoxide (Sigma-Aldrich). Bafilomycin A1
(Ba-A1) was purchased from Sigma-Aldrich. Fetal bovine serum (FBS)
and Gibco RPMI 1640 medium containing 50 U/ml and 50 µg/ml
streptomycin were purchased from Thermo Fisher Scientific, Inc.
(Waltham, MA, USA). Rabbit polyclonal antibodies against human
microtubule-associated protein 1 light chain 3 (LC3; cat. no.
NB100-2220; dilution, 1:1,000), beclin-1 (cat. no. NB500-249;
dilution, 1:1,000), ATG9A (cat. no. NB110-56893; dilution,
1:1,000), ATG7 (NB110-74811) and mouse anti-human p62 (cat. no.
H0008878; dilution, 1:1,000) were purchased from Novus Biologicals
LLC (Littleton, CO, USA). Horseradish peroxidase (HRP)-conjugated
goat anti-rabbit (cat. no. 7074; dilution, 1:3,000) and horse
anti-mouse (cat. no. 7072; dilution, 1:3,000) IgG secondary
antibodies were obtained from Cell Signaling Technology, Inc.
(Danvers, MA, USA). The Annexin V-fluorescein isothiocyanate (FITC)
apoptosis detection kit was purchased from BD Pharmingen (San
Diego, CA, USA). All other chemicals were from Sigma-Aldrich. The
GFP-LC3 plasmid was kindly provided by Dr Kim (Division of
Pulmonology, Yonsei University College of Medicine, Seoul, South
Korea).
Cell culture
The human leukemia cell line, U937, was obtained
from the American Type Culture Collection (Rockville, MD, USA). The
Ara-C-resistant U937 (U937/AR) cell line was established in our
laboratory by exposing parental U937 cells to stepwise increasing
concentrations of Ara-C (1 nM – 2 mM, usually at intervals of 10
passages) in complete RPMI 1640 medium supplemented with 10%
heat-inactivated FBS and 100 U/ml penicillin. Resistant sublines
were grown for longer than 6 months. To maintain exponential
growth, cells were seeded at 1×105 cells/ml and passaged
every 3 days.
Cell death assay
U937 and U937/AR cells were cultured on 12-well
plates (Corning Incorporated, Corning, NY, USA) at 2×105
cells/well in medium containing RPMI 1640, 10% FBS and 100 U/ml
penicillin and streptomycin for 24, 48 and 72 h. Cultured cells
were washed with phosphate-buffered saline (PBS; Gibco; Thermo
Fisher Scientific, Inc.) and incubated in 100 µl 1X binding
buffer containing 5 µl Annexin V-FITC, and the nuclei were
counterstained with 10 µl propidium iodide according the kit
manufacturer's instructions. The percentage of apoptotic cells was
determined using a FACSCalibur flow cytometer and analyzed using
CellQuest version 3.3 software (BD Immunocytometry Systems, San
Jose, CA, USA).
Western blot analysis
U937 and U937/AR cells were cultured in 12-well
plates at 2×105 cells/well in RPMI 1640 medium with or
without 10% FBS for 48 h. Cells were lysed in lysis buffer
containing 50 mM Tris-HCl (pH 7.5), 120 mM NaCl, 20 mM NaF, 1 mM
EDTA, 5 mM ethylene
glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid, 15 mM
sodium pyrophosphate, 1 mM benzamidine, 0.1 mM phenylmethylsulfonyl
fluoride, and 1% Nonidet P-40, and this was briefly sonicated. The
mitochondrial and cytosolic fractions were obtained using the QIA88
ProteoExtract Cytosolic/Mitochondria Fractionation kit (Oncogene
Research Products, La Jolla, CA, USA). Protein yields were
quantified using a detergent-compatible protein assay kit (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) according to the
manufacturer's instructions. Equivalent amounts of protein (10 mg)
were boiled for 10 min and separated by SDS-PAGE (15% for LC3; 12%
for beclin-1 and p62; and 10% for ATG9A and ATG7). Proteins were
subsequently transferred to nitrocellulose membranes (GE Healthcare
Bio-Sciences, Pittsburgh, PA, USA). Following blocking with 0.05%
Tris-buffered saline with Tween 20 (TBST) and 5% bovine serum
albumin solution, or 5% skim milk, for 1 h at room temperature,
blots were incubated with LC3, beclin-1, ATG9A, ATG7 and p62
primary antibodies overnight at 4°C. Blots were washed four times
for 5 min with TBST and incubated for 1 h at room temperature with
HRP-conjugated anti-rabbit or anti-mouse IgG secondary antibodies
(1:3,000). Subsequent to washing with TBST, the reactive proteins
were visualized using the enhanced chemiluminescence reagent
(Amersham; GE Healthcare Life Sciences). The density of the protein
bands on the membrane were scanned and analyzed with ImageJ
(imagej.nih.gov/ij/).
EGFP-LC3 puncta assay
U937 and U937/AR cell suspensions (2×106
cells) were immediately electroporated with pEGFP-LC3 cDNA using
the Nucleofector™ 2b system (program T-20; Lonza Cologne GmbH,
Cologne, Germany) according to the manufacturer's instructions.
Immediately following electroporation, cells were resuspended in
complete medium and incubated at 37°C in a humidified 5%
CO2 incubator. pEGFP vector transfection was performed
as control. After 24 h, cells were rinsed and resuspended in
serum-free medium and incubated at 37°C for 48 h. Cultured cells
were centrifuged at 800 × g onto glass slides. Fluorescence images
were analyzed using an LSM 700 laser-scanning confocal microscope
(Zeiss GmbH, Jena, Germany). The GFP-LC3 puncta in a single cell
were manually counted using confocal microscopy. For each group, 50
cells were randomly selected to estimate the average number of
GFP-LC3 puncta per cell. Data presented are one representative
experiment of a minimum of three independent repeats.
