Introduction
Liver fibrosis describes the excessive accumulation
of extracellular matrix, which occurs in the majority of chronic
liver diseases. Advanced fibrosis results in cirrhosis, portal
hypertension and liver failure, and often requires liver
transplantation (1–3). From another perspective, liver
fibrosis represents the wound-healing response to chronic injury,
which is a physiological response when tissues are 'under attack'
(3–5). When the tissue injury is severe or
repetitive, or the wound-healing response itself becomes
dysregulated, normal tissue repair can evolve into a progressively
irreversible fibrotic response. Under these circumstances,
excessive collagen deposition in and around the inflamed or damaged
tissue distorts normal tissue architecture, leading to
hepatocellular dysfunction and increased hepatic resistance to
blood flow This causes hepatic insufficiency, portal hypertension
and the eventual succumbing to mortality, as observed in end-stage
liver disease (2,3,5).
There has been encouraging evidence that fibrosis
exerts pivotal, but divergent, effects on the liver. Although it is
widely accepted that hepatic fibrosis results in deleterious
effects, as described above, increasing evidence suggests a more
favorable effect of liver fibrosis. In a mouse model of partial
bile duct ligation (PBDL), injured ligated lobes exhibit improved
tolerance to tumor necrosis factor (TNF)-α- and Fas-induced
hepatocyte apoptosis, compared with non-ligated lobes, preventing
mass hemorrhage and protecting mice from liver failure (6). In accordance with this, in
vitro and in vivo animal experiments, performed by
Bourbonnais et al, indicated that hepatocytes exposed to
type I collagen, or fibrotic mice induced with thioacetamide (TAA),
were less vulnerable to injury (7). However, the association between liver
fibrosis induced by CCl4 and subsequent acute injury
remains to be elucidated.
High-mobility group box (HMGB)1 is an evolutionarily
conserved protein, which is present in the nucleus of almost all
eukaryotic cells (8). The function
of HMGB1 is diverse and compartment-specific. As a DNA chaperone,
nuclear HMGB1 is engaged in several DNA-activity-associated events,
including DNA replication, recombination, transcription and repair.
HMGB1 can be actively secreted by innate immune cells or passively
released by dead, dying or injured cells. Extracellular HMGB1, as a
damage-associated molecular pattern (DAMP), is critically involved
in several pathophysiological processes, including infection,
tissue injury, inflammation, apoptosis and the immune response
(9–11). All these characteristics make HMGB1
a critical molecular target in several diseases, including
infection, ischemia-reperfusion injury, immune disorders and cancer
(11–16).
The profibrotic function of HMGB1 has been
demonstrated previously (9,10,17,18).
The upregulation of HMGB1 during liver fibrosis may be involved in
tissue remodeling and fibro-genesis through the direct activation
of hepatic stellate cells (HSCs). Therefore, inhibiting the
bioavailability of HMGB1 may constitute a therapeutic strategy for
the treatment of liver fibrosis (17,18).
Accumulating evidence has indicated that HMGB1 is critical in the
pathogenesis of acute liver injury/failure originating from a
variety of stimuli (19–22). HMGB1 has been reported as a
sensitive serum diagnostic and biomarker for the assessment of
severity in patients with acute liver injury (22,23).
To support the hypothesis, the translocation and
extracellular release of HMGB1 were compared between control and
fibrotic mice in response to D-GalN/LPS challenge, and inflammatory
response mediated by HMGB1 was analyzed. The present study aims to
demonstrate that hepatoprotection induced by liver fibrosis is
mediated by HMGB1. These findings will provide novel interpretation
for the pathogenesis of acute liver injury, in the setting of
hepatic fibrosis. This may, at least in part, account for the
reduced inflammatory response and alleviation of liver damage in
these mice.
Materials and methods
Animals
A total of 35 male Balb/c mice (6–8-week-old; 20–25
g) were purchased from Laboratory Animal Center, Academy of
Military Medical Sciences, Beijing, China. The animals were housed
in a specific pathogen-free environment under controlled conditions
(22–24°C, 12-h light/dark cycle) and fed an AIN93 diet with access
to water throughout the experiment. Experimental procedures were
approved by the Institutional Animal Care and Use Committee at
Beijing YouAn Hospital affiliated to Capital Medical University
(Beijing, China), according to the Guide for the Care and Use of
Laboratory Animals (24).
Experimental designs
To investigate the correlation between liver
fibrosis and injury tolerance, the following animal models were
developed: i) Induction of fibrosis: liver fibrosis was established
by intraperitoneal injection of CCl4 (Sinopharm Chemical
Reagent Beijing Co., Ltd., Beijing, China) in mineral oil (Amresco
LLC, Solon, OH, USA) twice weekly, for 6 weeks. The initial dose of
CCl4 was 0.2 µl/g, following which the doses
increased gradually, up to 3 µl/g ii) Acute challenge:
Control and fibrotic mice were sacrificed by a lethal dose of
hepatic toxins (1 mg/g D-GalN+50 ng/g LPS; Sigma-Aldrich, St.
Louis, MO, USA). The mice were divided as follows: Control 5,
D-GalN/LPS 10, fibrosis 10, and Fib+ D-GalN/LPS 10. Sera and liver
tissues were harvested from the mice at indicated time points for
analysis. Following harvesting, a section of the liver was fixed in
10% neutral-buffered formalin (Sinopharm Chemical Reagent Beijing
Co., Ltd.) for histological analysis and immunostaining. The
remaining liver was cut into pieces and snap-frozen for
homogenization to extract total liver RNA.
