Introduction
Obstructive jaundice (OJ) occurs due to occlusion of
the common bile duct, or complications associated with surgery,
tumor, trauma, gallstones, hepatitis, or idiopathic and metabolic
diseases, including primary biliary cirrhosis and sclerosing
cholangitis (1). Cholestatic liver
injury is accompanied by serious complications, which increase the
risk of mortality and morbidity (2). The mechanisms by which cholestasis
induce acute liver injury remain controversial; however,
intrahepatic accumulation of reactive oxygen species (ROS) is
thought to be an important contributory factor (3–5). ROS
are able to trigger the opening of mitochondrial permeability
transition (MPT) pores in the mitochondrial inner membrane, which
nonspecifically transport solutes up to a molecular mass of 1,500
Da (6). Opening of MPT pores lead
to cytochrome c release and apoptosis (7,8).
Previous studies have indicated that mitochondrial stress and
apoptosis have an important role in hepatic injury in OJ (8,9). In
particular, it has been demonstrated that, following cholestasis,
an accumulation of bile acids in hepatocytes contributes to cell
death and is one of the major pathogenic factors resulting in
chronic liver damage and fibrosis (10).
Hydrogen (H2), in the gaseous state or
dissolved in water, has been reported to have therapeutic value as
a selective antioxidant via its ability to reduce cytotoxic ROS and
to suppress inflammatory reactions (11–13).
Unlike other gaseous molecules, H2 can penetrate the
cell membrane to reach subcellular compartments, including
mitochondria, which are notoriously difficult to target. The
utilization of H2 gas-saturated physiological saline,
also known as hydrogen-rich saline (HS), is considered to be less
complicated and safer than H2 gas inhalation for
clinical application. Furthermore, we have previously demonstrated
that HS is able to attenuate bile duct ligation (BDL)-induced liver
damage by reducing hepatic oxidative stress and inflammation, and
can reduce apoptosis in neonatal brain tissue from a rat model of
hypoxia-ischemia (14,15). The present study specifically
assessed the impact of BDL-induced injury on the hepatic
mitochondria of mice, in order to investigate whether HS was able
to exert direct protective effects on the mitochondria, and thus
prevent mitochondrial damage and mitochondria-induced hepatocyte
apoptosis.
Materials and methods
HS production
HS was prepared as previously described (16). HS was freshly prepared on a weekly
basis to ensure that a concentration >0.6 mmol/l was
maintained.
Experimental protocol
Male C57BL/6 mice, weighing 22–25 g, were obtained
from the Experimental Animal Center of Chinese Academy of Sciences
(Shanghai, China). Mice received ad libitum access to
standard rodent chow and tap water, and were maintained under a
natural day/night cycle. All experimental procedures were approved
by the Institutional Animal Care and Use Committee of Nankai
University (Tianjin, China).
Mice were randomly divided into three experimental
groups, each containing 20 mice. Group 1 animals underwent a sham
operation and were treated with normal saline (NS; 10 ml/kg); group
2 animals underwent BDL and were treated with NS (10 ml/kg); and
group 3 animals underwent BDL and were treated with HS (10
ml/kg).
Prior to the operation, mice were fasted for 12 h
with ad libitum access to water. Each mouse was weighed and
anesthetized with pentobarbital (50 mg/kg; i.p.; Shanghai Reagent
Factory, Shanghai, China). Following a midline incision, the common
bile duct was exposed and a double-ligature was performed using 6-0
silk suture, causing the bile duct to be sectioned between the
ligatures. In sham-operated animals, the common bile duct was freed
from the surrounding soft tissue without ligation. A running suture
was used for abdominal closure using 2-0 nylon. NS or HS was
administered intraperitoneally at 14:00 every day, beginning 2 h
prior to the operation and continuing until 2 days after. Mice were
sacrificed via cervical dislocation according to protocol 3 days
after BDL.
Preparation of mitochondrial and
cytosolic fractions
Following homogenization of the liver tissues, liver
mitochondria and cytosol were prepared by differential
centrifugation using a mitochondria isolation kit (Pierce
Biotechnology, Inc., Rockford, IL, USA), according to
manufacturer's protocol. The resulting supernatant contained
soluble mitochondrial protein, which was used for western blot
analysis of B-cell lymphoma 2 (Bcl-2), Bcl-2-associated X protein
(Bax), and mitochondrial cytochrome c. Protein content was
determined using a bicinchoninic acid (BCA) protein assay kit
(Pierce Biotechnology, Inc.).
Determination of antioxidant and lipid
peroxidation levels in mitochondria
The mitochondrial suspension was acidified with 2%
3-[(3-cholamidopropyl)dimethylammonio]-1-pro-panesulfonate in
Tris-buffered saline (TBS) and centrifuged at 9,055.8 × g for 2 min
at room temperature, according to manufacturer's protocol for the
mitochondria isolation kit. The supernatant was analyzed for
mitochondrial malondialdehyde (mMDA), mitochondrial glutathione
(mGSH), mitochondrial glutathione disulfide (mGSSG), mitochondrial
superoxide dismutase (mSOD), mitochondrial catalase (mCAT), and
mitochondrial glutathione peroxidase (mGpx) levels. mMDA levels,
and reduced and oxidized mGSH levels were assessed
spectrophotometrically, according to previously described methods
(16,17). mSOD, mCAT and mGpx activities were
assessed using commercial enzyme-linked immunosorbent assay kits,
according to the manufacturer's protocols (Nanjing Jiangcheng
Bioengineering Institute, Nanjing, China). All readings were taken
using a spectrophotometer (Synergy 2; BioTek Instruments, Inc.,
Winooski, VT, USA). All assays were conducted in duplicate. Protein
content in each sample was determined using a BCA protein assay kit
(Pierce Biotechnology, Inc.).
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) staining
Following dehydration, permeation and methanol
fixation, TUNEL staining was performed on 5-μm-thick
paraffin-embedded sections using an In Situ Cell Death
Detection kit (Nanjing Keygen Biotech. Co. Ltd., Nanjing, China),
according to manufacturer's protocol, in order to detect apoptotic
hepatocytes. TUNEL-positive cells were detected using an Olympus
IX70 fluorescence microscope (Olympus Corporation, Tokyo,
Japan).
Fluorescence-activated cell sorting
(FACS) analysis
Single-cell suspensions were prepared using a Tissue
Dissociation kit (Nanjing Keygen Biotech. Co. Ltd.), according to
the manufacturer's protocol. Hepatocyte apoptosis was measured by
flow cytometric analysis using an Annexin V-fluorescein
isothiocyanate (FITC) assay (Nanjing Keygen Biotech. Co. Ltd.),
according to the manufacturer's protocol. Hepatocytes were stained
with Annexin V and propidium iodide (PI; BD Biosciences, San Diego,
CA, USA) (18) and apoptotic cells
were identified as Annexin V-positive/PI-negative. Analysis was
performed using the BD FACSAria flow cytometer (BD
Biosciences).
Caspase activity assay
Caspase 3, 8 and 9 activities were measured in liver
tissue using Caspase Assay kits (Promega Corporation, Madison, WI,
USA), according to the manufacturer's protocols. The luminescence
of each sample was measured using a spectrophotometer (Synergy 2;
BioTek Instruments, Inc.).
Mitochondrial swelling
Fresh liver mitochondria were isolated from the BDL
and sham-operated mice by differential centrifugation and incubated
with 100 μM CaCl2 prior to treatment with
cyclosporin A (CsA, 1 mM; Amresco, LLC, Solon, OH, USA) MPT
inhibitor to assess calcium-induced mitochondrial swelling, as
previously described (19).
