Introduction
Cardiovascular diseases are the leading contributor
to mortality rates worldwide. According to the World Health
Organization (WHO), by 2030, >23,000,000 individuals will
succumb to mortality from cardiovascular diseases annually.
Myocardial ischemia is one of the main consequences of
cardiovascular diseases and can be characterized into several forms
(1). Although timely reperfusion
is essential for recovery of the ischemia myocardium, sudden
restoration of blood flow may exaggerate myocardial injury
(2,3). A study in 1960 reported that the
reperfusion of ischemic myocardium may aggravate myocardial injury
in dogs, which is known as myocardial ischemia/reperfusion (I/R)
injury (4,5). Although the underlying mechanism
regulating myocardial injury induced by I/R remains to be fully
elucidated, previous evidence suggests that apoptosis is an
essential form of cell death in I/R injury (6). Myocardial apoptosis is initiated
shortly following ischemia, however, for substantial apoptotic cell
death to occur, reperfusion is necessary.
The endoplasmic reticulum (ER) is an organelle with
an essential role in multiple cellular processes, including sensing
oxidative stress, maintaining calcium homeostasis and triggering
apoptotic signaling (7,8). I/R injury impairs key functions of
ER, including the synthesis, folding and sorting of proteins, and
induces a stress state, which activates the evolutionarily
conserved unfolded protein response (UPR) (8). ER stress is initially sensed by three
ER transmembrane receptors, protein kinase RNA-like endoplasmic
reticulum kinase (PERK), activating transcription factor 6 (ATF6)
and inositol-requiring enzyme 1 (IRE1), which monitor the
homeostasis of the ER and trigger the UPR (9). The UPR rapidly attenuates general
protein synthesis, induces the expression of ER chaperone proteins
and enhances the degradation of misfolded/unfolded proteins.
Although the UPR is primarily an adaptive response, if the stress
persists, the ER stress receptors can also trigger pro-apoptotic
pathways to initiate cell death (10). Sustained ER stress, acting through
PERK, ATF6, and IRE1, induces expression of the pro-apoptotic
protein C/EBP homologous protein (CHOP). The overexpression of CHOP
induces apoptosis through a B cell lymphoma-2 (Bcl-2)-inhibiting
mechanism and c-Jun amino-terminal kinase (JNK) enhancing pathways
(10,11). Sufficient evidence indicates that
high protein levels of CHOP are associated with cell death by ER
stress-independent mechanisms. Inhibiting the apoptotic process
induced by the ER stress pathway may prevent the loss of
contractile cells, minimize cardiac injury induced by I/R, and slow
the occurrence of myocardial stunning and heart failure (12).
4-phenylbutyric acid (4-PBA) is a low molecular
weight, terminal aromatic substituted fatty acid and a non-toxic
pharmacological compound, which is currently approved for clinical
use as an ammonia scavenger in disorders of the urea cycle in
children (13). This molecule is
also used for the treatment of sickle cell disease due to its
capacity to activate β-globin transcription (14). Previously, 4-PBA was found acting
as a chemical chaperone in the ER, improving ER folding capacity
and facilitating the trafficking of mutant proteins. The underlying
mechanism predominantly relies on its physicochemical properties,
which allow the stabilization of peptide structures, improving the
luminal folding capacity and the traffic of aberrant proteins
(15–17). It has been reported that 4-PBA
shows protective effects against ER stress-induced neuronal and
myocardial cell death (18). Thus,
the use of 4-PBA may provide a therapeutic approach for inhibiting
the pathologic process of ER stress and cell death induced by
I/R.
However, the mechanism underlying the activation of
ER stress, as well as the function of 4-PBA in the injured
myocardium, remain to be elucidated. Understanding how 4-PBA
responds to ER stress is necessary to assess its contribution to
cardioprotection. The present study aimed to investigate the
effects of the chemical chaperone, 4-PBA, on ER stress and examine
the mechanisms underlying these effects in a model of myocardial
cell death induced by I/R injury.
Materials and methods
Reagents
The following reagents were purchased from
Sigma-Aldrich (St. Louis, MO, USA): 4-PBA,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
(MTT), 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl benzimidazolyl
carbocyanine iodide (JC-1) and dimethyl sulfoxide (DMSO).
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum
(FBS), and phosphate-buffered saline (PBS) were purchased from
Gibco; Thermo Fisher Scientific, Inc. (Waltham, MA, USA). A
terminal deoxynucleotidyl transferase-mediated biotinylated UTP
nick end-labeling (TUNEL) kit was purchased from Invitrogen; Thermo
Fisher Scientific, Inc. Primary antibodies against
glucose-regulated protein 78 (Grp78; sc-13968), ATF6 (sc-22799),
PERK (sc-13071), CHOP (sc-7351), JNK (sc-7345), phosphorylated
(P)-JNK (sc-6254), BAX (sc-493), Bcl-2 (sc-492), and β-actin
(sc-47778) were obtained from Santa Cruz Biotechnology, Inc. (Santa
Cruz, CA, USA). Secondary antibodies were purchased from Beijing
ComWin Biotech Co., Ltd. (Beijing, China).