Evaluation of liver injury
The liver tissues fixed in 10% formalin were
embedded in paraffin (Sinopharm Chemical Reagent Beijing Co.,
Ltd.), sectioned into 3 µm sections and stained with
hematoxylin and eosin (Sinopharm Chemical Reagent Beijing Co.,
Ltd.) for light microscopy. Histological severity of liver injury
was graded numerically, according to the pathological grading
criteria described by Lefkowitch (25). The parameters were graded with a
score between 0 and 6, with 0 indicating no abnormality, 1 or 2
indicating mild liver injury, 3 or 4 moderate injury, and 5 or 6
severe injury.
SYBR Green
reverse-transcription-quantitative polymerase chain reaction
Frozen liver tissue (~50 mg) was cut into pieces,
and homogenized in 1 ml of TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) using an electric
homogenizer (Tissue Tearor, BioSpec Products Inc., Bartlesville,
OK, USA) on ice. Total RNA was extracted according to the
manufacturer's protocol. cDNA was synthesized from 2.5 µg
RNA using random primers and an AMV Retrotranscriptase system
(Takara Biotechnology Co., Ltd., Dalian, China) using the following
temperature protocol: 30°C for 10 min; 42°C for 30 min and 95°C for
5 min. The SYBR Green RT-qPCR was performed using the ABI StepOne
Plus and software (Applied Biosystems; Thermo Fisher Scientific,
Inc.). All reactions were performed in triplicate. In a final
reaction volume of 20 µl, the following were added: 1X SYBR
Green (Takara Biotechnology Co., Ltd.) cDNA, 0.5 mM each primer and
ROX (Takara Biotechnology Co., Ltd.). The conditions of the qPCR
reaction were as follows: 50°C (2 min), 95°C (5 min), followed by
40 cycles of 95°C (15 sec) and 60°C (30 sec). The primers used were
designed using Primer version 3.0 (26) and the sequences are listed in
Table I. The relative expression
levels of the target genes were calculated and normalized to the
expression of GAPDH, a housekeeping gene.
 | Table IPrimer sequences used for reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction analysis.
| Gene | Sense | Anti-sense |
|---|
| GAPDH |
5′-AACTTTGGCATTGTGGAAGG-3′ |
5′-ACACATTGGGGGTAGGAACA-3′ |
| IL-1β |
5′-GCCCATCCTCTGTGACTCAT-3′ |
5′-AGGCCACAGGTATTTTGT-3′ |
| IL-6 |
5′-AGTTGCCTTCTTGGGACTGA-3′ |
5′-TCCACGATTTCCCAGAGAAC-3′ |
| IL-12p40 |
5′-CAGCTTCTTCATCAGGGACAT-3′ |
5′-CTTGAGGGAGAAGTAGGAATGG-3′ |
| TNF-α |
5′-GCCTCTTCTCATTCCTGCTTGT-3′ |
5′-TTGAGATCCATGCCGTTG-3 |
Immunofluorescence
The liver tissues were snap-frozen in liquid
nitrogen (Haotian Corporation, Beijing, China) and embedded in