Transmission electron microscopy
(TEM)
For TEM evaluation, U937 and U937/AR cells were
seeded in 12-well plates at 2×105 cells/well and
cultured with or without FBS for 48 h at 37°C. Following
incubation, the cells were collected and fixed for 2 h at 4°C with
ice-cold 2% glutaraldehyde-paraformaldehyde (EMD Millipore,
Billerica, MA, USA) and post-fixed at 4°C with 1% OsO4
(KeyGen Biotech Co. Ltd.; dissolved in 0.1 M PBS) for 2 h and
dehydrated through serial dilutions of ethanol (50, 70, 90 and 100%
for 15 min and then three times at 100%) and infiltrated with
propylene oxide (Sigma-Aldrich) prior to embedding in epoxy resin
using a Poly/Bed 812 kit (Polysciences, Inc., Warrington, PA, USA)
at 65°C electron microscope oven (TD-700; Dosaka EM, Kyoto, Japan)
for 24 h. The embedded sections were cut into sections of 250–250
nm for staining with toluidine blue (Sigma-Aldrich) and observation
under a light microscope and into ultrathin sections (70 nm) were
double-stained with 7% uranyl acetate for 20 min and lead citrate
(Thermo Fisher Scientific, Inc.) for 10 min for contrast staining.
The sections were cut with an ultramicrotome (Leica EM UC7; Leica
Microsystems GmbH, Wetzlar, Germany) and mounted on copper and
nickel grids. Sections were viewed using a JEM-1011 system (JEOL
USA, Inc., Peabody, MA, USA) at an acceleration voltage of 80 kV
(Camera Megaview, Soft Imaging System, Berlin, Germany). Photoshop
(Adobe Systems, Inc., San Jose, CA, USA) was used to further
quantify the TEM images.
Statistical analysis
The statistical analysis was performed using the
two-tailed Student's t-test. Unless otherwise indicated, data are
expressed as the mean ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference.
Results
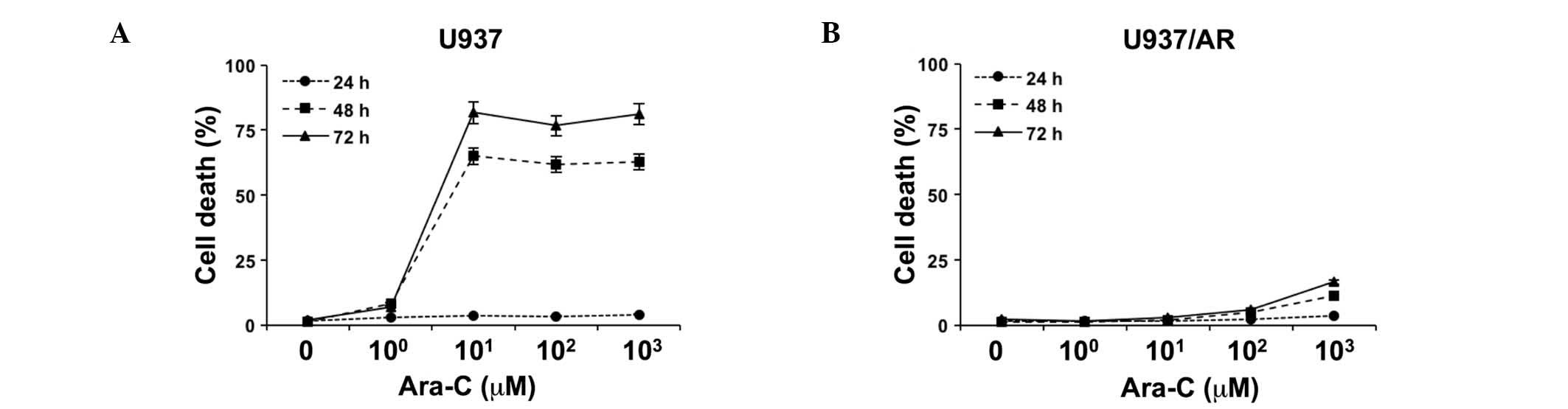
Sensitivity of U937 and U937/AR leukemia
cell lines to Ara-C
The relative sensitivity of U937 and U937/AR to
Ara-C was assessed in vitro as a baseline study. Following
treatment with logarithmically scaled concentrations of Ara-C
(100–103 µM), cell death was assessed
using an Annexin V binding assay, combined with flow cytometry. As
demonstrated in Fig. 1A, cell
death was induced in a time-dependent manner in the Ara-C U937
cells. Ara-C (101 µM) induced cell death in
65.0±1.0 and 81.7±0.8% of U937 cells after 48 and 72 h,
respectively. The U937/AR cells were resistant to Ara-C and the
maximum level of cell apoptosis observed was 16.6±0.2% at 72 h
after treatment with the highest concentration of Ara-C (Fig. 1B).