Cell line and cell culture
Cells of the rat H9c2 cardiomyocyte cell line were
cultured in DMEM with 10% FBS and 1% antibiotic-antimycotic
(penicillin and streptomycin; Gibco; Thermo Fisher Scientific,
Inc.). When cell confluency reached to 70–80%, the H9c2 cells,
maintained at 37°C with 5% CO2, were used for
experiments. For all the treatment groups, the H9c2 cells (100,000
cells/ml) were pre-incubated with or without different
concentrations (0.5, 1, 2.5 and 5 mM) of 4-PBA, dissolved in PBS,
for 24 h prior to initiating I/R. A simulated I/R cell model was
used, as described previously (19) with modifications. The H9c2 cells
were exposed to ischemia by replacing the DMEM with ischemic buffer
(DMEM without D-glucose; pH 6.3; Gibco; Thermo Fisher Scientific,
Inc.). Subsequently, the cells were incubated in a Modular
Incubator Chamber (Billups-Rothenberg, Del Mar, CA, USA) in an
atmosphere of 95% nitrogen and 5% CO2 at 37°C for 6 h.
Following ischemia, reperfusion was initiated by incubating the
cells in DMEM at 37°C with 5% CO2 for different
durations. In the normoxic control groups, the cells were incubated
with DMEM in an atmosphere of 5% CO2 at 37°C.
Cell viability analysis
Cell viability was determined using an MTT assay and
cell death was measured using a lactate dehydrogenase (LDH) assay,
as previously reported (20).
Briefly, the H9c2 cardiomyocytes were seeded at a density of
5×103 cells per well into 96-well plates and, at the end
of the respective treatments, the cells were incubated with MTT
solution (1 mg/ml final concentration stock solution in PBS per
well) at 37°C for 4 h. The medium was then removed, and the
formazan crystals were dissolved with 150 µl DMSO. The
absorbance at 570 nm was determined using a microplate reader
(Spectra Fluo, Tecan, Sunrise, Austria). The experiments were
repeated in triplicate and data were expressed as the percentages
of the control. The LDH assay was used to evaluate myocardial cell
death. Following I/R treatment with or without 4-PBA incubation,
the medium of the cardiomyocytes was collected to measure LDH
release using LDH assay kits, according to the manufacturer's
protocol.
Determination of cardiomyocyte
apoptosis
Myocardial apoptosis was assessed using TUNEL
staining, according to the manufacturer's protocol. The apoptotic
index was expressed as the number of apoptotic cells of all
cardiomyocytes per field, following the assessment of 20
randomly-selected fields per specimen. Positive (DNase-treated) and
negative (no addition of terminal transferase) control tissue
sections were incorporated into each assay. Individual nuclei were
visualized at 200× magnification, and all measurements were
performed in a blinded-manner.
Measurement of mitochondrial membrane
potential (Ψm)
The changes in Ψm were detected using JC-1 staining.
The H9c2 cardiomyocytes were cultured (1×105 cells/well)
in poly-L-lysine-coated six-well plates. Following the
pretreatment, the cells were incubated with JC-1 (2 µM final
concentration) at 37°C in the dark for 15 min. The cells were then
washed three times with PBS and examined under a Nikon TE300
confocal fluorescence microscope equipped with an argon laser
(Nikon Coproration, Tokyo, Japan).
Western blotting
For immunoblotting, the cells were lysed with RIPA
buffer, containing 50 mM Tris-HCl (pH 7.5), 100 mM NaCl and 1%
Triton X-100, with protease inhibitors (aprotinin, leupeptin,
phenylmethylsulfonyl fluoride and pepstatin) and phosphatase
inhibitor (Sigma cocktail; Sigma-Aldrich). Following the lysate
centrifugation for 15 min at 12,000 × g at 4°C, the supernatants
were collected by removing insoluble materials. Protein was
quantified using a BCA Protein Assay Kit (Beijing ComWin Biotech
Co, Ltd.). For Western blot analysis, an equal quantity of protein
(50–100 µg) was loaded in each well and separated by 10–15%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The
separated proteins were then transferred from the gel onto
polyvinylidinene fluoride membranes (EMD Millipore, Bedford, MA,
USA) and blocked in 5% non-fat dry milk prepared in 1X
Tris-buffered saline with 20% Tween-20 (TBST; Beijing ComWin
Biotech Co., Ltd.) for 2 h. The membranes were incubated overnight
at 4°C with the following primary antibodies: Polyclonal rabbit
anti-Grp78 (1:500), monoclonal mouse anti-CHOP (1:200), polyclonal
rabbit anti-BAX (1:200), polyclonal rabbit anti-Bcl-2 (1:200),
polyclonal rabbit anti-PERK (1:500), polyclonal rabbit anti-ATF6
(1:500), monoclonal mouse anti-P-JNK (1:200), monoclonal mouse
anti-JNK (1:200) and monoclonal mouse anti-β-actin (1:1,000).
Following washing of the membranes three times with 1X TBST, the
membranes were probed with the following secondary antibodies
(1:1,000) for 2 h at room temperature: Horseradish peroxidase
(HRP)-conjugated goat anti-rabbit IgG (CW0103M) and HRP-conjugated
goat anti-mouse IgG (CW0102M). Following 30 min of washing with
TBST, the membranes were visualized using an ECL Plus detection
system (Beyotime Institute of Biotechnology, Haimen, China) and the
relative band densities were measured using a Fluor Chem FC2 system
(NatureGene Corp., Beijing, China).