Western blot analysis
Mitochondrial lysates were used for western blot
analysis of cytochrome c, Bcl-2 and Bax. Cytosolic fractions
were used for western blot analysis of cytochrome c, and
were processed according to the manufacturer's protocol (Abcam,
Cambridge, MA, USA). Proteins (2.5 μg/μl) were
separated by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and transferred onto a polyvinylidene difluoride
membrane (Bio-Rad Laboratories, Hercules, CA, USA). Following
blocking with 5% skimmed milk, the membranes were washed three
times in TBS with Tween 20 (TBST) for 5 min, and subsequently
incubated with primary monoclonal antibodies against cytochrome
c (ab13575), Bcl-2 (ab32503) and Bax (all 1:1,000; ab117115)
for 2 h at room temperature. Following washing three times with
TBST for 5 min, the membranes were subsequently incubated with goat
anti-mouse and rabbit secondary antibodies (1:2,000; DC02L-200UG;
Calbiochem, LaJolla, CA, USA) for 2 h at room temperature. For all
determinations, mouse monoclonal anti-glyceraldehyde 3-phosphate
dehydrogenase antibody (1:5,000; ab9485; Abcam)) was used as a
loading control and goat anti-manganese superoxide dismutase
antibody (1:1,000; 13194; Cell Signaling Technology, Inc., Danvers,
MA, USA) was used as a mitochondrial loading control. Blots were
visualized using a Beyotime enhanced chemiluminescence Plus
substrate system (Beyotime Institute of Biotechnology, Haimen,
China) and analyzed with Quantity One 4.62 software (Bio-Rad
Laboratories). It should be noted that it has previously been
suggested that cytoplasmic cytochrome c may exist in
polymeric forms, appearing as a 58–60 kD protein-sized band in
western blots, rather than in monomeric form, which migrates at a
reduced molecular weight of 15 kD (20).
Transmission electron microscopy
(TEM)
Liver samples were fixed in a 2% solution of
glutaraldehyde and post-fixed in osmium tetroxide, prior to
embedding in epoxy resin for TEM. Ultrathin sections (40–50 nm)
were stained with uranyl acetate and lead citrate, and were then
examined under a TEM (Hitachi H-7650; Hitachi, Tokyo, Japan).
Determination of mitochondrial adenosine
triphosphate (ATP) content
ATP content was measured using the ATP
Bioluminescent Assay kit (Sigma-Aldrich Canada, Oakville, ON,
Canada), according to the manufacturer's protocol.
Determination of mitochondrial
respiratory function
Mitochondrial respiratory function was determined
using the Clark Oxygen Electrode system (Oxygraph™, Hansatech
Instruments, Ltd., King's Lynn, UK), according to previously
described methods (21). The
mitochondrial respiratory control ratio (RCR) and adenosine
diphosphate (ADP) to oxygen ratio (ADP/O) can reflect mitochondrial
respiratory function and integrity of the respiratory chain.
Mitochondrial RCR is the ratio of state 3 and 4 respiration rates.
ADP/O is the ratio of ADP and state 3 oxygen consumption. State 3
is the respiration rate after the addition of 1 mM ADP; whereas
state 4 is the oxygen consumption rate after the complete
phosphorylation of ADP.
Statistical analysis
All experiments were performed in triplicate and
statistical analyses were conducted using SPSS 22.0 software (IBM
SPSS, Armonk, NY, USA). Data are presented as the mean ± standard