Tissue-Tek OCT (Sakura Finetek USA Inc., Torrance, CA, USA)
compound. For immunofluorescence staining, the liver sections were
fixed and stained with the following primary antibodies at 4°C
overnight: Rabbit anti-mouse HMGB1 monoclonal antibody (1:100; cat.
no ab79823, Abcam, Cambridge, MA, USA), goat anti-mouse collagen
type I (1:200; cat. no. 1310-01, Southern Biotech, San Diego, CA,
USA) and DAPI (EMD Millipore, Billerica, MA, USA). For indirect
immunofluorescence staining, liver sections were incubated with the
following secondary antibodies at 37°C for 30 min: fluorescein
isothiocyanate-conjugated donkey anti-rabbit IgG for HMGB1 (1:500;
cat. no. sc-2090; Santa Cruz Biotechnology, Inc., Dallas, TX, USA)
and Cy3-conjugated rabbit anti-goat IgG for collagen type I (1:500;
cat. no. C2821, Sigma-Aldrich) were used. The results were
visualised and quantified using a Inverted Flourescence Microscope
ECLIPSE Ti and NIS-Elements F 3.0 software (Nikon Corporation,
Tokyo, Japan)
HMGB1 immunohistochemical staining
Following deparaffinization and rehydration, the
embedded liver sections were treated with 3%
H2O2 for 15 min, followed by microwave
antigen retrieval for a further 15 min in citrate buffer
(Sino-pharm Chemical Reagent Beijing Co., Ltd.). The nonspecific
proteins were blocked with 10% goat serum for 30 min. For HMGB1
staining, the specimens were incubated with rabbit anti-mouse HMGB1
monoclonal antibody (cat. no. ab79823; 1:200; Abcam) overnight at
4°C, followed by 30 min incubation with
horseradish-peroxidase-conjugated goat anti-rabbit secondary
antibody (cat. no. ZB-2301; 1:200; Zhongshan Golden Bridge
Biotechnology Co,. Ltd., Beijing, China). The sections were
incubated with diaminobenzidine (Zhongshan Golden Bridge
Biotechnology Co., Ltd.) for 5 min as a chromogenic substrate and
were counterstained with hematoxylin. The tissue sections were then
dehydrated and stabilized with mounting medium (Zhongshan Golden
Bridge Biotechnology Co., Ltd.). Images were captured using Bx51
microscope (Olympus America, Inc., Melville, NY, USA) and cellSens
software (version 1.4.1.; Olympus Corporation, Tokyo, Japan)
Statistical analysis
Graphpad Prism version 5.0 (GraphPad Software, San
Diego, CA, USA) was used for data processing and analysis. Results
are expressed as the mean ± standard error of the mean. Group
comparisons were performed using one-way analysis of variance or
Student's t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Survival of fibrotic mice following
exposure to a lethal dose of D-GalN/LPS
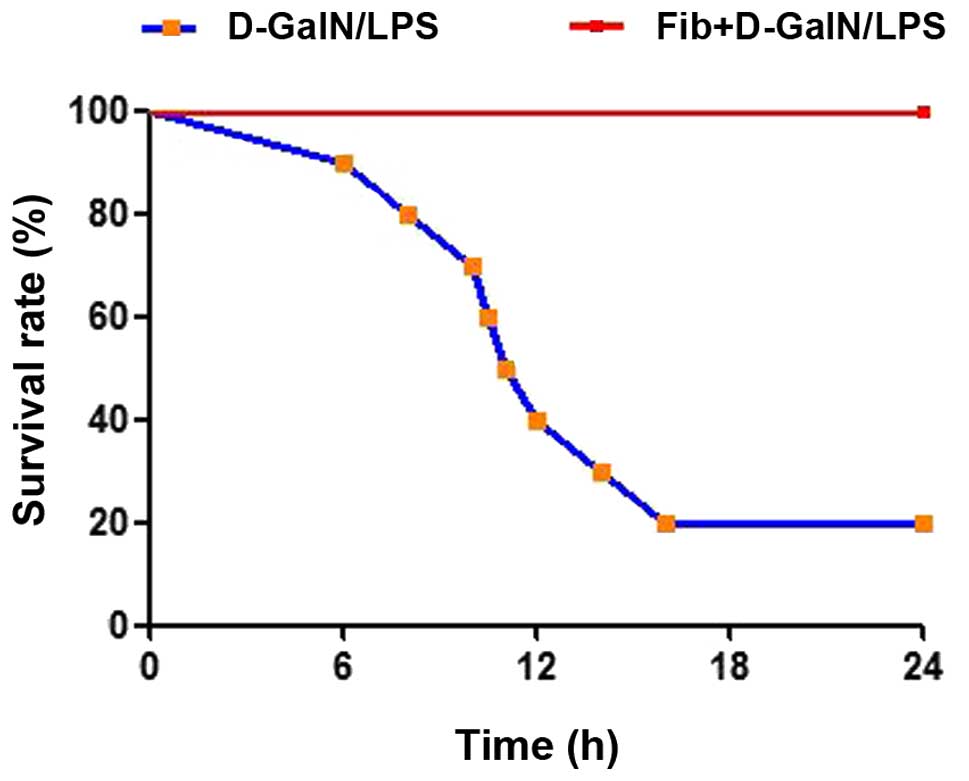