Statistical analysis
The mean values were calculated from the data
obtained from three investigations in each group and at least three
separate in vitro experiments. GraphPad Prism version 5.0
(GraphPad Software, Inc., La Jolla, CA, USA) was used to perform
all statistical analyses. The results were compared using one-way
analysis of variance and Tukey's post-hoc test to identify specific
differences between groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
I/R-induced cell death is dependent on
the duration of reperfusion
In order to investigate whether 4-PBA was able to
protect against cardiac injury induced by I/R in vitro, the
present study first determined the effect of reperfusion duration
on cell death. The H9c2 cardiomyocytes were exposed to hypoxia for
6 h, as previously reported (19),
followed by reoxygenation for different periods of time (0.5, 1, 2,
4, 8 and 12 h) to mimic in vitro I/R conditions. Cell
viability was then detected using an MTT assay and cell death was
evaluated using an LDH assay. As shown in Fig. 1A, the cells in control group were
considered 100% viable. Hypoxia and reoxygenation caused a decrease
in cell viability in a time-dependent manner. The viability of the
cardiomyocytes at 12 h post-reoxygenation was ~57%. In addition,
LDH leakage was measured as a biomarker of cell death. As shown in
Fig. 1B, reoxygenation induced the
release of LDH in a time-dependent manner, compared with the
untreated control group. LDH leakage increased rapidly between 0.5
and 8 h post-reoxygenation and reached a peak at 12 h, which was
~8-fold of that in the untreated control group. Based on these
results, ischemia for 6 h and reperfusion for 12 h were selected
for the in vitro I/R conditions in the following
experiments.
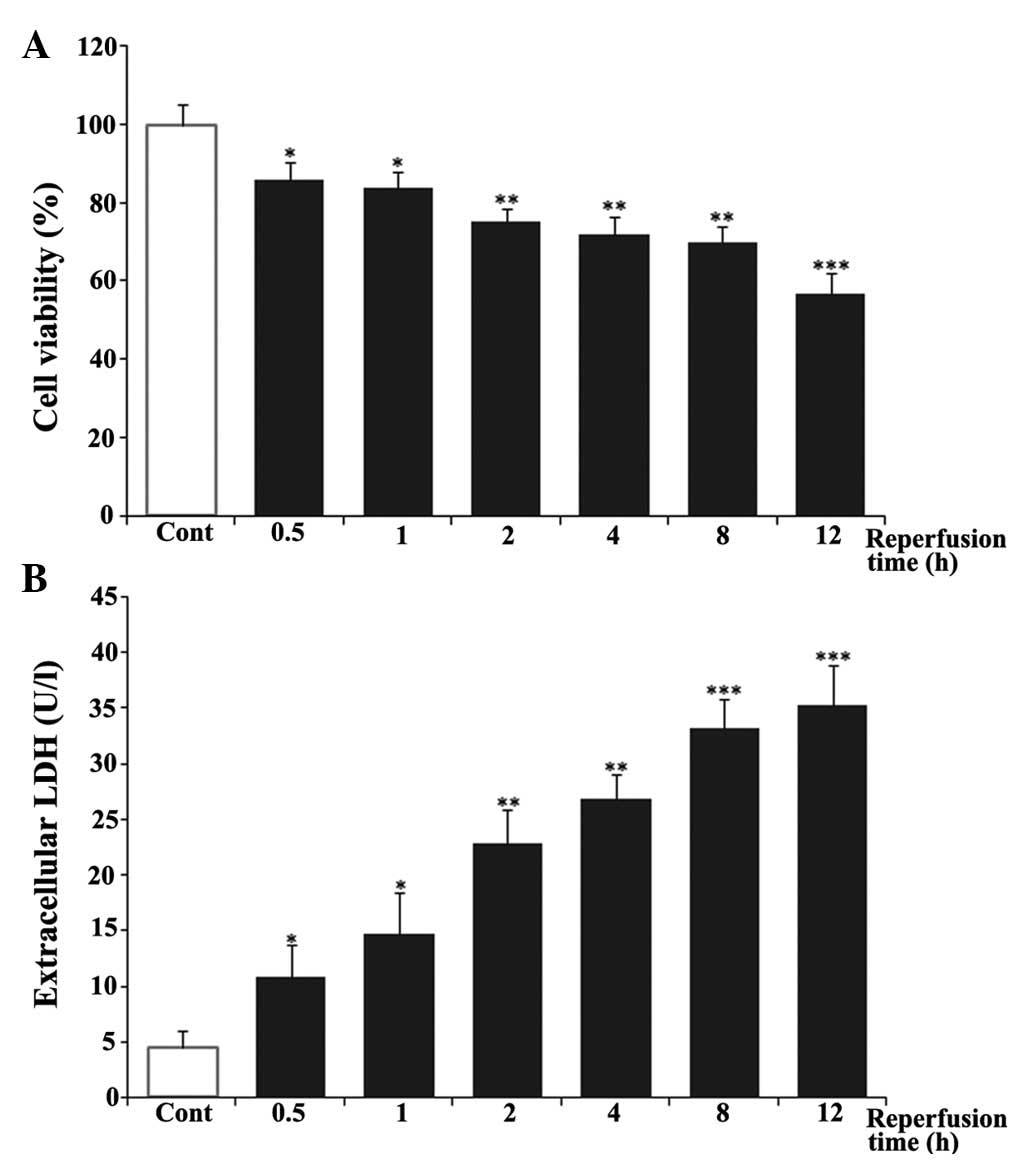 | Figure 1Effects of reperfusion time on cell
viability and LDH release in H9c2 cardiomyocytes. H9c2
cardiomyocytes were exposed to 6 h of hypoxia, followed by
reoxygenation for 0.5, 1, 2, 4, 8 or 12 h. (A) Cell viability was
then examined using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl
tetrazolium bromide assay. (B) Cell death was measured using an LDH
assay kit. The results are represented as the mean ± standard
deviation from three independent experiments.