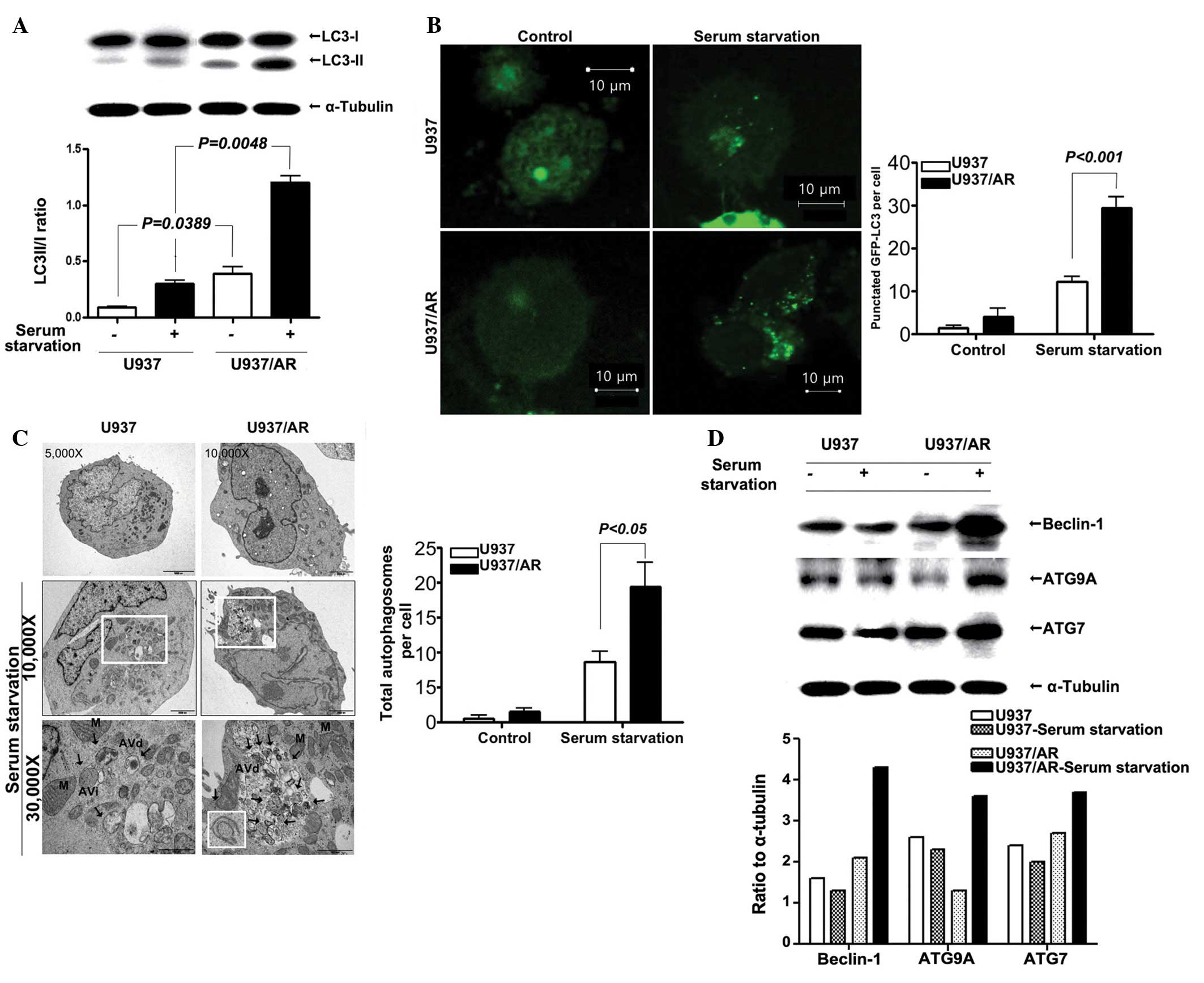
Status of autophagy in U937 and U937/AR
cells according to the culture conditions
Following standard cell culture conditions (media
supplemented with FBS) and serum starvation, the status of
autophagy was assessed. Western blot analysis results demonstrated
that the LC3-II/I ratio in the U937/AR cells was significantly
higher compared with that of the Ara-C sensitive U937 cells, under
the standard culturing condition with FBS (0.39±0.08 vs. 0.09±0.01;
P=0.0389; Fig. 2A) and under serum
starvation (1.21±0.08 vs. 0.30±0.04; P=0.0048; Fig. 2A). The fractional increases in the
LC3-II/I ratio under serum starvation were ~3-fold in the two cell
lines and thus, were not observed to be significantly different
(P>0.05).
LC3 expression was assessed using the GFP-LC3 puncta
assay. The results demonstrated a significant increase in the
number of GFP-LC3 puncta per cell in the U937/AR cells compared
with the U937 cells, regardless of culturing conditions (4.0±2.0
vs. 1.5±0.5 puncta per cell with FBS; P=0.0239 or 29.5±2.5 vs.
12.2±1.2 puncta per cell without FBS; P<0.001; Fig. 2B). Furthermore, the total number of
autophagosomes per cell was counted using TEM (Fig. 2C), and the results were similar to
those from western blot and GFP-LC3 puncta assays. As demonstrated
in Fig. 2C, the number of
autophagosomes significantly increased when cells were cultured
without FBS. The number of autophagosomes in U937/AR cells cultured
without FBS was significantly higher than that in the U937 cells
(19.3±3.5 vs. 8.7±1.5; P<0.05; Fig.
2C).
In addition, the expression of several
autophagy-associated genes was determined. As demonstrated in
Fig. 2D, the increase in beclin-1,
ATG9A, ATG7 and p62/SQSTM1 protein expression levels was more
prominent in the U937/AR cells (ratio to α-tubulin, 4.3, 2.6 and
3.7, respectively) compared with the U937 cells (ratio to
α-tubulin, 1.3, 2.3 and 2.0, respectively) when cultured without
FBS.