deviation. Statistical analyses were performed using one-way
analysis of variance (ANOVA) for the comparison of three groups.
When ANOVA exhibited significant differences, pairwise comparisons
between means were tested by Student-Newman-Keuls post-hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
HS prevents mitochondrial oxidative
stress and increases antioxidant activities in BDL mice
The levels of mMDA were significantly increased
(99.6%) in the livers of BDL mice compared with in the
sham-operated mice. Conversely, treatment with HS markedly
prevented the elevation of lipid peroxidation in mitochondria
(Fig. 1A; P=0.006). The potential
antioxidative properties of HS were determined by measuring the
levels of mGSH and mGSSG. In NS-treated BDL mice, mGSH levels were
reduced to 58.9% of normal levels and mGSSG levels were increased
by 1.276-fold; however, HS significantly attenuated the altered
mGSH and mGSSG levels (Fig. 1B and
C; P=0.005 and P=0.004, respectively). The results also
indicated that an increase in mitochondrial oxidative stress was
accompanied by a significant decrease in the activities of the
antioxidant enzymes SOD, CAT and Gpx in the hepatic mitochondria of
BDL mice, whereas HS treatment markedly increased antioxidant
activities in BDL mice (Fig. 1D–F;
P=0.006, P=0.005 and P=0.009, respectively).
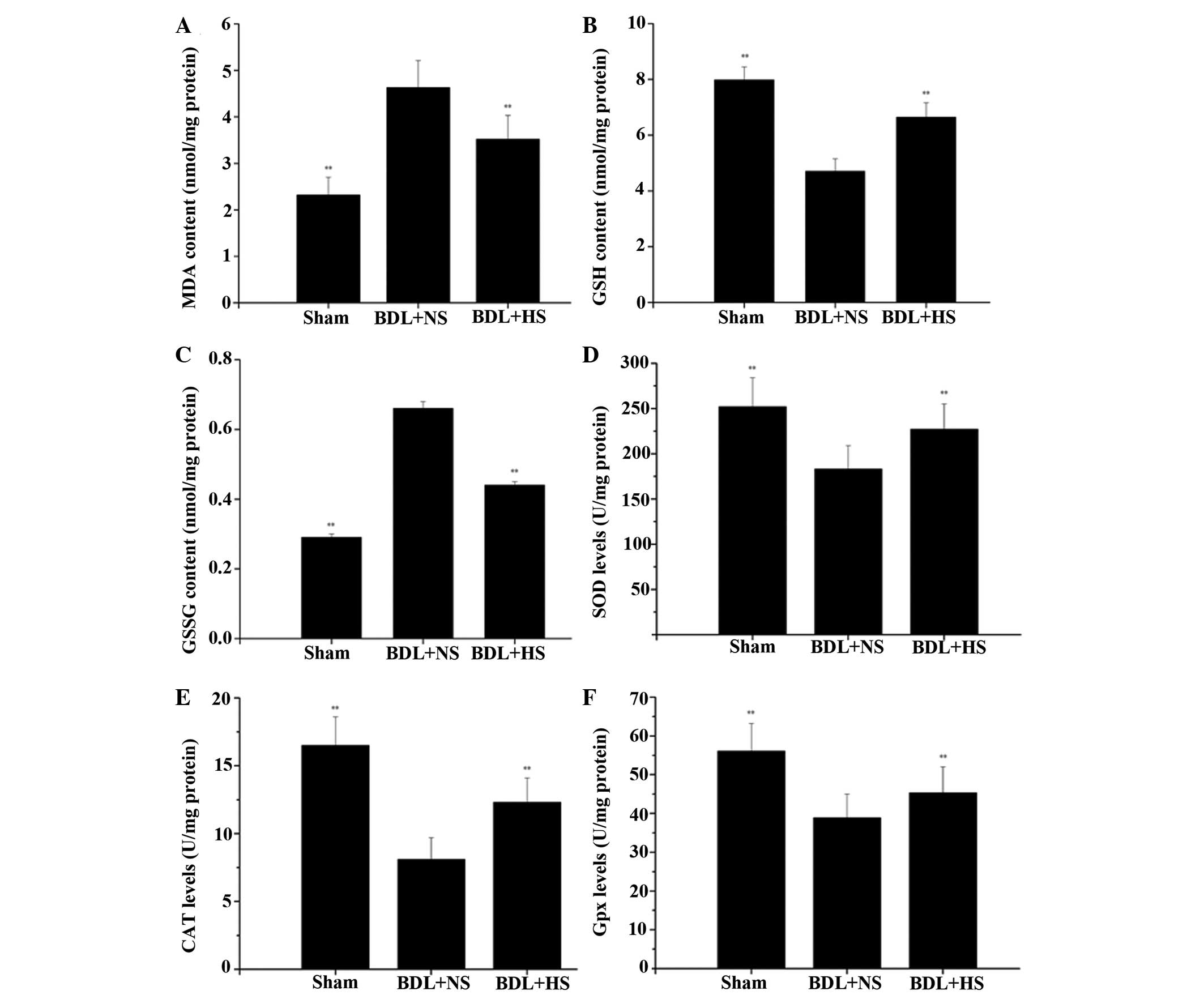 | Figure 1Effects of HS on liver mitochondrial
enzyme levels and activities following BDL in mice. (A)
Mitochondrial MDA levels; (B) mitochondrial GSH levels; (C)
mitochondrial GSSG levels; (D) mitochondrial SOD activities; (E)
mitochondrial CAT activities and (F) mitochondrial Gpx activities
were measured spectrophotometrically, according to the
manufacturers' protocols. Data are presented as the mean ± standard
deviation. **P<0.01 vs. the BDL + NS group. HS,
hydrogen-rich saline; BDL, bile duct ligation; NS, normal saline;
MDA, malondialdehyde; GSH, glutathione; GSSG, glutathione
disulfide; SOD, superoxide dismutase; CAT, catalase; Gpx,
glutathione peroxidase. |
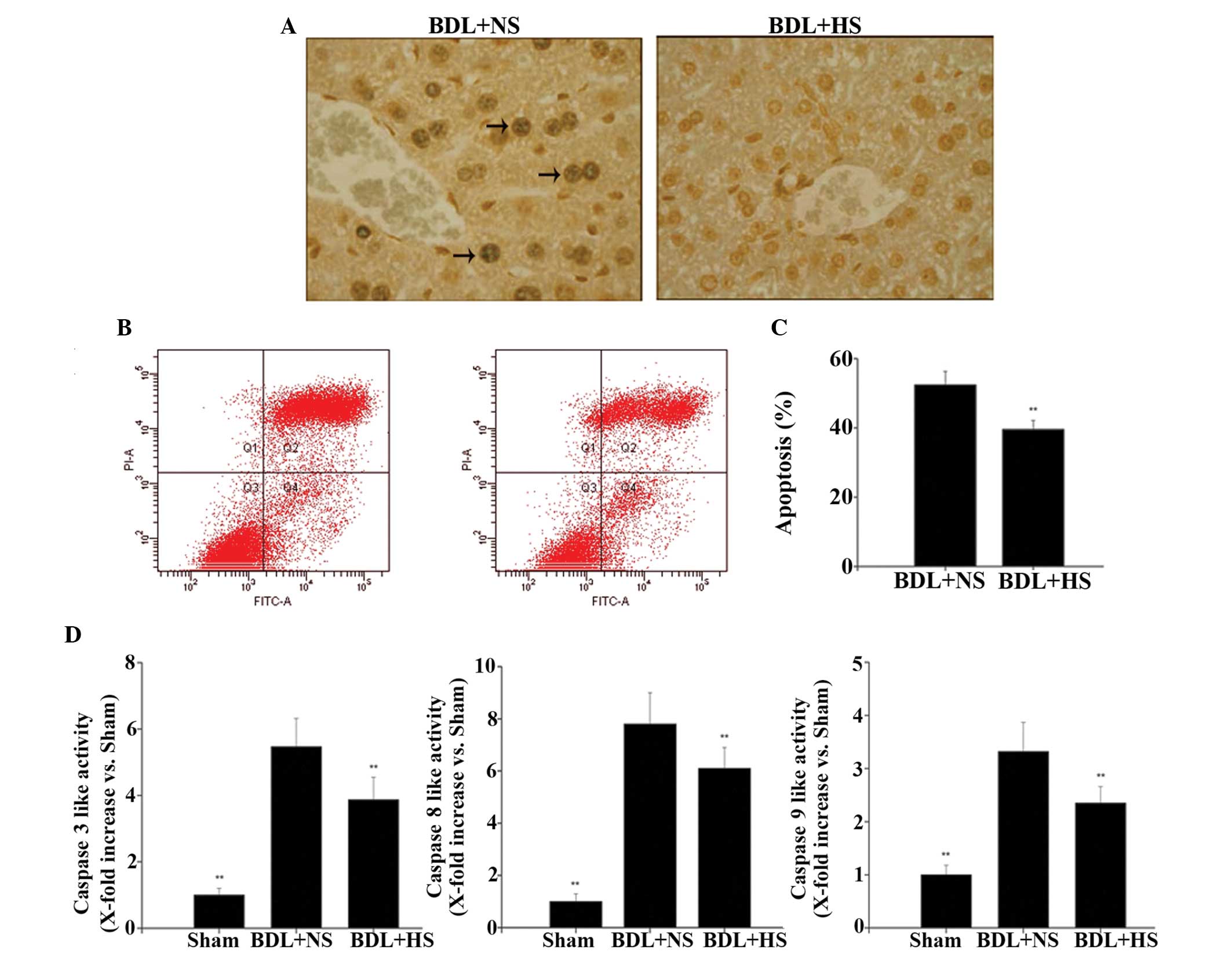
HS decreases apoptosis in hepatocytes
from BDL mice
HS markedly reduced the number of TUNEL-positive
cells in the liver compared with the NS-treated group (Fig. 2A), thus suggesting that HS is able
to inhibit BDL-induced hepatocyte apoptosis. To further confirm
that HS affected hepatocyte apoptosis, flow cytometric analysis was
conducted using Annexin V-FITC and PI staining, in order to
discriminate between apoptotic and necrotic cells. In BDL mice,
treatment with HS induced significantly lower levels of apoptosis
(39.8%) compared with in the NS-treated group (52.5%) (Fig. 2B and C; both P=0.008).
HS inhibits caspase activities in the
livers of BDL mice
Since caspase activation has a key role in apoptotic
cell death, the present study further investigated whether caspase
activities could be altered following treatment with HS. BDL
triggered a significant increase in hepatic caspase 3, 8 and 9
activities, whereas HS administration significantly reduced caspase
activities (Fig. 2D; P=0.007,
P=0.008 and 0.007, respectively). These findings were consistent
with the results of the apoptosis analysis.