*P<0.05, **P<0.01 and
***P<0.001, vs. Cont. Cont, untreated control; LDH,
lactate dehydrogenase. |
Treatment with 4-PBA prevented I/R injury-mediated
myocardial cell death. To evaluate the effect of 4-PBA (Fig. 2A) on I/R injury on the rat H9c2
cells, the cardiomyocytes were incubated with 4-PBA at a range of
concentrations between 0 and 5 mM for 24 h, prior to mimicking I/R
injury in vitro. MTT and LDH assays were performed to
determine the role of 4-PBA in the prevention of I/R-induced
damage. The data demonstrated that cell viability was significantly
improved following treatment with 4-PBA at concentrations between
0.5 and 5 mM, in a dose-dependent manner, in triplicate experiments
(Fig. 2B). No statistically
significant differences were found in cell viability or cell death
between the cells incubated with 5 mM 4-PBA for 24 h and the
control group (Fig. 2B). The level
of LDH leakage in the I/R group was significantly higher, compared
with that in the control group (P<0.001), whereas 4-PBA
pretreatment decreased the levels of LDH (Fig. 2C). The results confirmed that 4-PBA
pretreatment had the ability to inhibit the H9c2 cell death induced
by I/R, and pretreatment of the cells with 4-PBA at 5 mM for 24 h
was used in the subsequent experiments.
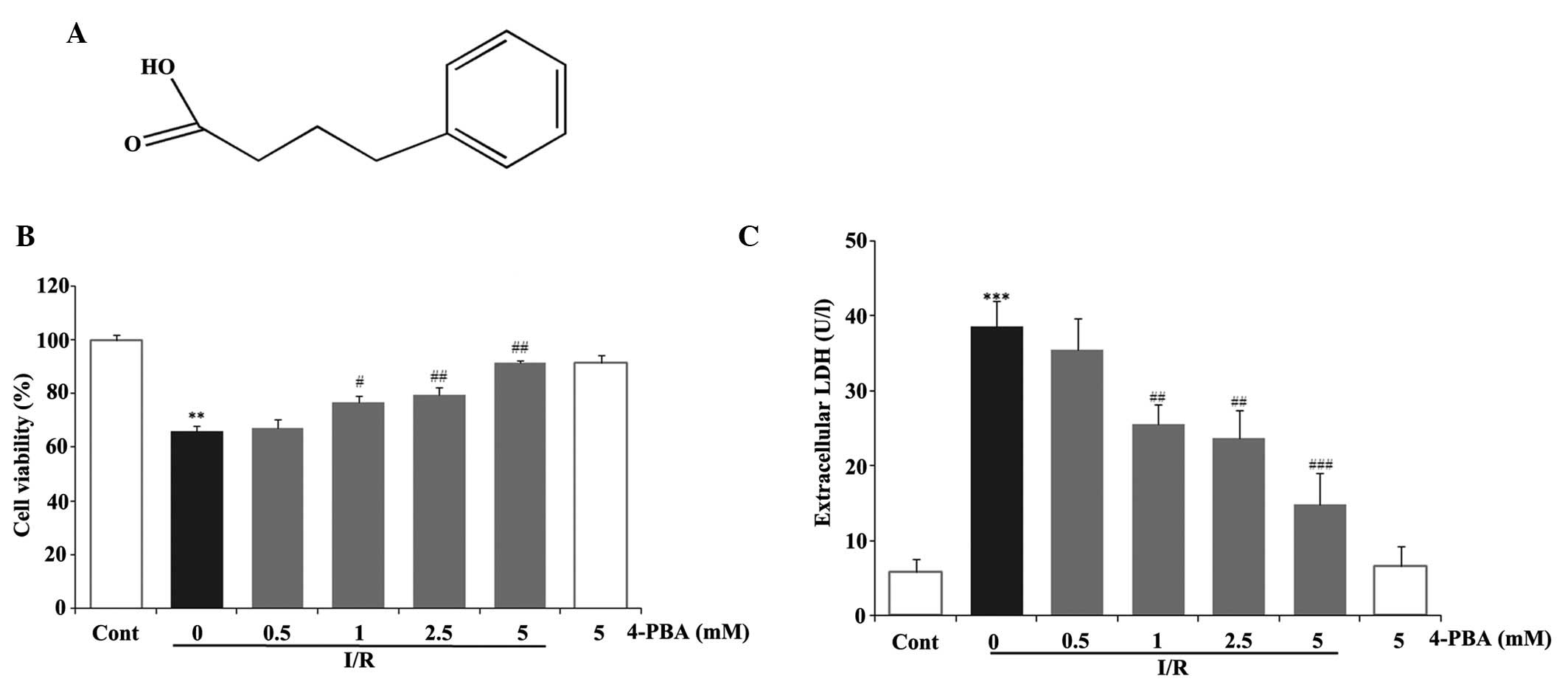 | Figure 2Effects of 4-PBA on cell viability and
LDH release in I/R-induced H9c2 cells. H9c2 cardiomyocytes were
incubated with different concentrations of 4-PBA (0.5, 1, 2.5 and 5
mM) for 2 h prior to exposure to hypoxia for 6 h and reoxygenation
for 12 h. (A) Chemical structure of 4-PBA. (B) Cell viability was
determined using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl
tetrazolium bromide assay and expressed as a relative percentage of
the control group. (C) LDH release was measured to determine cell
death. The results are represented as the mean ± standard deviation
from three independent experiments. **P<0.01 and
***P<0.001, vs. Cont; #P<0.05,
##P<0.01 and ###P<0.001, vs.