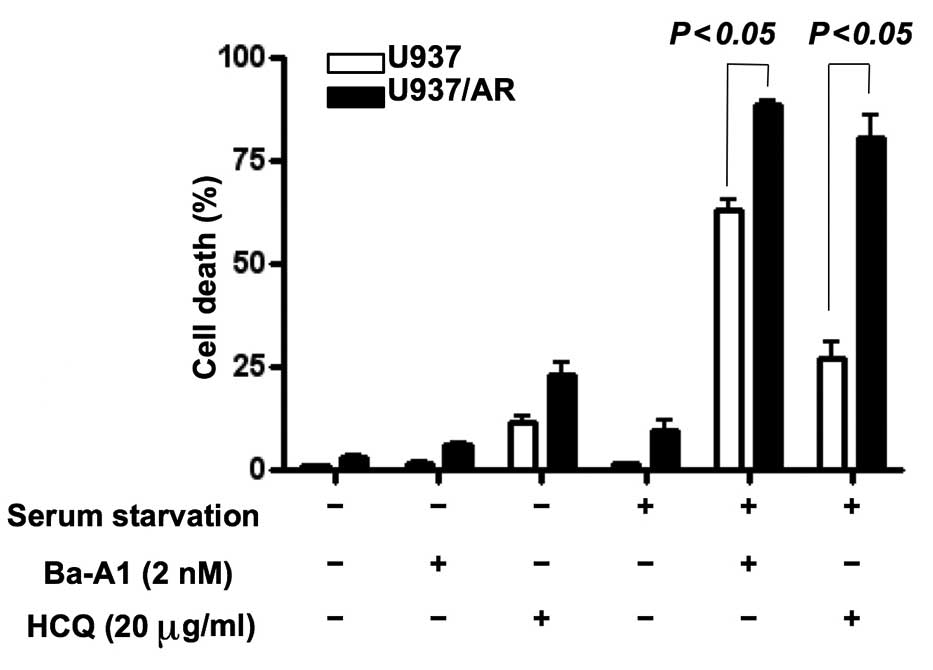
Effect of autophagy inhibitors in U937
and U937/AR cells cultured without serum
Following cell treatment with or without serum, the
mechanism of action of the autophagy inhibitors affecting apoptosis
was investigated. The U937 and U937/AR cells were treated with two
autophagy inhibitors separately, Ba-A1 (2 nM) and HCQ (20
µg/ml) for 48 h. Untreated cell lines grow when cultured
with FBS, but autophagic activity is required to prolong cell
survival when cultured without FBS. As demonstrated in Fig. 3, the autophagy inhibitors induced
cell death in <25% of the cells cultured with serum. In the
cultures with FBS, the autophagy inhibitors increased cell death in
the U937/AR cells compared with the U937 cells as follows: Ba-A1,
6.2±0.5 vs. 1.6±0.4% (P<0.05); HCQ, 23.0±3.0 vs. 11.7±1.6%
(P<0.05). These results were consistent with the status of
autophagy in the U937 and U937/AR cells treated with FBS (Fig. 2).
Furthermore, in a comparison of FBS-cultured and
serum starved cells, the autophagy inhibitors induced cell death in
a significantly higher percentage of serum starved cells, compared
with the FBS-treated cells as follows: Ba-A1, 62.9±2.7 vs. 1.6±0.4%
in U937 (P<0.05) and 88.7±0.9 vs. 6.2±0.5% in U937/AR
(P<0.05); HCQ, 27.0±3.9 vs. 11.7±1.6% in U937 (P<0.05) and
80.8±5.0 vs. 23.0±3.0% in U937/AR (P<0.05). In addition, the
difference in cell death between U937 and U937/AR induced by the
autophagy inhibitors was consistent and unaffected by culture
conditions or the type of autophagy inhibitor used.
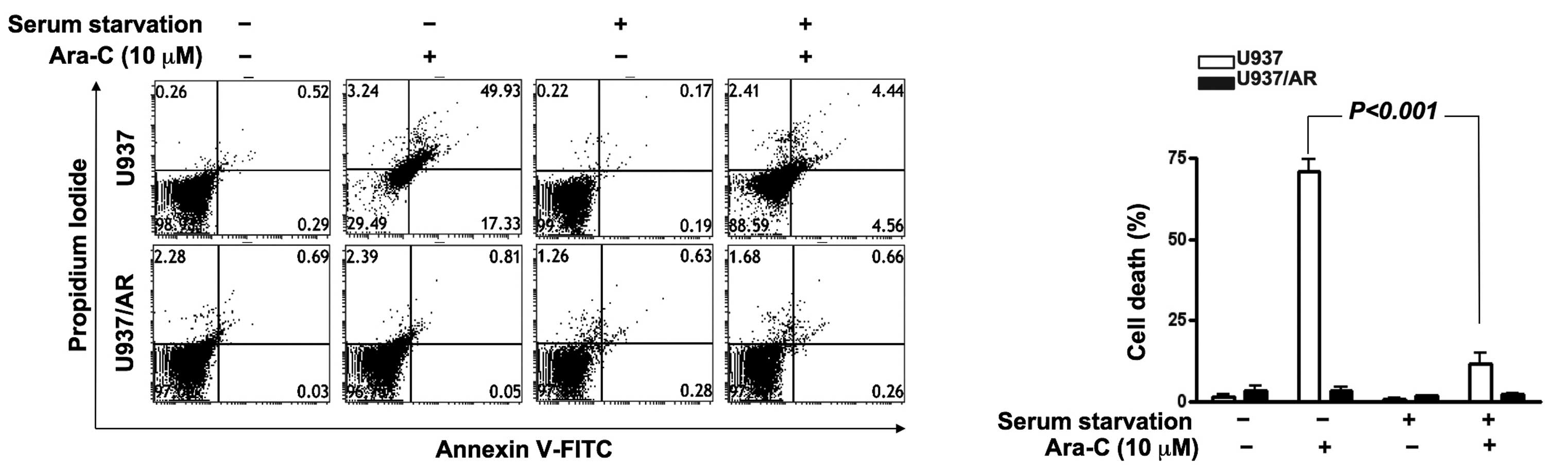
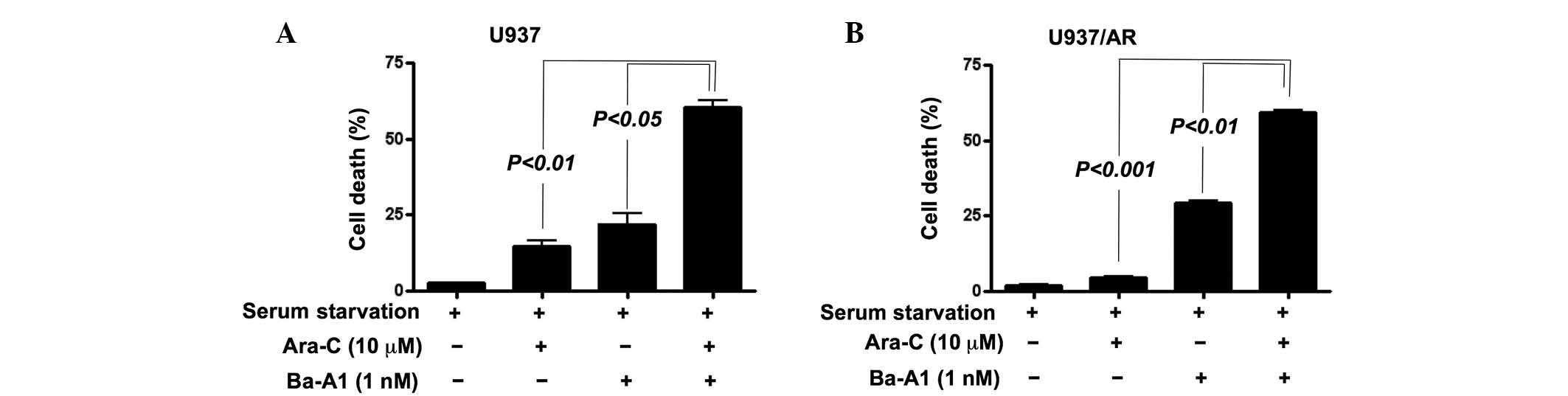
Induction of Ara-C resistance by culture
condition, and overcoming Ara-C resistance by co-treatment with
Ara-C and autophagy inhibitors
The association of autophagic activity and Ara-C
resistance was investigated. As demonstrated in Fig. 4, U937/AR cells were Ara-C resistant
in all culture conditions. However, Ara-C treatment induced cell
death in the U937 cell line with FBS. The Ara-C-sensitive U937
cells acquired Ara-C resistance upon culturing without FBS
(Fig. 4; cell death rate,
70.9±3.8% with FBS vs. 11.9±3.0% without FBS; P<0.001).
Cell death was then assessed in the U937 and U937/AR
cell lines following culturing without FBS and co-treatment with
Ara-C and Ba-A1. To evaluate whether the co-treatment had an
additive or synergistic effect on inducing cell death, a lower
concentration of Ba-A1 (1 nM) was used to induce moderate cell
death. Resistance to Ara-C in Ara-C-sensitive U937 cells was
demonstrated upon serum starved culture and co-treatment with Ba-A1