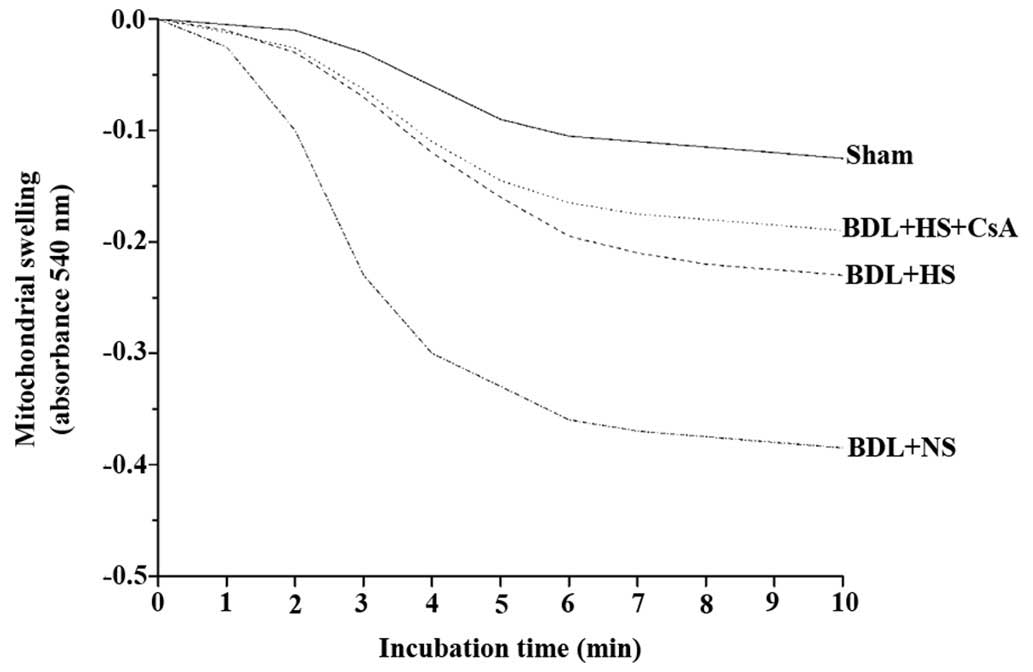
HS inhibits mitochondrial swelling
Mitochondria from the sham-operated mice tolerated
Ca2+ at a concentration of 100 μM without
undergoing MPT, as assessed by a mitochondrial swelling assay
(22). Conversely, mitochondria
from the NS-treated BDL mice were much more sensitive to MPT
induction. A large-amplitude swelling was observed in mitochondria
isolated from NS-treated BDL mice compared with in the mitochondria
from sham-operated mice. In addition, treatment with HS prevented
BDL-induced mitochondrial swelling (Fig. 3).
HS inhibits mitochondrial cytochrome c
release in the liver of BDL mice
Western blot analysis (Fig. 4) indicated that NS-treated BDL mice
exhibited a significant reduction in the protein expression levels
of mitochondrial cytochrome c, which was accompanied by a
release into the cytoplasm, as reflected by an increase in
cytosolic cytochrome c expression levels (Fig. 4A and C; P=0.008). Treatment with HS
significantly inhibited the release of cytochrome c from the
mitochondria to the cytoplasm (Fig. 4A
and C; P=0.006).
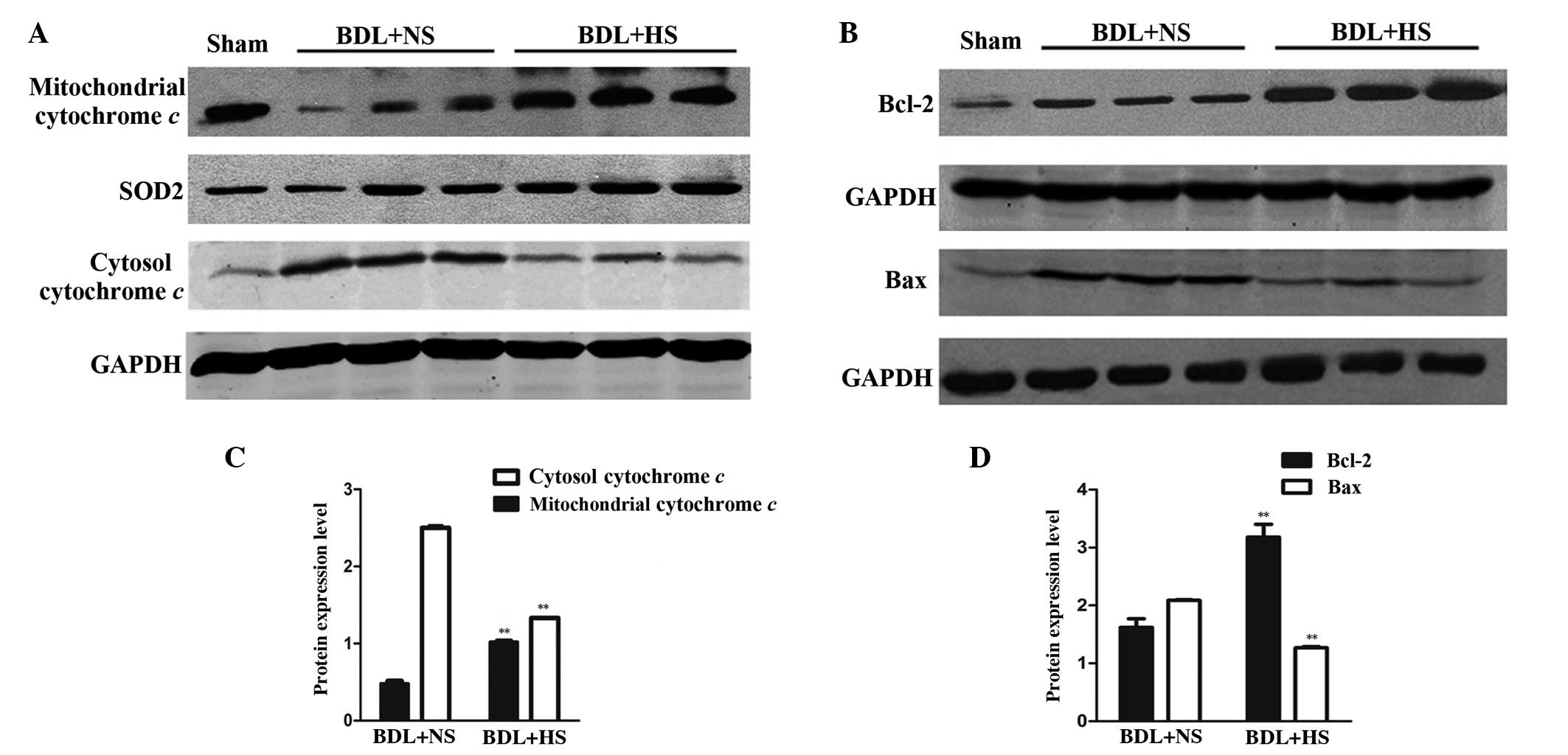 | Figure 4Western blot analysis of cytochrome
c, Bcl-2 and Bax protein expression levels in BDL mice. (A)
Mitochondrial and cytosolic lysates were subjected to western
blotting using cytochrome c-specific antibodies. Treatment
with HS significantly suppressed the release of cytochrome c
from the mitochondria into the cytosol. Images are representative
of four independent experiments. SOD2 and GAPDH were used as the
mitochondrial and cytosolic loading controls, respectively. (B)
Liver lysates were subjected to western blot analysis using Bcl-2
and Bax-specific antibodies. The protein expression levels of Bax
were increased, whereas Bcl-2 expression was decreased in livers
from HS-treated mice compared with those obtained from NS-treated
mice. Images are representative of four independent experiments.
GAPDH was used as a loading control for normalization. (C and D)
Western blotting quantification. Data are presented as the mean ±
standard deviation. **P<0.01 vs. the BDL + NS group.
HS, hydrogen-rich saline; BDL, bile duct ligation; NS, normal
saline; SOD2, manganese superoxide dismutase; GAPDH, glyceraldehyde
3-phosphate dehydrogenase; Bcl-2, B-cell lymphoma 2; Bax,
Bcl-2-associated X protein. |
HS prevents alterations in mitochondrial
Bcl-2 family protein expression in the liver of BDL mice
Pro-apoptotic and anti-apoptotic members of the
Bcl-2 protein family have critical roles in regulating the MPT and
cytochrome c release (23).