I/R-treated cells. 4-PBA, 4-phenylbutyric acid; I/R,
iscehemia/reperfusion; Cont, untreated control; LDH, lactate
dehydrogenase. |
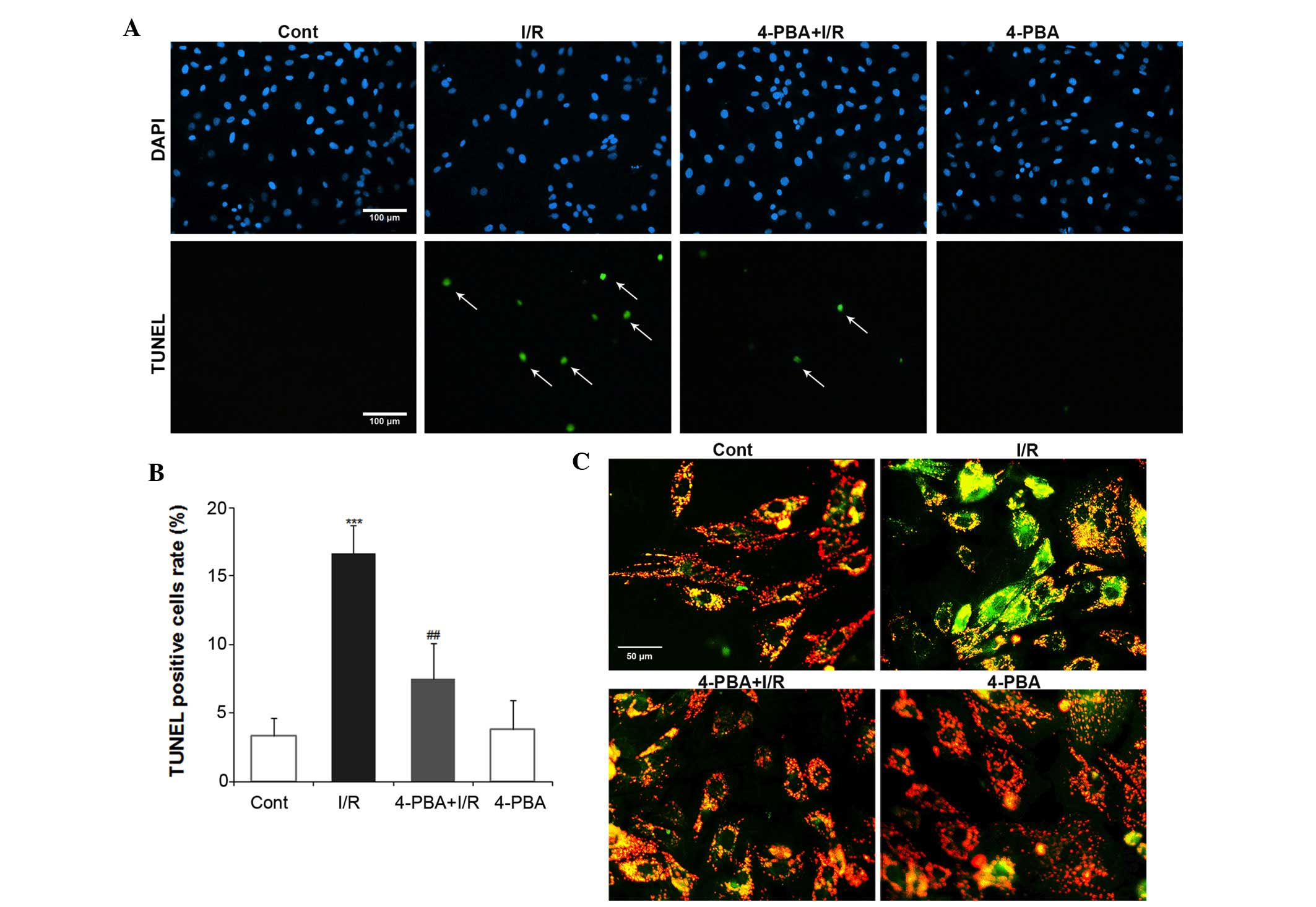
Treatment with 4-PBA prevented the cell apoptosis
induced by myocardial I/R injury. To examine whether 4-PBA has any
effect on I/R-induced apoptosis in H9c2 cells, the present study
performed a TUNEL assay and JC-1 staining. The nuclei of normal
cells were stained by propidium iodide and those of apoptotic cells
were stained green. The number of TUNEL-positive cells was manually
counted in five randomly-selected fields, with the apoptotic index
expressed as a percentage of the total counted cells (Fig. 3A). TUNEL staining showed that I/R
increased the apoptotic index between 5.6 and 16.7% (P<0.01),
compared with the control group. However, 5 mM 4-PBA pretreatment
reduced this value to 7.5% (P<0.01, compared with I/R group;
Fig. 3B). No significant
differences were identified in apoptotic index between the
4-PBA-only treated group and the control group. As the change in Ψm
is one of the first signs of apoptosis, the present study
determined the levels of mitochondrial depolarization using JC-1
staining (Fig. 3C). I/R
significantly decreased the mitochondrial red:green fluorescence
intensity ratio, indicating altered Ψm and depolarization.