and Ara-C (cell death rate, 14.8±2.5% for Ara-C monotherapy vs.
60.5±3.2% for Ara-C and Ba-A1; P<0.01; Fig. 5A). Furthermore, the acquired Ara-C
resistance was of higher significance in the U937/AR cells (cell
death rate, 4.5±0.8% for Ara-C monotherapy vs. 59.2±1.2% for Ara-C
and Ba-A1; P<0.001; Fig.
5B).
Discussion
Autophagy is a catabolic process that recycles
intracellular components and selectively eliminates organelles for
regulation and maintenance of quality control (1). The first step of autophagy is the
formation of an isolation membrane and phagophore, the expansion of
the membrane. The edges of the phagophore fuse to form an
autophagosome, which further fuses with a lysosome to make an
autolysosome, while the inner membrane and materials are degraded
(2). Physiologically, autophagy
defends against various forms of metabolic stress, including
nutrient deprivation, growth factor depletion and hypoxia.
Autophagic degradation generates free amino acids and fatty acids
used by the tricarboxylic acid cycle to maintain cellular ATP
production (3). Autophagy performs
cellular functions to eliminate defective proteins or organelles,
prevent abnormal protein aggregate accumulation and remove
intracellular pathogens. In addition, previous studies demonstrated
that autophagy may act as a guardian of the genome to limit DNA
damage and chromosomal instability (16–18).
This role may be associated with the preventive effects of
autophagy against the initiation and progression of cancer. By
contrast, defective autophagy has been proposed as a contributor to
various human diseases. For example, patients with
neurodegenerative diseases, such as Alzheimer's disease,
Parkinson's and Huntington's diseases, have abnormal accumulation
of autophagosomes in the brain (19,20),
and germline or somatic mutations in genes, such as dynactin
subunit p150 glued or ceroid-lipofuscinosis neuronal 3, have been
associated with neurologic diseases (21). In addition, the liver, muscles and
heart may be damaged by defective autophagy, and several inherited
diseases, such as Danon or Pompe diseases, demonstrate the role of
defective autophagy in their pathogenesis (22–24).
Abnormalities in the autophagic process may present
in cancer. Liang et al (7)
demonstrated that the ATG gene, beclin-1, was a candidate tumor
suppressor gene in 1999, and previous studies have indicated that
defective autophagy is closely associated with the initiation or
progression of cancer (25–27).
Certain mutations in genes affecting autophagy, such as beclin-1,
Akt, PI3K, p53 and Bcl-2, serve a role in the pathogenesis of
malignant lymphoma and breast, ovarian and prostate cancer
(1).