Therefore, the present study evaluated the mitochondrial expression
levels of Bcl-2 and Bax in BDL livers. Western blotting
demonstrated a significant increase in Bax protein expression, and
a concomitant reduction in Bcl-2 protein expression in the
mitochondria of BDL mice (Fig. 4B and
D; P=0.006). These BDL-induced effects were prevented by HS
treatment (Fig. 4B and D;
P=0.008).
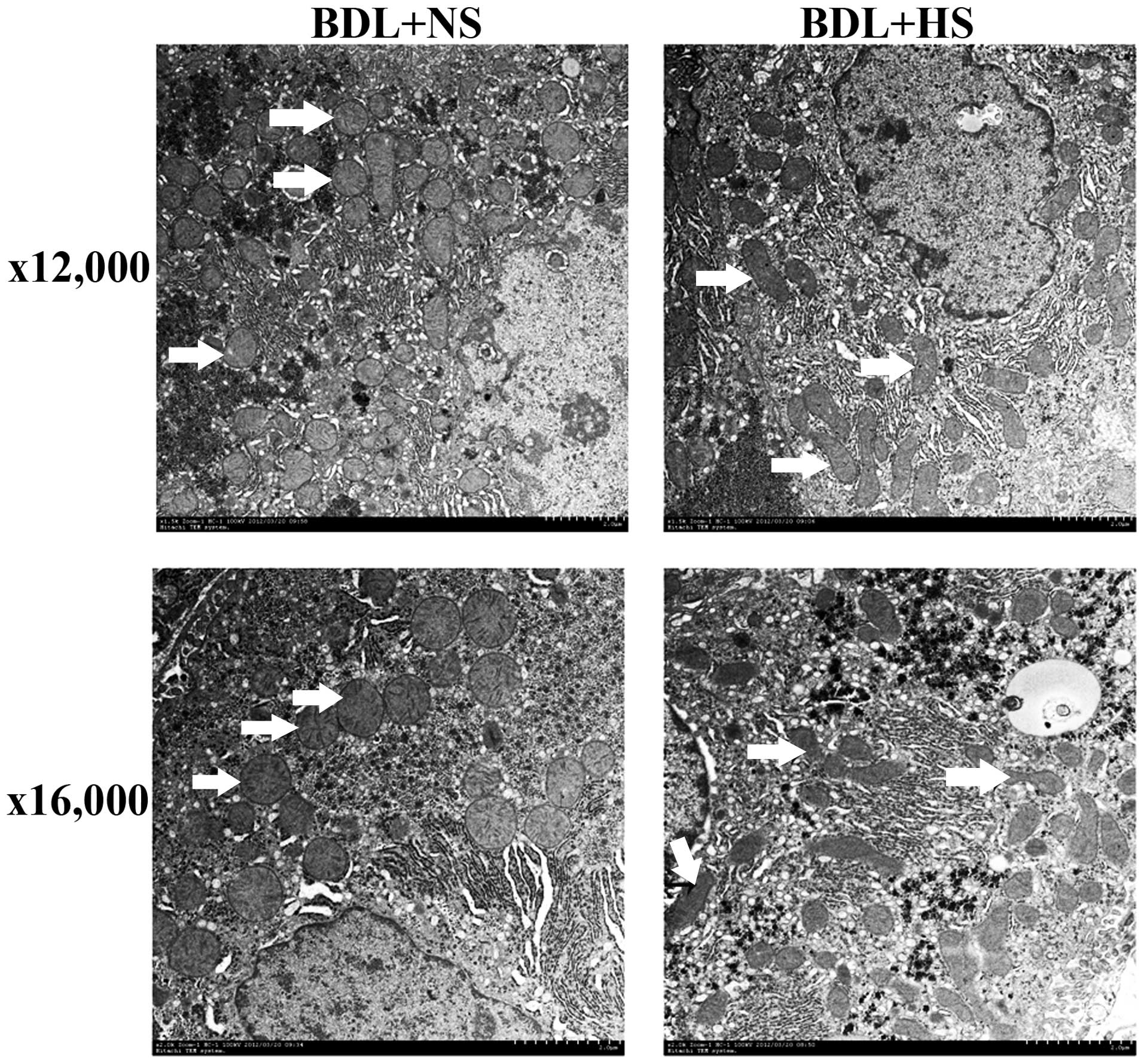
HS protects mitochondrial ultrastructure
in BDL mice
TEM analysis of liver tissue indicated that
mitochondrial ultrastructural alterations occurred in the
hepatocytes of NS-treated BDL mice in the present study. In
addition to the observed swelling, TEM analysis clearly showed
impaired mitochondria, with an absent double membrane; distorted
cristae, which were far fewer than in the sham mice; and an
appreciable reduction in electron-dense granules in the
intramitochondrial matrix. All of these ultrastructural
modifications were markedly alleviated following treatment with HS
(Fig. 5).
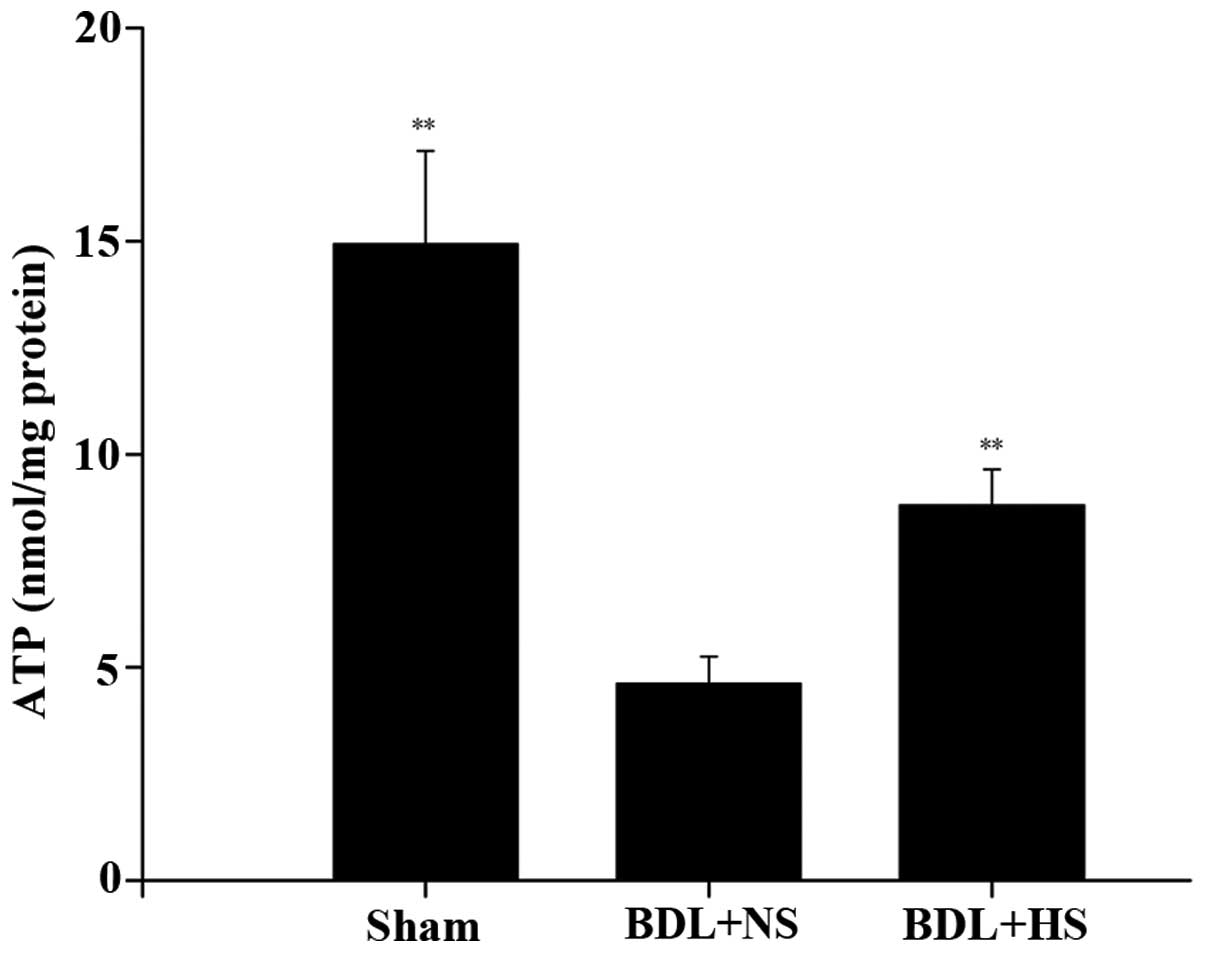
HS prevents the depletion of ATP levels
in hepatocytes of BDL mice
The present study examined the effects of HS on ATP
levels, which is a sensitive parameter of mitochondrial function.