Treatment with 4-PBA partly recovered the mitochondrial red:green
fluorescence intensity ratio, suggesting that it provided
protection against the mitochondrial depolarization induced by I/R.
In conclusion, these data confirmed that exposure of cardiomyocytes
to I/R induced apoptotic cell death, however, pretreatment with
4-PBA reversed this effect and protected the cardiomyocytes.
ER stress-induced apoptosis was involved in the
protective effect of 4-PBA on the myocardial I/R injury. To
investigate whether the cardioprotective effect of 4-PBA is
associated with the apoptosis induced by ER stress signaling during
I/R injury, the protein levels of pro-apoptotic CHOP, BAX, JNK, and
P-JNK, anti-apoptotic Bcl-2, and the ER stress response proteins,
Grp78, PERK and ATF6, were measured using Western blot analysis.
CHOP is a critical pro-apoptotic factor in ER stress-associated
apoptosis. As shown in Fig. 4A and
B, the present study found that the myocardial protein
expression of CHOP in the I/R group was significantly upregulated,
compared with that in the control group (P<0.01). However, 4-PBA
attenuated the I/R-induced upregulation in the expression of CHOP,
compared with the I/R group (P<0.01). In addition, the
I/R-induced increases in the expression levels of BAX (Fig. 4C) and P-JNK (Fig. 4D) were significantly inhibited by
4-PBA treatment. However, the inhibition of I/R on the protein
expression of anti-apoptotic Bcl-2 (Fig. 4E) was attenuated by 4-PBA. To
further understand the mechanism underlying the effect of 4-PBA on
ER stress, the present study also examined the effect of 4-PBA on
Grp78, PERK and ATF6, the major transcription factors involved in
ER stress. The immunoblotting results indicated that the protein
levels of Grp78, ATF6 and PERK were significantly increased