The present study examined the effect of fibrosis on
the survival rates of mice in response to hepatic toxins. Control
and fibrotic mice were subjected to lethal doses of D-GalN (1
µg/g) plus LPS (50 ng/g) as an acute insult. During 24 h
observation, 8/10 of the mice died in the control group treated
with D-GalN/LPS (20% survival rate), however, no fibrotic mice
succumbed to mortality when exposed to the same dose of D-GalN/LPS
(survival rate 100%; Fig. 1). This
provided direct and macroscopic evidence of the advantageous effect
of fibrosis. The extent of hepatic damage was analyzed and compared
between the control and fibrotic mice treated with D-GalN/LPS.
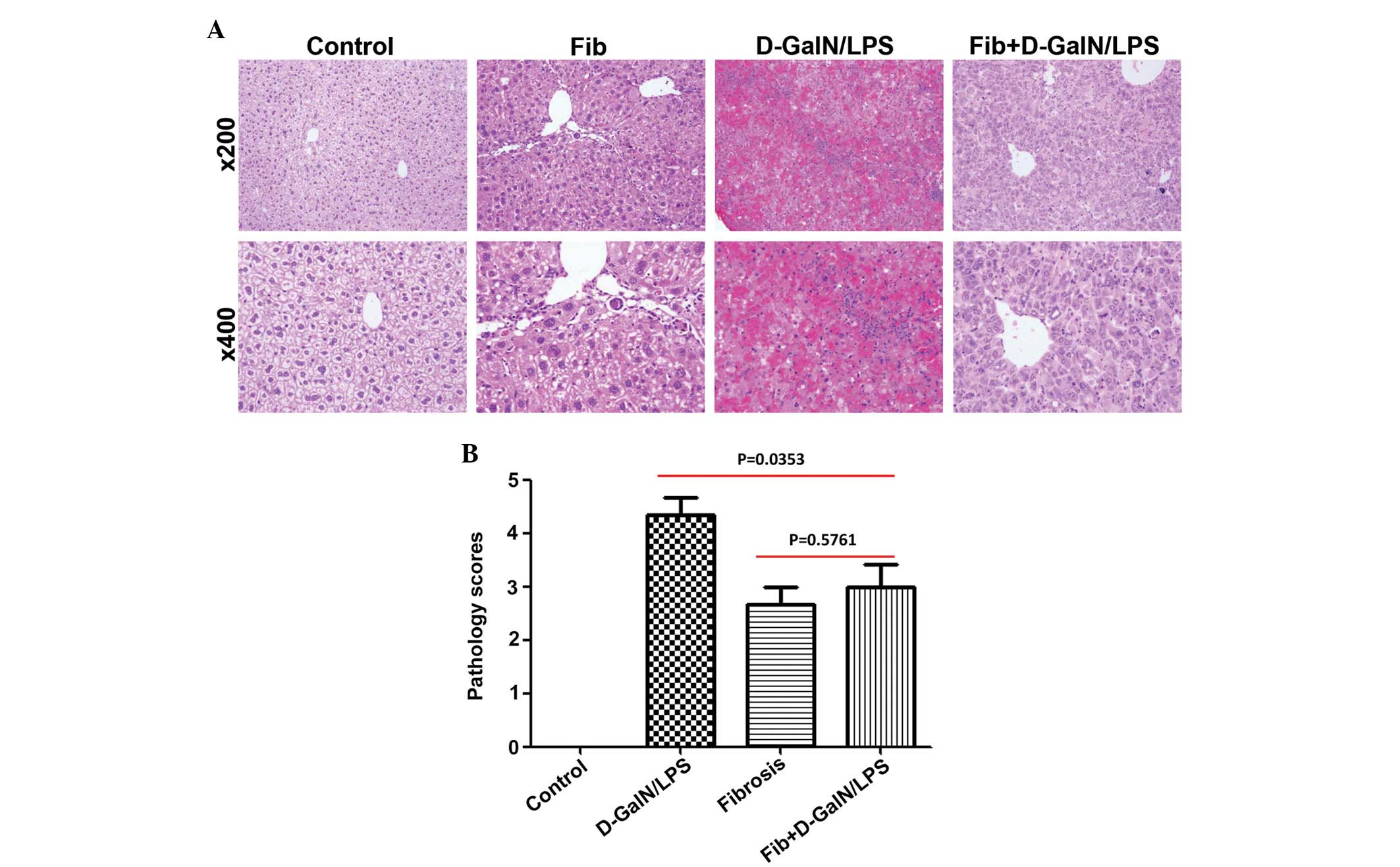
Microscopic examination of liver tissues from control mice treated
with D-GalN/LPS revealed pronounced hepatocyte destruction with
mass hemorrhage. By contrast, marked improvements in histology were
noted in the CCl4-induced fibrotic mice treated in the
same way (Fig. 2A). Histological
grading provided further quantitative information of the extent of
liver damage, and the difference between the control and the
fibrotic mice in response to D-GalN/LPS was significant (P=0.0353;
Fig. 2B). Improved survival rates
and preservation of liver architecture provided compelling evidence
that fibrosis may result in decreased susceptibility of hepatocytes
to lethal doses of D-GalN/LPS.
Translocation and extracellular release
of HMGB1 is inhibited in fibrotic mice treated with D-GalN/LPS
The present study evaluated the immunoreactive
expression of HMGB1, which is important in the initiation and
progression of proinflammatory processes (12). As the functions of HMGB1 are
diverse and compartment-specific (9–11),
the cellular localization of HMGB1 in the liver was determined in
the present study. The liver tissues were obtained from control
mice (at any time point), control mice upon D-GalN/LPS challenge
(at the time of death), fibrotic mice (24 h after the last
CCl4 injection), and fibrotic mice upon D-GalN/LPS
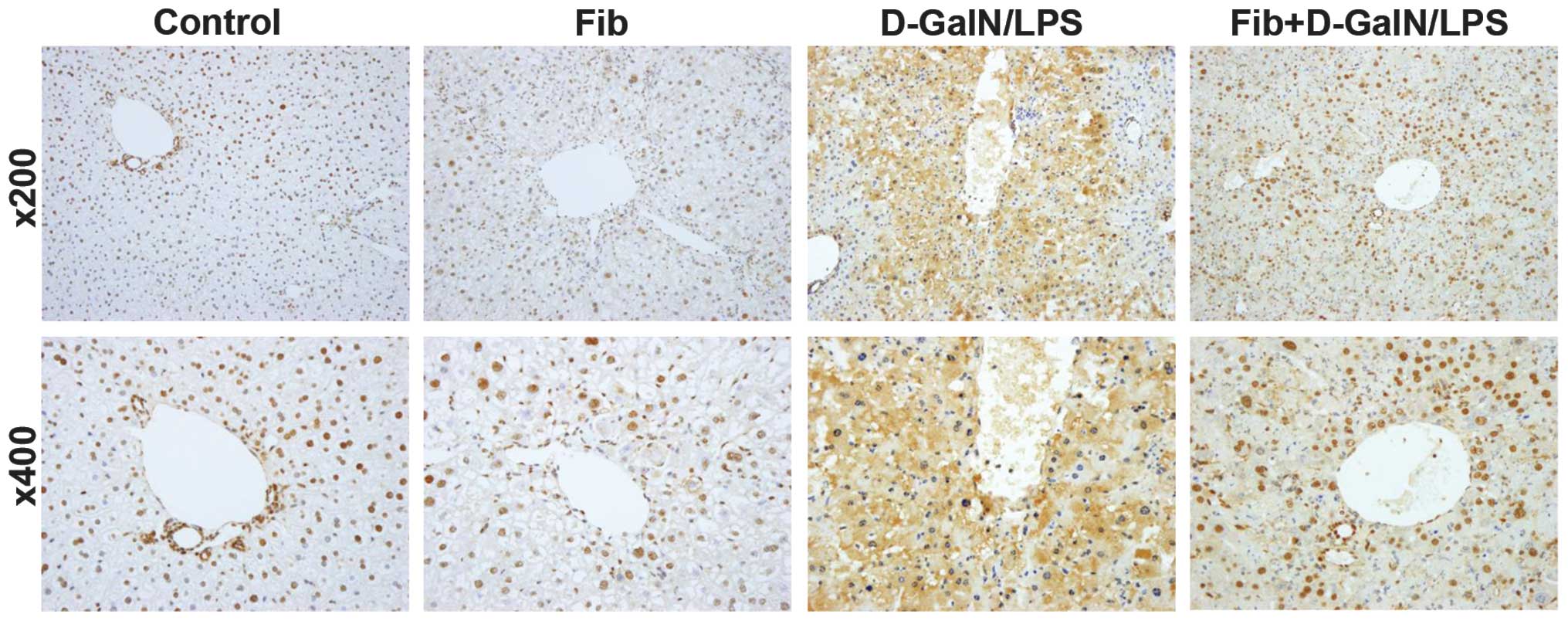
challenge (24 h after D-GalN/LPS challenge). Immunohistochemical
staining with antibodies against HMGB1 revealed a distinct
expression pattern in paraffin sections retrieved from control and
fibrotic mice, with or without D-GalN/LPS challenge. HMGB1 was
localized in the nucleus of the majority of the hepatocytes in the
control mice. In the fibrotic liver tissues, a relative increase in
the expression of HMGB1 was observed, and extranuclear
HMGB1-positive staining was visible in a number of hepatocytes.