ATP levels were decreased (67.1%) in BDL mice compared with in the
sham-operated mice. Conversely, treatment with HS significantly
increased ATP levels in the hepatocytes of BDL mice (Fig. 6; P=0.007).
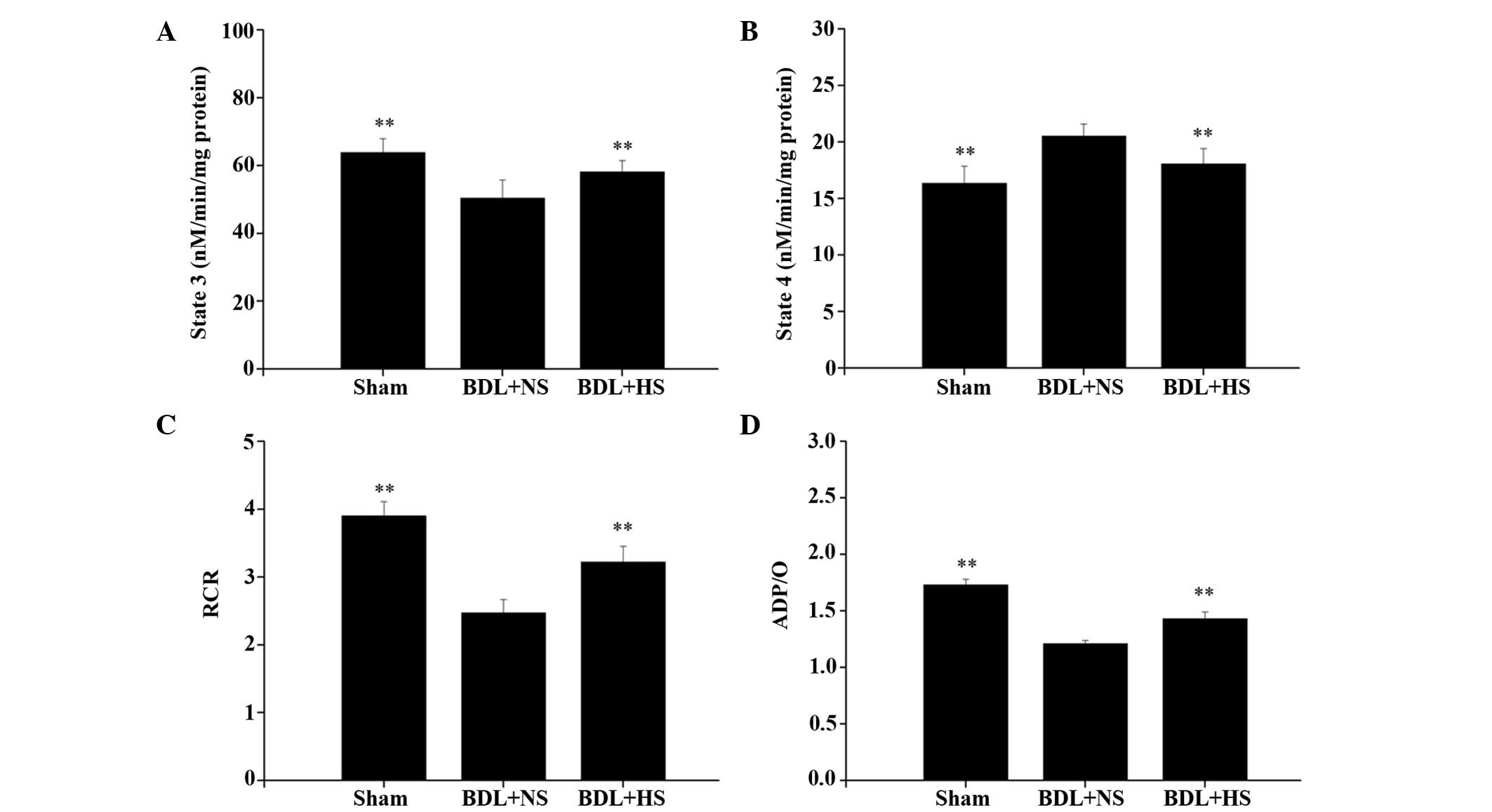
HS improves the BDL-induced decline in
mitochondrial respiratory function
BDL induced a significant reduction in RCR (31.3%)
compared with in the sham-operated mice (P<0.01). This decrease
in RCR could be principally attributed to the observed significant
decrease (18.5%) of State 3 (Fig.
7A; P=0.009), as opposed to the 19.3% increase of State 4
(Fig. 7B; P= 0.009). HS promoted a
significant increase in RCR compared with the NS group (Fig. 7C; P=0.08). This protection was
principally related to an increase of State 3 and a decrease of
State 4 (Fig. 7A and B). ADP/O was
also significantly increased (17.4%) following HS treatment
(Fig. 7D; P=0.009).
Discussion
OJ induces ROS generation and tumor necrosis
factor-α expression in the liver, both of which can induce
depolarization of the mitochondrial membrane and eventually
initiate apoptosis (24). Our
previous study demonstrated that HS was able to ameliorate
BDL-induced liver injury by reducing oxidative stress and
inflammatory cascades in liver tissue (14). Theoretically, H2 is
highly diffusible and could potentially reach mitochondria, which
are targets of excessive ROS and central mediators of apoptosis
(25,26). Therefore, the aim of the present
study was to evaluate whether HS, a selective antioxidant, has the
capacity to reduce mitochondrial oxidative stress, and thus protect
against mitochondrial dysfunction and inhibit mitochondrial
apoptosis, in order to prevent BDL-induced liver injury.
Mitochondria are the major source and target of
excessive ROS (26). Mitochondrial
GSH scavenges free oxygen radicals that are generated by the
mitochondrial respiratory chain (27) and antioxidant enzymes, including
SOD, CAT and Gpx, work synergistically to cope with oxidative
stress. Accumulated exposure to ROS leads to an oxidative stress
burden in the mitochondria, which can induce an apparent increase
in ROS generation by the electron transfer chain and suppress the
mitochondrial antioxidant system (28). In the present study, mMDA was
significantly increased by BDL. In addition, OJ was revealed to
impair the activities of mSOD, mCAT and mGpx by 36.1, 53.8 and
32.2%, respectively, and mGSH was also depleted by BDL-induced
oxidative stress. Treatment with HS markedly decreased mMDA levels,
increased mGSH levels, and elevated the activities of mSOD, mCAT
and mGpx, thus suggesting that H2 may reduce
mitochondrial lipid peroxidation, boost antioxidant capacity and
maintain the mitochondrial redox balance.