following I/R treatment, which were attenuated by 4-PBA (Fig. 5). These data suggested that the ER
stress response was activated acutely in I/R injury, whereas
treatment with 4-PBA inhibited the activation of ER
stress-associated proteins, compared with the respective control
group. Taken together, these results demonstrated that the
protective role of 4-PBA in I/R injury was associated with the
inhibition of apoptosis signaling pathways induced by ER
stress.
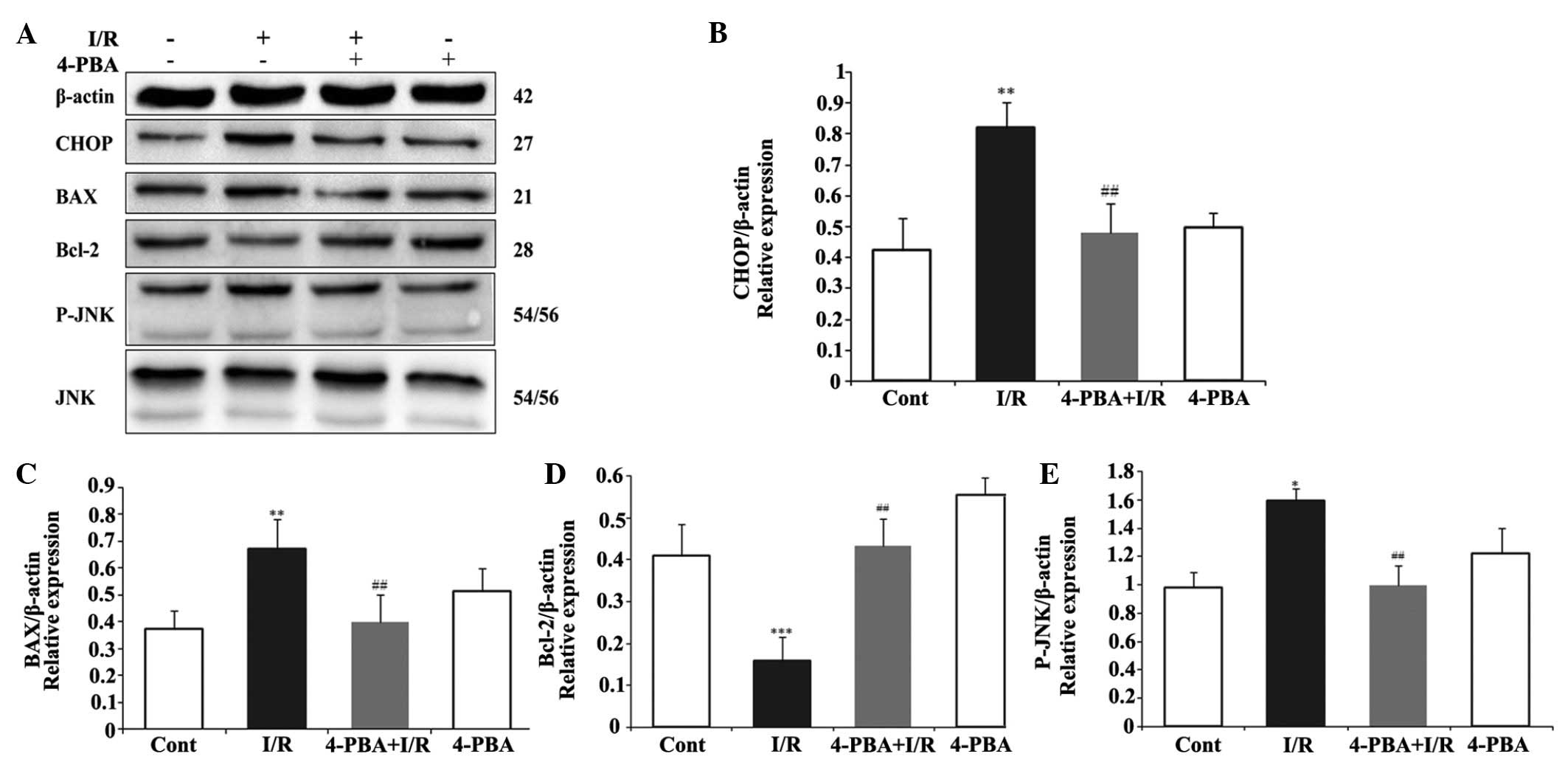 | Figure 4Effects of the expression of 4-PBA on
apoptosis-associated proteins in I/R-induced H9c2cells. The H9c2
cells were exposed or not exposed to I/R, with or without 5 mM
4-PBA incubation. (A) 4-PBA suppressed the I/R-induced upregulation
of CHOP, BAX and P-JNK, and downregulation of Bcl-2. The normalized
ratio of (B) CHOP, (C) BAX and (D) Bcl-2 to β-actin, and (E) P-JNK
to JNK were determined by densitometry. The results are expressed
as the mean ± standard deviation of three independent experiments.
*P<0.05, **P<0.01 and
***P<0.001 vs. Cont; ##P<0.01 vs. I/R.
4-PBA, 4-phenylbutyric acid; I/R, iscehemia/reperfusion; Cont,
untreated control; CHOP, C/EBP homologous protein, Bcl-2, B cell
lymphoma-2; BAX, Bcl-2-associated X protein; JNK, c-Jun N-terminal
kinase; P-JNK, phosphorylated JNK. |
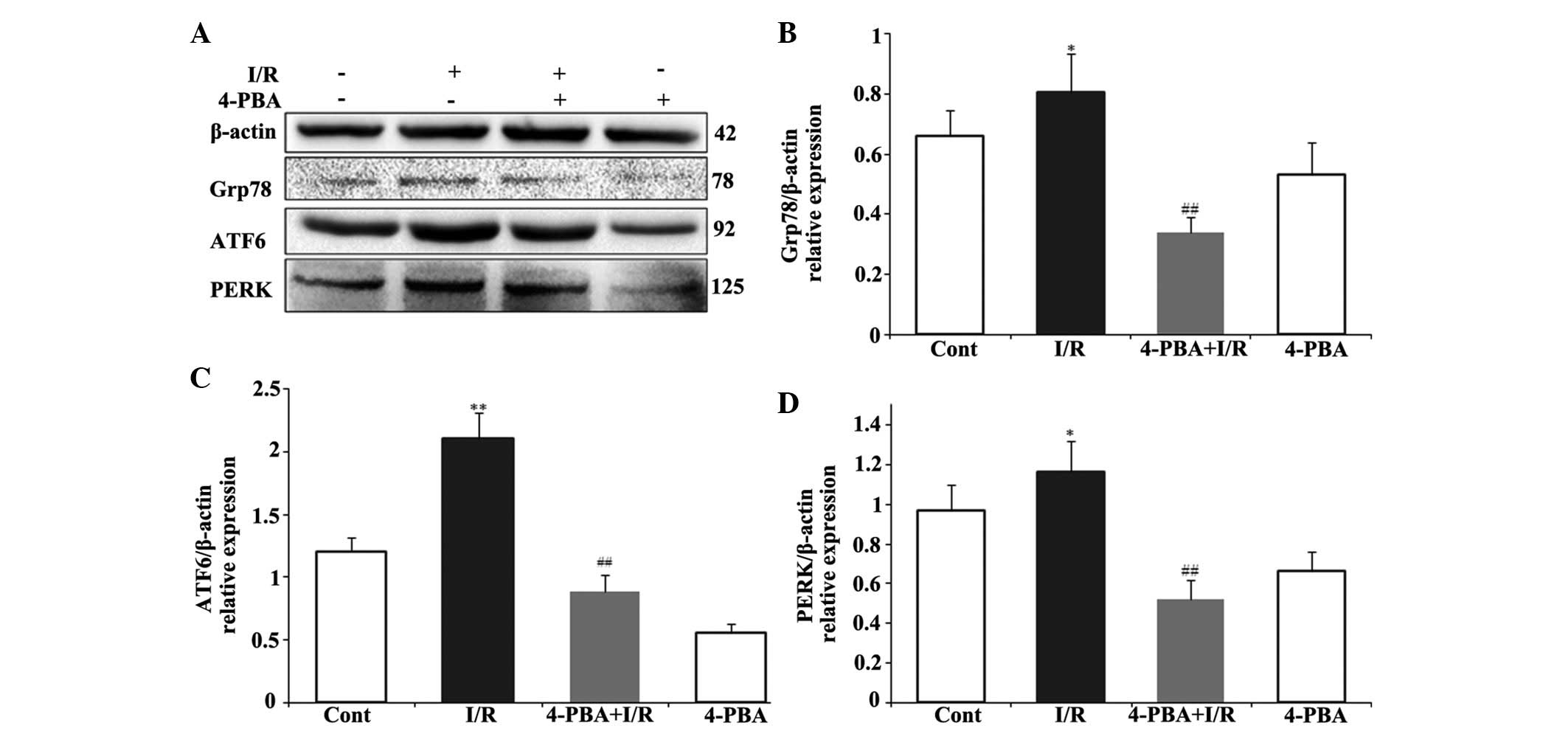 | Figure 5Involvement of ER stress signaling
pathway in the cardioprotective effect of 4-PBA in I/R-induced H9c2
cells. The H9c2 cardiomyocytes were pretreated with or without
4-PBA (5 mM) for 2 h prior to I/R exposure. (A) Expression levels
of Grp78, ATF6 and PERK were detected by immunoblotting. The
normalized ratio of (B) Grp78, (C) ATF6 and (D) PERK to β-actin,
were determined by densitometry. The results are presented as the
mean ± standard deviation from three independent experiments.