Following D-GalN/LPS challenge, HMGB1-positive staining was
significantly enhanced, however, immunoreactivity for HMGB1 in the
nucleus was markedly reduced, and translocation and aberrant
extracellular expression of HMGB1 were observed. The expression of
HMGB1 and its translocation into extranuclear and extracellular
milieu were significantly inhibited in the fibrotic mice treated
with the same dose of D-GalN/LPS (Fig.
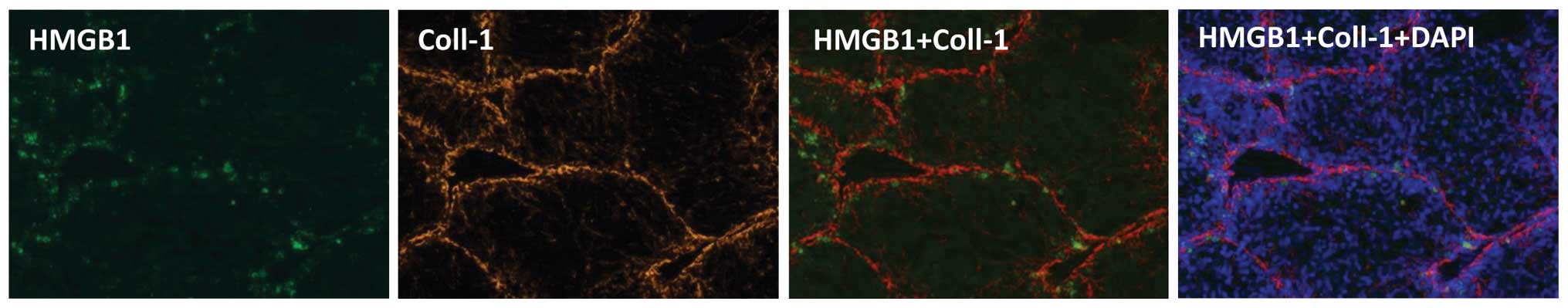
3). The distribution of HMGB1 coincided with that of type I
collagen in the fibrotic tissues on visualization following
immunofluorescent staining, supporting the close correlation
between fibrosis and the expression of HMGB1 (Fig. 4). Accordingly, fibrosis may inhibit
the expression and release of HMGB1 triggered by D-GalN/LPS,
leading to alleviated liver damage.
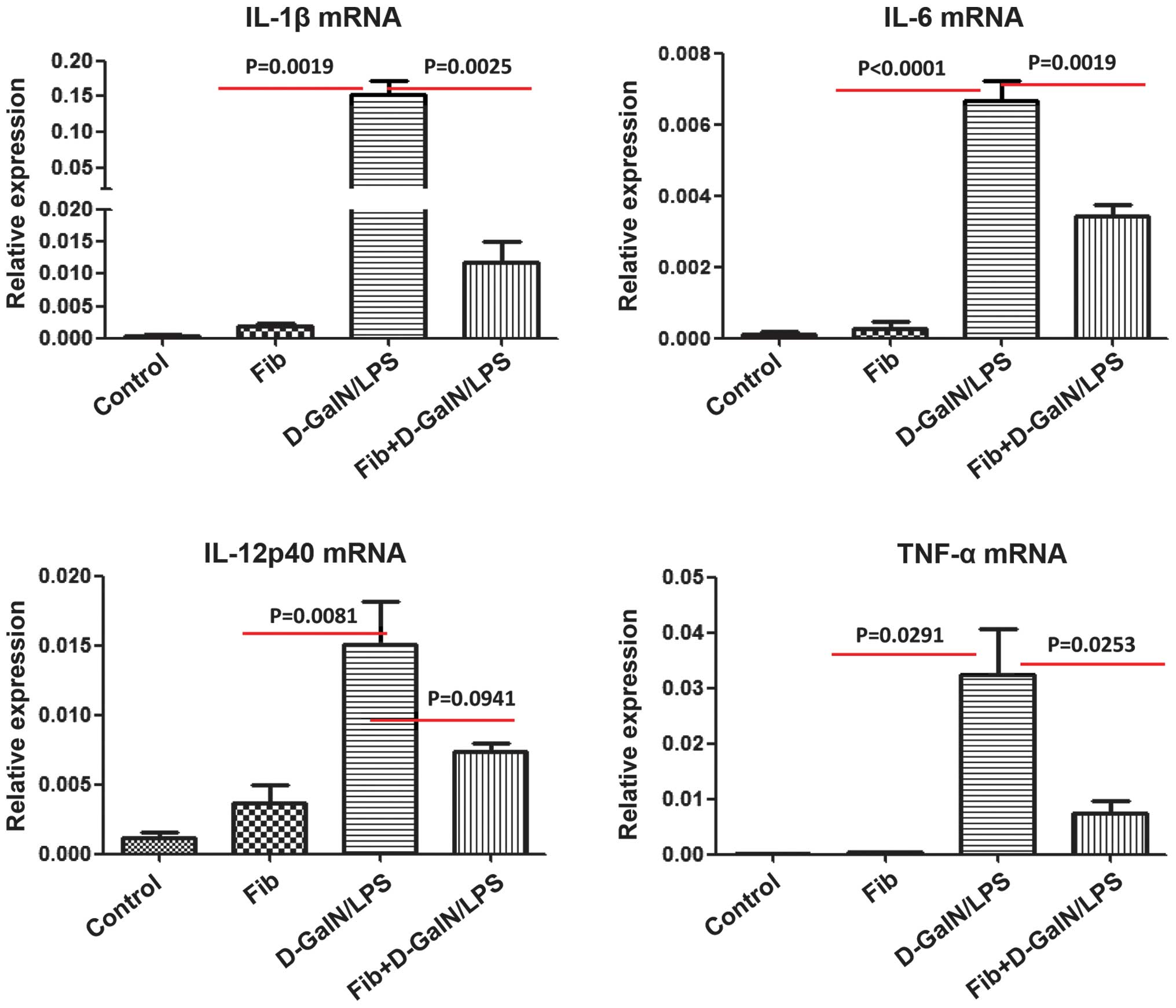
HMGB1-mediated inflammatory responses are
reduced in fibrotic mice treated with D-GalN/LPS
Increasing evidence suggests that HMGB1 can
selectively bind multiple receptors, for example in receptors for
advanced glycation end-products (RAGE) and Toll-like receptors, to
activate different types of liver cells, including macrophages,
neutrophils, dendritic cells and T cells, to produce cytokines,
including TNF-α, IL-1β, IL-6, IL-10 and IL-12 (12). To further examine the molecular and
immunological mechanisms involved in the development of reduced
liver injury in fibrotic mice treated with D-GalN/LPS, the present
study analyzed the mRNA expression levels of these HMGB1-triggered
proinflammatory cytokines using RT-qPCR. The mRNA levels of IL-1β,
IL-6, TNF-α and IL-12P40 were significantly increased in the liver
tissues retrieved from the control mice, which were exposed to
lethal doses of D-GalN/LPS. The upregulation of these inflammatory
cytokines was suppressed in the liver tissues retrieved from the
fibrotic mice, which were exposed to D-GalN/LPS (Fig. 5). These data provided additional
evidence that the HMGB1-mediated inflammatory and immune responses
triggered by D-GalN/LPS were inhibited by liver fibrosis.
Discussion
The present study provided the first evidence, to
the best of our knowledge, that inhibiting the expression,