Mitochondrial oxidative stress triggers the opening
of the MPT pore, and the simultaneous collapse of the mitochondrial
membrane potential (MMP) (29–31).
MPT further increases the permeability of the mitochondrial outer
membrane and induces mitochondrial swelling (30,31).
Furthermore, cytochrome c, a mitochondrial intermembrane
protein, is released into the cytosol via specific channels,
including the MPT pore and Bax channel, and may further activate
the downstream caspase pathway to induce irreversible apoptosis
(31).
The present study used 100 μM
CaCl2 as an MPT inducer, and demonstrated that
HS-treated BDL mice exhibited a decrease in mitochondrial swelling
compared with the NS-treated mice, thus indicating that HS inhibits
the opening of the MPT pore and prevents the onset of MPT. Similar
results were obtained following an intraperitoneal injection of CsA
for MPT protection. CsA, which is a specific MPT inhibitor, was
able to significantly protect against BDL-induced mitochondrial
swelling. The combination of CsA and HS offered more efficient
protection than HS or CsA alone against BDL-induced mitochondrial
swelling. Therefore, it may be hypothesized that part of the
mechanism involved in HS-induced protection is via inhibition of
MPT. The present study also detected leakage of cytochrome c
into the cytosol in BDL mice, which is correlated with MPT pore
opening. Conversely, treatment with HS prevented the release of
cytochrome c into the cytosol.
Mitochondria are the central control point of
apoptosis (32). OJ induces
apoptotic cell death, which is associated with mitochondrial
oxidative stress, MMP alteration and cytochrome c release
(33,34). In the present study, BDL resulted
in the accumulation of TUNEL-positive and Annexin-V-positive cells.
The TUNEL-positive and Annexin-V-positive cells were markedly
decreased in the HS-treated group, thus suggesting that HS may
provide hepatic protection via its anti-apoptotic activity.
Members of the Bcl-2 family are key players in the
mitochondrial intrinsic pathway of apoptosis (35). The Bcl-2 family consists of pro-
and anti-apoptotic proteins that work together to mediate
mitochondrial integrity and maintain a dynamic balance between cell
survival and cell death. Bax integrates with the permeability
transition pore complex and forms specific proteolipid channels in
the outer membrane, in order to promote cytochrome c
release, whereas Bcl-2 directly binds to Bax to inhibit formation
of the proteolipid pore (36–38).
In the present study, BDL increased the expression of Bax and
decreased the expression of Bcl-2 in the liver, whereas treatment
with HS markedly attenuated BDL-induced elevation of Bax expression
and reduction of Bcl-2 expression. These data suggested that HS may
markedly suppress downstream apoptotic events by modulating the
expression levels of Bcl-2 family members.
The caspase family consists of cysteine proteases
that can cleave target proteins at specific aspartate residues.
Previous studies have reported that caspases have important roles
in the initiation, regulation and execution of apoptosis of
hepatocytes in response to BDL-induced injury (38–40).
One of the main consequences following mitochondrial cytochrome
c release is the activation of caspase 3 through the
apoptosome, which consists of cytochrome c, apoptotic
protease activating factor-1 and procaspase 9 (9). Caspases 3 and 9 are downstream
effectors in the caspase-dependent intrinsic apoptosis pathway,
whereas caspase 8 is an essential part of the extrinsic pathway.
The present study demonstrated that activation of caspases 3, 8 and
9 was significantly suppressed in HS-treated mice, thus indicating
that HS may inhibit BDL-induced apoptosis via both intrinsic and
extrinsic pathways.
The results of the present study demonstrated that
HS was able to protect mitochondrial respiratory function and
attenuate the depletion of mitochondrial ATP in BDL mice. In
addition, HS was shown to attenuate mitochondrial ultra-structural
injury, as evidenced by TEM observations. These results strongly
suggested that HS may effectively prevent BDL-induced mitochondrial
dysfunction.
In conclusion, the present study provides some of
the first evidence to suggest that HS is able to ameliorate
BDL-induced acute liver damage by reducing mitochondrial ROS
production, protecting against mitochondrial dysfunction, and
inhibiting mitochondria-mediated apoptosis. These findings provide
evidence regarding the possible mechanisms underlying the
protective role of HS. Furthermore, H2 treatment appears
to decrease liver fibrosis, cirrhosis, and overall mortality during
long-term cholestasis (unpublished data). Since mitochondria are
known to be involved in numerous signaling pathways and there are
limited data from clinical trials involving HS treatment, it
remains to be elucidated whether the regulatory effects of HS on
gene pathways associated with apoptosis and oxidative stress have
direct clinical value.
Abbreviations:
|
OJ
|
obstructive jaundice
|
|
ROS
|
reactive oxygen species
|
|
MPT
|
mitochondrial permeability
transition
|
|
H2
|
hydrogen
|
|
HS
|
hydrogen-rich saline
|
|
BDL
|
bile duct ligation
|
|
mMDA
|
mitochondrial malondialdehyde
|
|
mGSH
|
mitochondrial glutathione
|
|
mGSSG
|
mitochondrial glutathione
disulfide
|
|
mSOD
|
mitochondrial superoxide dismutase
|
|
mCAT
|
mitochondrial catalase
|
|
mGpx
|
mitochondrial glutathione
peroxidase
|
|
TUNEL
|
terminal deoxynucleotidyl transferase
dUTP nick end labeling
|
|
HBSS
|
Hank's balanced salt solution
|
|
PBS
|
phosphate-buffered saline
|
|
TEM
|
transmission electron microscopy
|
|
SOD2
|
superoxide dismutase
|
|
CsA
|
cyclosporin A
|
|
MPT
|
mitochondrial potential transition
|
|
RCR
|
respiratory control ratio
|
|
ADP/O
|
oxidative phosphorylation
|
|
ANOVA
|
analysis of variance
|
|
NS
|
normal saline
|
|
MMP
|
mitochondrial membrane potential
|
Acknowledgments
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no.
31170926).
References
|
1
|
Poupon R, Chazouillères O and Poupon RE:
Chronic cholestatic diseases. J Hepatol. 32(Suppl 1): 129–140.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Greig JD, Krukowski ZH and Matheson NA:
Surgical morbidity and mortality in one hundred and twenty-nine
patients with obstructive jaundice. Br J Surg. 75:216–219. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vendemiale G, Grattagliano I, Lupo L,
Memeo V and Altomare E: Hepatic oxidative alterations in patients
with extra-hepatic cholestasis. Effect of surgical drainage J
Hepatol. 37:601–605. 2002.
|
|
4
|
Assimakopoulos SF, Vagianos CE,
Zervoudakis G, Filos KS, Georgiou C, Nikolopoulou V and Scopa CD:
Gut regulatory peptides bombesin and neurotensin reduce hepatic
oxidative stress and histological alterations in bile duct ligated
rats. Regul Pept. 120:185–193. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu TZ, Lee KT, Chern CL, Cheng JT, Stern
A and Tsai LY: Free radical-triggered hepatic injury of
experimental obstructive jaundice of rats involves overproduction
of proinflammatory cytokines and enhanced activation of nuclear
factor kappaB. Ann Clin Lab Sci. 31:383–390. 2001.PubMed/NCBI
|
|
6
|
Kantrow SP, Tatro LG and Piantadosi CA:
Oxidative stress and adenine nucleotide control of mitochondrial
permeability transition. Free Radic Biol Med. 28:251–260. 2000.
View Article : Google Scholar
|
|
7
|
Kim JS, He L, Qian T and Lemasters JJ:
Role of the mitochondrial permeability transition in apoptotic and
necrotic death after ischemia/reperfusion injury to hepatocytes.