Autophagy protects against cell starvation and
hypoxia, which are the hallmarks of the tumor microenvironment. A
previous study suggested that autophagy serves an important role in
the chemoresistance of cancer to therapeutics that typically induce
apoptosis (9). Numerous clinical
trials evaluating autophagy in various solid cancers, including
breast (28), lung (29), melanoma (30), rectal/colon (31), renal cell carcinoma (32), prostate (33) and pancreatic cancer (34), have demonstrated inconsistent
results regarding the anticancer effect of autophagy manipulation
(35). Certain previous studies
demonstrated that autophagic cell death observed during
chemotherapy acted as an anticancer machinery (8,36),
however other studies suggested that autophagy prevents apoptosis
of cancer cells from chemotherapy (9,37).
These inconsistent results may be due to the dynamic nature of
autophagy and the diversity of molecules or organelles targeted by
it. Autophagy may target tumor-initiating proteins developed in
normal cells, therefore suppressing tumor activity. In addition,
cancer cells may preserve themselves using autophagy during
chemotherapy, thus promoting cancer cell survival.
The myeloid leukemias are a heterogeneous group of
diseases characterized by neoplastic cells that infiltrate the
blood, bone marrow and other tissues of the hematopoietic system.
In previous studies, induction of autophagy was demonstrated to be
important for the death of leukemic cells (11–14,38),
and the induction of autophagy by various drugs, such as imatinib
mesylate, arsenic trioxide, everolimus, brevinin-2R and eupalinin
A, was attempted. Autophagy may be an important pathway of cell
death during various chemotherapeutic modalities, and manipulation
of autophagy may be a useful clinical application for targeting
multidrug-resistant leukemia. Although triggering autophagy may be
a potential therapeutic strategy to overcome drug resistance,
inhibiting autophagy may be another therapeutic strategy to improve
the outcome of anticancer treatments. For example, autophagy acts
as a prosurvival mechanism and contributes to drug resistance in
various types of leukemia (38).
By contrast, inhibition of autophagy was documented to enhance the
therapeutic benefit of tyrosine kinase inhibitors in Philadelphia
(Ph)-positive leukemias, and the tumor anti-leukemic effect of the
histone deacetylase inhibitor, SAHA, was augmented by co-treatment
with an autophagy inhibitor (11).
Overcoming drug resistance by manipulating autophagy was primarily
attempted for Ph-positive types of leukemia, such as chronic
myeloid leukemia, Ph-positive acute lymphoblastic leukemia and
acute promyelocytic leukemia (39,40).
However, the role of autophagy in association with drug resistance
in AML remains unclear.
In the current study, the status of autophagy in AML
cell lines was assessed according to the resistance against Ara-C,
a chemotherapeutic agent used to induce remission. In addition, an
attempt was made to overcome the Ara-C resistance by combination
treatment with an autophagy inhibitor and Ara-C. As demonstrated in
Fig. 2, specific characteristics
of autophagy in the U937 and U937/AR cell lines were identified,
including LC3-I-to-LC3-II conversion (Fig. 2A), formation of EGFP-LC3 puncta
(Fig. 2B) and acidic
autophagolysosomes (Fig. 2C). The
three assays demonstrated a consistent highly active autophagic
status in the U937/AR cells compared with the U937 cells. To verify
the autophagic activity at the molecular level, the expression of
autophagy-associated molecules was investigated. Following
autophagy initiation by the ATG1-ATG13 protein complex, which is
activated by the absence of signaling of the nutrient-sensing
kinase mTOR, class III PI3K-beclin-1 complexes promote formation of
the isolation membrane. Elongation of the isolation membrane is
then mediated by two ubiquitin-like conjugation systems as follows:
i) ATG7 and ATG10 act to conjugate ATG5 to ATG12; ii) the
ATG5-ATG12 conjugate acts with ATG7 and ATG3 to conjugate ATG8 to
phosphatidylethanolamine in the membrane of the growing
autophagosome (41). In addition,
the expression of beclin-1, ATG9A, ATG7 and p62/SQSTM1 was
determined. The autophagy-associated molecules were expressed in
similar patterns as observed in the LC3-I-to-LC3-II conversion,
EGFP-LC3 puncta and autophagosome assays. The results of the
current study demonstrated that the U937/AR cells had higher levels
of autophagic activity compared with the U937 cells, and that
autophagic activity increased after Ara-C treatment, thus the
ability of an autophagy inhibitor to overcome Ara-C resistance in
the U937/AR cells and to improve the antileukemic effect of Ara-C
in the U937 cells was assessed. As the Ara-C-resistant U937/AR cell
line was established by exposing parental U937 cells to increasing
concentrations of Ara-C in culture media with FBS, the increasing
autophagic activity in U937/AR cells may be induced by Ara-C. Ara-C
is an anti-metabolite and other anti-metabolites, such as
5-fluorouracil or 6-thioguanine have been demonstrated to induce
autophagy (42,43). The current study demonstrated that
the U937/AR cells had a higher level of autophagic activity than
U937 cells when the cells were treated with Ara-C (Fig. 2). The augmented autophagic activity
in Ara-C-treated U937/AR cells may be one of the mechanisms leading
to the U937/AR cells resistance to higher concentrations of Ara-C
with longer treatment durations (Fig.
1B). The evidence of the anti-apoptotic role of autophagy in
the U937 cells during metabolic stress was supported by the results
demonstrated when U937 cells were cultured with autophagy
inhibitors under starvation (Fig.
3). As U937 is a leukemia cell line, serum starvation alone
cannot induce sufficient cell death, however, when the cell lines
were cultured without FBS and treated with autophagy inhibitors,
sufficient cell death was induced. Furthermore, culturing the cells
without FBS may transform Ara-C sensitive U937 cells to Ara-C
resistant U937 cells with no further manipulations (Fig. 4). Although the underlying mechanism
of this effect was not fully investigated, the maintenance of
autophagic activity may be associated with the acquisition of Ara-C
resistance, even in the absence of Ara-C. Combination treatment
with a low dose of the autophagy inhibitor and Ara-C improved the
anti-leukemic activity in the U937 cells (Fig. 5A) and overcame Ara-C resistance in
the U937/AR cells.