translocation and release of HMGB1 at least partially alleviated
hepatic injury in CCl4-induced fibrotic mice exposed to
D-GalN/LPS. The present study is the first to link the
translocation and release of HMGB1 with resistance to
fibrosis-based acute injury.
The identification of fibrosis as a key event in the
alleviation of liver damage in response to insult is not
unprecedented. Resistance to acute liver damage induced by TNF-α or
Fas has been demonstrated in PBDL- and TAA-based models of fibrosis
(6,7). However, it remains important to
verify and analyze this association in additional fibrosis models
with different insults. The present study investigated the effects
of CCl4-induced liver fibrosis, another well-established
animal model for liver fibrosis, on the secondary challenge of
D-GalN/LPS, which is widely used as an inducer for acute hepatic
injury/failure. The fibrosis induced by CCl4 conferred
significant protection against lethal challenge with D-GalN/LPS, as
demonstrated by improved survival rates and improved preservation
of liver architecture. Previous clinical data confirmed the ongoing
fibrosis and HSC activation in the progression of acute liver
failure, supporting the hypothesis that fibrosis in acute liver
failure may be a physiological, and possibly beneficial, response
by the liver (27,28).
The molecular basis for the hepatoprotective
response induced by liver fibrosis has been discussed in previous
reports. Much attention has been paid to the balance between the
survival and apoptosis of hepatocytes. In this regard, enhanced
cell survival and liver regeneration, along with attenuated
hepatocyte apoptosis, mediated by the activation of AKT and
extracellular signal-regulated kinase signaling, have been
documented (6,7). However, inflammation is a major
component of the pathology of drug-induced liver injury and liver
fibrosis, and advances in the biology of inflammation have revealed
that specific cytokines are important and effective pathogenic
mediators (12). For this reason,
it is imperative to determine whether HMGB1, an important
proinflammatory mediator, functions in acute hepatic injury
occurring in the setting of fibrosis, and to investigate the
underlying mechanism.