Curr Mol Med. 3:527–535. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Miyoshi H, Rust C, Roberts PJ, Burgart LJ
and Gores GJ: Hepatocyte apoptosis after bile duct ligation in the
mouse involves Fas. Gastroenterology. 117:669–677. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Canbay A, Feldstein A, Baskin-Bey E, Bronk
SF and Gores GJ: The caspase inhibitor IDN-6556 attenuates hepatic
injury and fibrosis in the bile duct ligated mouse. J Pharmacol Exp
Ther. 308:1191–1196. 2004. View Article : Google Scholar
|
|
10
|
Wang L, Hartmann P, Haimerl M, Bathena SP,
Sjöwall C, Almer S, Alnouti Y, Hofmann AF and Schnabl B: Nod2
deficiency protects mice from cholestatic liver disease by
increasing renal excretion of bile acids. J Hepatol. 60:1259–1267.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ohsawa I, Ishikawa M, Takahashi K,
Watanabe M, Nishimaki K, Yamagata K, Katsura K, Katayama Y, Asoh S
and Ohta S: Hydrogen acts as a therapeutic antioxidant by
selectively reducing cytotoxic oxygen radicals. Nat Med.
13:688–694. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zheng X, Mao Y, Cai J, Li Y, Liu W, Sun P,
Zhang JH, Sun X and Yuan H: Hydrogen-rich saline protects against
intestinal ischemia/reperfusion injury in rats. Free Radic Res.
43:478–484. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kajiya M, Silva MJ, Sato K, Ouhara K and
Kawai T: Hydrogen mediates suppression of colon inflammation
induced by dextran sodium sulfate. Biochem Biophys Res Commun.
386:11–15. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu Q, Shen WF, Sun HY, Fan DF, Nakao A,
Cai JM, Yan G, Zhou WP, Shen RX, Yang JM and Sun XJ: Hydrogen-rich
saline protects against liver injury in rats with obstructive
jaundice. Liver Int. 30:958–968. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cai J, Kang Z, Liu K, Liu W, Li R, Zhang
JH, Luo X and Sun X: Neuroprotective effects of hydrogen saline in
neonatal hypoxia-ischemia rat model. Brain Res. 1256:129–137. 2009.
View Article : Google Scholar
|
|
16
|
Zhuge J and Cederbaum AI: Serum
deprivation-induced HepG2 cell death is potentiated by CYP2E1. Free
Radic Biol Med. 40:63–74. 2006. View Article : Google Scholar
|
|
17
|
Zhuge J and Cederbaum AI: Increased
toxicity by transforming growth factor-beta 1 in liver cells
overexpressing CYP2E1. Free Radic Biol Med. 41:1100–1112. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sherwood SW and Schimke RT: Cell cycle
analysis of apoptosis using flow cytometry. Methods Cell Biol.
46:77–97. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu D and Cederbaum A: Cytochrome P4502E1
sensitizes to tumor necrosis factor alpha-induced liver injury
through activation of mitogen-activated protein kinases in mice.
Hepatology. 47:1005–1017. 2008. View Article : Google Scholar
|
|
20
|
Rickmann M, Vaquero EC, Malagelada JR and
Molero X: Tocotrienols induce apoptosis and autophagy in rat
pancreatic stellate cells through the mitochondrial death pathway.
Gastroenterology. 132:2518–2532. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang L and Yu T: Prolonged donor heart
preservation with pinacidil: The role of mitochondria and the
mitochondrial adenosine triphosphate-sensitive potassium channel. J
Thorac Cardiovasc Surg. 139:1057–1063. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gogvadze V, Orrenius S and Zhivotovsky B:
Analysis of mitochondrial dysfunction during cell death. Curr
Protoc Cell Biol Chapter. 18:Unit 18.5. 2003. View Article : Google Scholar
|
|
23
|
Youle RJ and Strasser A: The BCL-2 protein
family: Opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View Article : Google Scholar
|
|
24
|
Palmeira CM and Rolo AP:
Mitochondrially-mediated toxicity of bile acids. Toxicology.
203:1–15. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sun Q, Kang Z, Cai J, Liu W, Liu Y, Zhang
JH, Denoble PJ, Tao H and Sun X: Hydrogen-rich saline protects
myocardium against ischemia/reperfusion injury in rats. Exp Biol
Med (Maywood). 234:1212–1219. 2009. View Article : Google Scholar
|
|
26
|
Mantena SK, King AL, Andringa KK,
Eccleston HB and Bailey SM: Mitochondrial dysfunction and oxidative
stress in the pathogenesis of alcohol- and obesity-induced fatty
liver diseases. Free Radic Biol Med. 44:1259–1272. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cederbaum AI, Lu Y and Wu D: Role of
oxidative stress in alcohol-induced liver injury. Arch Toxicol.
83:519–548. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial ROS-induced ROS release: An update and review.
Biochim Biophys Acta. 1757:509–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zorov DB, Filburn CR, Klotz LO, Zweier JL
and Sollott SJ: Reactive oxygen species (ROS)-induced ROS release:
A new phenomenon accompanying induction of the mitochondrial
permeability transition in cardiac myocytes. J Exp Med.
192:1001–1014. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Petronilli V, Costantini P, Scorrano L,
Colonna R, Passamonti S and Bernardi P: The voltage sensor of the
mitochondrial permeability transition pore is tuned by the
oxidation-reduction state of vicinal thiols. Increase of the gating
potential by oxidants and its reversal by reducing agents. J Biol
Chem. 269:16638–16642. 1994.PubMed/NCBI
|
|
31
|
Kim JS, He L and Lemasters JJ:
Mitochondrial permeability transition: A common pathway to necrosis
and apoptosis. Biochem Biophys Res Commun. 304:463–470. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Adams JM and Cory S: The Bcl-2 protein
family: Arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Malhi H, Gores GJ and Lemasters JJ:
Apoptosis and necrosis in the liver: A tale of two deaths?
Hepatology. 43(2 Suppl 1): S31–S44. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wei MC, Zong WX, Cheng EH, Lindsten T,
Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB and
Korsmeyer SJ: Proapoptotic BAX and BAK: A requisite gateway to
mitochondrial dysfunction and death. Science. 292:727–730. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chaudhary K, Liedtke C, Wertenbruch S,
Trautwein C and Streetz KL: Caspase 8 differentially controls
hepatocytes and non-parenchymal liver cells during chronic
cholestatic liver injury in mice. J Hepatol. 59:1292–1298. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tiao MM, Lin TK, Liou CW, Wang PW, Chen
JB, Kuo FY, Huang CC, Chou YM and Chuang JH: Early transcriptional
deregulation of hepatic mitochondrial biogenesis and its consequent
effects on murine cholestatic liver injury. Apoptosis. 14:890–899.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kahraman A, Barreyro FJ, Bronk SF,
Werneburg NW, Mott JL, Akazawa Y, Masuoka HC, Howe CL and Gores GJ:
TRAIL mediates liver injury by the innate immune system in the bile
duct-ligated mouse. Hepatology. 47:1317–1330. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yoon JH and Gores GJ: Death
receptor-mediated apoptosis and the liver. J Hepatol. 37:400–410.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Marin JJ, Hernandez A, Revuelta IE,
Gonzalez-Sanchez E, Gonzalez-Buitrago JM and Perez MJ:
Mitochondrial genome depletion in human liver cells abolishes bile
acid-induced apoptosis: Role of the Akt/mTOR survival pathway and
Bcl-2 family proteins. Free Radic Biol Med. 61:218–228. 2013.
View Article : Google Scholar : PubMed/NCBI
|