Previous studies have emphasized the role of
autophagy in AML. One study demonstrated that overexpression of
melanoma differentiation-associated gene-7/interleukin-24 inhibited
autophagy and strongly augmented the anti-leukemia activity in an
AML cell line and in a mouse model of leukemia (44). Another study demonstrated that
bone-morphogenetic protein 4, a member of the TGF-β super-family,
has a role in promoting chemoresistance through the activation of
autophagy and subsequent inhibition of apoptosis in leukemic cells
(45). S100A8, a member of the
S100 calcium-binding protein family, has also been proposed to be
involved in the development of chemoresistance in leukemic cells by
regulating autophagy, and has been suggested as a novel target for
improving leukemia therapy (12,46).
In conclusion, the role of autophagy as a protector
against cellular stress may be important for the survival of AML
cells when treated with chemotherapeutics. Chemoresistant AML cells
demonstrated increased autophagic activity that augmented the
anti-leukemic activity of Ara-C and overcame Ara-C resistance in
vitro. Prior to validating the clinical benefit of autophagy
inhibition, the genetic and epigenetic mechanisms of autophagy that
protect leukemic cells and promote resistance to chemotherapeutics
should be further investigated.
Acknowledgments
The current study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education (grant no. NRF-2012R1A1A2
009114).
References
|
1
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang Z and Klionsky DJ: Mammalian
autophagy: Core molecular machinery and signaling regulation. Curr
Opin Cell Biol. 22:124–131. 2010. View Article : Google Scholar :
|
|
3
|
Murrow L and Debnath J: Autophagy as a
stress-response and quality-control mechanism: Implications for
cell injury and human disease. Annu Rev Pathol. 8:105–137. 2013.
View Article : Google Scholar
|
|
4
|
Cully M, You H, Levine AJ and Mak TW:
Beyond PTEN mutations: The PI3K pathway as an integrator of
multiple inputs during tumorigenesis. Nat Rev Cancer. 6:184–192.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schwartz RA, Fernández G, Kotulska K and
Jóźwiak S: Tuberous sclerosis complex: Advances in diagnosis,
genetics, and management. J Am Acad Dermatol. 57:189–202. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ligresti G, Militello L, Steelman LS,
Cavallaro A, Basile F, Nicoletti F, Stivala F, McCubrey JA and
Libra M: PIK3CA mutations in human solid tumors: Role in
sensitivity to various therapeutic approaches. Cell Cycle.
8:1352–1358. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Notte A, Leclere L and Michiels C:
Autophagy as a mediator of chemotherapy-induced cell death in
cancer. Biochem Pharmacol. 82:427–434. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu
Y, Xie M, Yin X, Livesey KM, Lotze MT, et al: HMGB1-induced
autophagy promotes chemotherapy resistance in leukemia cells.
Leukemia. 25:23–31. 2011. View Article : Google Scholar
|
|
10
|
Pan Y, Gao Y, Chen L, Gao G, Dong H, Yang
Y, Dong B and Chen X: Targeting autophagy augments in vitro and in
vivo antimyeloma activity of DNA-damaging chemotherapy. Clin Cancer
Res. 17:3248–3258. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Carew JS, Nawrocki ST, Kahue CN, Zhang H,
Yang C, Chung L, Houghton JA, Huang P, Giles FJ and Cleveland JL:
Targeting autophagy augments the anticancer activity of the histone
deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug
resistance. Blood. 110:313–322. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang L, Yang M, Zhang H, Wang Z, Yu Y, Xie
M, Zhao M, Liu L and Cao L: S100A8-targeting siRNA enhances arsenic
trioxide-induced myeloid leukemia cell death by down-regulating
autophagy. Int J Mol Med. 29:65–72. 2012.
|
|
13
|
Zhu S, Cao L, Yu Y, Yang L, Yang M, Liu K,
Huang J, Kang R, Livesey KM and Tang D: Inhibiting autophagy
potentiates the anticancer activity of IFN1α/IFNα in chronic
myeloid leukemia cells. Autophagy. 9:317–327. 2013. View Article : Google Scholar :
|
|
14
|
Amrein L, Soulières D, Johnston JB and
Aloyz R: p53 and autophagy contribute to dasatinib resistance in
primary CLL lymphocytes. Leuk Res. 35:99–102. 2011. View Article : Google Scholar
|
|
15
|
Larson RA: New agents for induction and
postremission therapy of acute myeloid leukemia. Leukemia.
15:675–676. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ryan KM: p53 and autophagy in cancer:
Guardian of the genome meets guardian of the proteome. Eur J
Cancer. 47:44–50. 2011. View Article : Google Scholar
|
|
18
|
García-Arencibia M, Hochfeld WE, Toh PP
and Rubinsztein DC: Autophagy, a guardian against
neurodegeneration. Semin Cell Dev Biol. 21:691–698. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Scheper W, Nijholt DA and Hoozemans JJ:
The unfolded protein response and proteostasis in Alzheimer
disease: Preferential activation of autophagy by endoplasmic
reticulum stress. Autophagy. 7:910–911. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Martinez-Vicente M and Cuervo AM:
Autophagy and neurodegeneration: When the cleaning crew goes on
strike. Lancet Neurol. 6:352–361. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Puls I, Oh SJ, Sumner CJ, Wallace KE,
Floeter MK, Mann EA, Kennedy WR, Wendelschafer-Crabb G, Vortmeyer
A, Powers R, et al: Distal spinal and bulbar muscular atrophy
caused by dynactin mutation. Ann Neurol. 57:687–694. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Czaja MJ, Ding WX, Donohue TM Jr, Friedman
SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, Ou JH, et
al: Functions of autophagy in normal and diseased liver. Autophagy.