HMGB1, which is constitutively expressed in the
nucleus of the majority of cells under basal conditions, functions
as a structural co-factor that is critical for proper
transcriptional regulation. In the past decade, the active
secretion of extracellular HMGB1 by innate immune cells in response
to pathogenic products, and its release by injured or dying cells,
has been identified to occupy a central role in the pathogenesis of
sterile and infectious inflammation (12). Mechanistically, extracellular HMGB1
binds to pattern recognition receptors, including Toll- like
receptor 4 and RAGE, and acts as a DAMP molecule to activate
intracellular signals, including nuclear factor-κB and the
mitogen-activated protein kinase pathway, which regulate the gene
expression of various immune and inflammatory mediators, including
TNF-α, IL-1β, IL-6, IL-10 and IL-12. In addition, HMGB1 functions
as an immune adjuvant, to trigger the activation of immune cells,
including T cells, dendritic cells and endothelial cells, and the
secretion of HMGB1, which forms a positive feedback loop that
potentially amplifies local inflammatory responses by enhancing the
release of cytokines and chemokines (11,12,29).
HMGB1 is translocated from the nucleus to the
cytoplasm, and is subsequently released into the extracellular
milieu, which has been noted under certain conditions, including
ischemia and reperfusion injury, hepatitis C virus or hepatitis B
viral infections, and drug-induced acute liver failure (20,21,23,25,30).
Although the profibrotic function of HMGB1 is established, the
effect of liver fibrosis on the translocation and extracellular
release of HMGB1 remains to be fully elucidated. In the present
study, lethal doses of D-GalN/LPS triggered the translocation and
excessive release of HMGB1, accompanied by a significant
upregulation in the gene expression levels of proinflammatory
IL-1β, IL-6, TNF-α and IL-12P40. By contrast, the increase in HMGB1
release and proinflammatory gene expression were markedly inhibited
in fibrotic mice exposed to D-GalN/LPS. In addition, the
distribution of HMGB1 detected by immuno-fluorescence staining was
in accordance with that of type I collagen, suggesting that
fibrosis was closely associated with the expression of HMGB1. Thus,
the present study hypothesized that fibrosis inhibits the
translocation and release of HMGB1, and the inflammatory response
triggered by HMGB1. In addition, the release and activity of HMGB1
were restored following the resolution of liver fibrosis,
supporting the inhibitory action of hepatic fibrosis on HMGB1
translocation (unpublished data).
Depending on the inducing stimulus, the mechanism
underlying the secretion and release of HMGB1 can vary. In response
to exogenous pathogen-associated molecular patterns, for example
endotoxin, or endogenous inflammatory stimuli, for example DAMP
molecules, HMGB1 is modified by different post-transcriptional
modifications, including acetylation and phosphorylation, which
impedes its re-entry into the nucleus, with subsequent migration
into the cytoplasm and release into the extracellular milieu
(31,32). In the present study, the liver
fibrosis induced by CCl4 may have altered the function
of HMGB1 through effects on its post-transcriptional
modifications.
Although current data are only associative,
CCl4-induced fibrosis may function as a critical event
in the inhibition of HMGB1 release and the resulting alleviation of
liver injury. Understanding the physiological and beneficial roles
of fibrosis in the progression of acute liver injury may reveal
novel opportunities for the treatment of liver failure,
particularly of acute-on-chronic liver failure.
Acknowledgments
This study was funded by the National Science and
Technology Key Project of China on 'Major Infectious Diseases such
as HIV/AIDS, Viral Hepatitis Prevention and Treatment' (grant no.
2012ZX10002004-006, 2012ZX10004904-003-001 and 2013ZX10002002-006),
the High Technical Personnel Training Item in Beijing Health System
(grant nos. 2011-3-083 and 2013-3-071), the Special Fund for
Clinical Medicine Development of Beijing Municipal Administration
of Hospitals (grant no. XM201308), the National Key Subject
Construction Project (grant nos. WJWYA-2014-002 and WJWYA-2014-004)
and the Basic-Clinical Cooperation Project of Capital Medical
University (grant no. 14JL72, 14JL73).
References
|
1
|
Friedman SL: Mechanisms of hepatic
fibrogenesis. Gastroenterology. 134:1655–1669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Friedman SL: Evolving challenges in
hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 7:425–436. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bataller R and Brenner DA: Liver fibrosis.
J Clin Invest. 115:209–218. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
White ES and Mantovani AR: Inflammation,
wound repair and fibrosis: Reassessing the spectrum of tissue
injury and resolution. J Pathol. 229:141–144. 2013. View Article : Google Scholar
|
|
5
|
Wynn TA and Ramalingam TR: Mechanisms of
fibrosis: Therapeutic translation for fibrotic disease. Nat Med.
18:1028–1040. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Osawa Y, Hannun YA, Proia RL and Brenner
DA: Roles of AKT and sphingosine kinase in the antiapoptotic
effects of bile duct ligation in mouse liver. Hepatology.
42:1320–1328. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bourbonnais E, Raymond VA, Ethier C,
Nguyen BN, El-Leil MS, Meloche S and Bilodeau M: Liver fibrosis
protects mice from acute hepatocellular injury. Gastroenterology.
142:130–139.e4. 2012. View Article : Google Scholar
|
|
8
|
Javaherian K, Liu JF and Wang JC:
Nonhistone proteins HMG1 and HMG2 change the DNA helical structure.
Science. 199:1345–1346. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen R, Hou W, Zhang Q, Kang R, Fan XG and
Tang D: Emerging role of high-mobility group box 1 (HMGB1) in liver
diseases. Mol Med. 19:357–366. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li LC, Gao J and Li J: Emerging role of
HMGB1 in fibrotic diseases. J Cell Mol Med. 18:2331–2339. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao
L, Huang J, Yu Y, Fan XG, Yan Z, et al: HMGB1 in health and
disease. Mol Aspects Med. 40:1–116. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Andersson U and Tracey KJ: HMGB1 is a
therapeutic target for sterile inflammation and infection. Annu Rev
Immunol. 29:139–162. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang H, Nace GW, McDonald KA, Tai S,
Klune JR, Rosborough BR, Ding Q, Loughran P, Zhu X, Beer-Stolz D,
et al: Hepatocyte-specific high-mobility group box 1 deletion
worsens the injury in liver ischemia/reperfusion: A role for
intracellular high-mobility group box 1 in cellular protection.