9:1131–1158. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Katsetos CD, Koutzaki S and Melvin JJ:
Mitochondrial dysfunction in neuromuscular disorders. Semin Pediatr
Neurol. 20:202–215. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sugimoto S: A novel vacuolar myopathy with
dilated cardiomyopathy. Autophagy. 3:638–639. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Edinger AL and Thompson CB: Defective
autophagy leads to cancer. Cancer Cell. 4:422–424. 2003. View Article : Google Scholar
|
|
26
|
Karantza-Wadsworth V and White E: Role of
autophagy in breast cancer. Autophagy. 3:610–613. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sheen JH, Zoncu R, Kim D and Sabatini DM:
Defective regulation of autophagy upon leucine deprivation reveals
a targetable liability of human melanoma cells in vitro and in
vivo. Cancer Cell. 19:613–628. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rouschop KM, van den Beucken T, Dubois L,
Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W,
Voncken JW, et al: The unfolded protein response protects human
tumor cells during hypoxia through regulation of the autophagy
genes MAP1LC3B and ATG5. J Clin Invest. 120:127–141. 2010.
View Article : Google Scholar :
|
|
29
|
Karpathiou G, Sivridis E, Koukourakis M,
Mikroulis D, Bouros D, Froudarakis M and Giatromanolaki A:
Light-chain 3A autophagic activity and prognostic significance in
non-small cell lung carcinomas. Chest. 140:127–134. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Savaraj N, You M, Wu C, Wangpaichitr M,
Kuo MT and Feun LG: Arginine deprivation, autophagy, apoptosis
(AAA) for the treatment of melanoma. Curr Mol Med. 10:405–412.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Koukourakis MI, Giatromanolaki A, Sivridis
E, Pitiakoudis M, Gatter KC and Harris AL: Beclin 1 over- and
underexpression in colorectal cancer: Distinct patterns relate to
prognosis and tumour hypoxia. Br J Cancer. 103:1209–1214. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Turcotte S, Chan DA, Sutphin PD, Hay MP,
Denny WA and Giaccia AJ: A molecule targeting VHL-deficient renal
cell carcinoma that induces autophagy. Cancer Cell. 14:90–102.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim RH, Bold RJ and Kung HJ: ADI,
autophagy and apoptosis: Metabolic stress as a therapeutic option
for prostate cancer. Autophagy. 5:567–568. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Boone BA, Bahary N, Zureikat AH, Moser AJ,
Normolle DP, Wu WC, Singhi AD, Bao P, Bartlett DL, Liotta LA, et
al: Safety and biologic response of pre-operative autophagy
inhibition in combination with gemcitabine in patients with
pancreatic adenocarcinoma. Ann Surg Oncol. 22:4402–4410. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Levy JM and Thorburn A: Targeting
autophagy during cancer therapy to improve clinical outcomes.
Pharmacol Ther. 131:130–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fanzani A, Zanola A, Rovetta F, Rossi S
and Aleo MF: Cisplatin triggers atrophy of skeletal C2C12 myotubes
via impairment of Akt signalling pathway and subsequent increment
activity of proteasome and autophagy systems. Toxicol Appl
Pharmacol. 250:312–321. 2011. View Article : Google Scholar
|
|
37
|
Zhao D, Yuan H, Yi F, Meng C and Zhu Q:
Autophagy prevents doxorubicin-induced apoptosis in osteosarcoma.
Mol Med Rep. 9:1975–1981. 2014.PubMed/NCBI
|
|
38
|
Ekiz HA, Can G and Baran Y: Role of
autophagy in the progression and suppression of leukemias. Crit Rev
Oncol Hematol. 81:275–285. 2012. View Article : Google Scholar
|
|
39
|
Helgason GV, Karvela M and Holyoake TL:
Kill one bird with two stones: Potential efficacy of BCR-ABL and
autophagy inhibition in CML. Blood. 118:2035–2043. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Orfali N, McKenna SL, Cahill MR, Gudas LJ
and Mongan NP: Retinoid receptor signaling and autophagy in acute
promyelocytic leukemia. Exp Cell Res. 324:1–12. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Watson AS, Mortensen M and Simon AK:
Autophagy in the pathogenesis of myelodysplastic syndrome and acute
myeloid leukemia. Cell Cycle. 10:1719–1725. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
O'Donovan TR, O'Sullivan GC and McKenna
SL: Induction of autophagy by drug-resistant esophageal cancer
cells promotes their survival and recovery following treatment with
chemotherapeutics. Autophagy. 7:509–524. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zeng X, Yan T, Schupp JE, Seo Y and
Kinsella TJ: DNA mismatch repair initiates 6-thioguanine-induced
autophagy through p53 activation in human tumor cells. Clin Cancer
Res. 13:1315–1321. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang C, Tong Y, Ni W, Liu J, Xu W, Li L,
Liu X, Meng H and Qian W: Inhibition of autophagy induced by
overexpression of mda-7/interleukin-24 strongly augments the
antileukemia activity in vitro and in vivo. Cancer Gene Ther.
17:109–119. 2010. View Article : Google Scholar
|
|
45
|
Zhao X, Liu J, Peng M, Liu J and Chen F:
BMP4 is involved in the chemoresistance of myeloid leukemia cells
through regulating autophagy-apoptosis balance. Cancer Invest.
31:555–562. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yang M, Zeng P, Kang R, Yu Y, Yang L, Tang
D and Cao L: S100A8 contributes to drug resistance by promoting
autophagy in leukemia cells. PLoS One. 9:e972422014. View Article : Google Scholar : PubMed/NCBI
|