Hepatology. 59:1984–1997. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kamo N, Ke B, Ghaffari AA, Shen XD,
Busuttil RW, Cheng G and Kupiec-Weglinski JW: ASC/caspase-1/IL-1β
signaling triggers inflammatory responses by promoting HMGB1
induction in liver ischemia/reperfusion injury. Hepatology.
58:351–362. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sims GP, Rowe DC, Rietdijk ST, Herbst R
and Coyle AJ: HMGB1 and RAGE in inflammation and cancer. Annu Rev
Immunol. 28:367–388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yanai H, Ban T, Wang Z, Choi MK, Kawamura
T, Negishi H, Nakasato M, Lu Y, Hangai S, Koshiba R, et al: HMGB
proteins function as universal sentinels for nucleic-acid-mediated
innate immune responses. Nature. 462:99–103. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Albayrak A, Uyanik MH, Cerrah S, Altas S,
Dursun H, Demir M and Uslu H: Is HMGB1 a new indirect marker for
revealing fibrosis in chronic hepatitis and a new therapeutic
target in treatment? Viral Immunol. 23:633–638. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang FP, Li L, Li J, Wang JY, Wang LY and
Jiang W: High mobility group box-1 promotes the proliferation and
migration of hepatic stellate cells via TLR4-dependent signal
pathways of PI3K/Akt and JNK. PLoS One. 8:e643732013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gong Q, Zhang H, Li JH, Duan LH, Zhong S,
Kong XL, Zheng F, Tan Z, Xiong P, Chen G, et al: High-mobility
group box 1 exacerbates concanavalin A-induced hepatic injury in
mice. J Mol Med (Berl). 88:1289–1298. 2010. View Article : Google Scholar
|
|
20
|
Zhou RR, Zhao SS, Zou MX, Zhang P, Zhang
BX, Dai XH, Li N, Liu HB, Wang H and Fan XG: HMGB1 cytoplasmic
translocation in patients with acute liver failure. BMC
Gastroenterol. 11(21)2011. View Article : Google Scholar
|
|
21
|
Kuroda N, Inoue K, Ikeda T, Hara Y, Wake K
and Sato T: Apoptotic response through a high mobility box 1
protein-dependent mechanism in LPS/GalN-induced mouse liver failure
and glycyrrhizin-mediated inhibition. PLoS One. 9:e928842014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Antoine DJ, Jenkins RE, Dear JW, Williams
DP, McGill MR, Sharpe MR, Craig DG, Simpson KJ, Jaeschke H and Park
BK: Molecular forms of HMGB1 and keratin-18 as mechanistic
biomarkers for mode of cell death and prognosis during clinical
acetaminophen hepatotoxicity. J Hepatol. 56:1070–1079. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tsung A, Klune JR, Zhang X, Jeyabalan G,
Cao Z, Peng X, Stolz DB, Geller DA, Rosengart MR and Billiar TR:
HMGB1 release induced by liver ischemia involves toll-like receptor
4 dependent reactive oxygen species production and calcium-mediated
signaling. J Exp Med. 204:2913–2923. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press; Washington (DC): 2011
|
|
25
|
Lefkowitch JH: Pathologic diagnosis of
liver disease. Hepatology - A Textbook of Liver Disease. Zakim D
and Boyer TD: W.B. Saunders Company; Philadelphia: pp. 844–871.
1996
|
|
26
|
Untergrasser A, Cutcutache I, Koressaar T,
Ye J, Faircloth BC, Remm M and Rozen SG: Primer 3 - new
capabilities and interfaces. Nucleic Acids Res. 40:e1152012.
View Article : Google Scholar
|
|
27
|
Dechêne A, Sowa JP, Gieseler RK, Jochum C,
Bechmann LP, El Fouly A, Schlattjan M, Saner F, Baba HA, Paul A, et
al: Acute liver failure is associated with elevated liver stiffness
and hepatic stellate cell activation. Hepatology. 52:1008–1016.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
He Y, Jin L, Wang J, Yan Z, Chen T and
Zhao Y: Mechanisms of fibrosis in acute liver failure. Liver Int.
35:1877–1885. 2015. View Article : Google Scholar
|
|
29
|
Yang H, Hreggvidsdottir HS, Palmblad K,
Wang H, Ochani M, Li J, Lu B, Chavan S, Rosas-Ballina M, Al-Abed Y,
et al: A critical cysteine is required for HMGB1 binding to
Toll-like receptor 4 and activation of macrophage cytokine release.
Proc Natl Acad Sci USA. 107:11942–11947. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jung JH, Park JH, Jee MH, Keum SJ, Cho MS,
Yoon SK and Jang SK: Hepatitis C virus infection is blocked by
HMGB1 released from virus-infected cells. J Virol. 85:9359–9368.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Youn JH and Shin JS: Nucleocytoplasmic
shuttling of HMGB1 is regulated by phosphorylation that redirects
it toward secretion. J Immunol. 177:7889–7897. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bonaldi T, Talamo F, Scaffidi P, Ferrera
D, Porto A, Bachi A, Rubartelli A, Agresti A and Bianchi ME:
Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect
it towards secretion. EMBO J. 22:5551–5560. 2003. View Article : Google Scholar : PubMed/NCBI
|