*P<0.05 and **P<0.01, vs. Cont;
##P<0.01, vs. I/R. 4-PBA, 4-phenylbutyric acid; I/R,
iscehemia/reperfusion; Cont, untreated control; Grp78,
glucose-regulated protein 78; ATF6, activating transcription factor
6; PERK, protein kinase RNA-like endoplasmic reticulum kinase. |
Discussion
In the present study, it was demonstrated that
4-PBA, an agent used clinically in disorders of the urea cycle in
children and in sickle cell disease, exhibited cardioprotective
effects during I/R through mediation of specific ER stress-induced
cell apoptosis pathways. The involvement of apoptosis in cell loss,
and the apoptotic signaling pathways in myocardial I/R are
relatively novel and controversial phenomena (3,6,21),
which remain to be fully elucidated. The present study examined
whether I/R induced the UPR and whether induction of the UPR during
I/R activates ER stress-associated cell death pathways. The
resulting data demonstrated that I/R significantly increased
apoptotic cardiomyocyte death by activating a series of
pro-apoptotic proteins (Fig. 4).
In addition, the expression levels of Grp78, ATF6 and PERK, which
are important molecular indicators of ER stress, were significantly
upregulated in the I/R group (Fig.
5), indicating that ER stress was induced by I/R. However, the
overexpression of the ER stress molecules was significantly
suppressed by 4-PBA treatment at an appropriate dose.
4-PBA is a chemical chaperone with approved in
vivo safety and has already been approved by the US Food and
Drug Administration for clinical use (22). Numerous studies have demonstrated
that 4-PBA inhibits ER stress and UPR signaling. A previous study
showed that 4-PBA significantly downregulates the expression levels
of fibrosis-associated genes and protects against cardiac fibrosis
in an animal model to withstand pressure overload (23). According to another report, the
attenuation of ER stress using 4-PBA prevents oproterenol-induced
cardiac fibrosis (24). The aim of
the present study was to confirm the hypothesis that 4-PBA protects
cardiomyocytes from I/R injury by inhibiting the accumulated
misfolded protein-induced ER stress response and the associated
apoptosis.
The ER, as a vast membranous organelle, is
responsible for the homeostasis of cellular Ca2+, and
the synthesis, maturation and trafficking of a wide range of
proteins (25). The capacity of
the ER is important for the cell due to its capabilities in
managing synthetic, metabolic and other adverse conditions
(26). Recognition of the
contribution of ER to apoptotic cell death has been developed. It
was reported in 2000 for the first time that treatment with calcium
ionophores, a sarcoplasmic/ER-calcium ATPase pump inhibitor
(thapsigargin), or an inhibitor of N-linked glycosylation
(tunicamycin) can initiate a form of apoptosis referred to as ER
stress-mediated apoptosis (27).
In the ER stress signaling cascade, the chaperone, Grp78, is the
first element, which is involved in protein folding and,
consequently, is responsible for activating the three PERK, ATF6
and IRE1 UPR sensors (8,28). Increased numbers of unfolded
proteins within the ER prompt Grp78 to 'undock' from IRE1, PERK and
ATF6. ATF6 undergoes proteolytic activation in the Golgi apparatus,
followed by nuclear translocation, where it dimerizes with basic
leucine zipper transcription factors, including X-box binding
protein 1, which regulate ER stress response genes (29). PERK oligomerization activates its
intrinsic kinase activity, resulting in eukaryotic translation
initiation factor 2, subunit 1 α (eIF2α) phosphorylation, which
suppresses translation. The phosphorylation of eIF2α promotes
pro-survival (early) and pro-apoptotic (late) transcriptional
programs (30). Several studies
have documented activation of the UPR in the myocardium (31). A common element following
activation of these three sensors is the expression of the protein
CHOP, which has been documented to mediate apoptosis following ER
stress and to be involved in several ER-stress-associated diseases
(12,32). In the present study, it was
demonstrated that the expression levels of Grp78, ATF6 and PERK
were significantly elevated in the I/R group. Of note, it was found
that 4-PBA effectively inhibited ER stress-mediated apoptosis
pathways. 4-PBA treatment not only mediated the protein expression
levels of BAX and Bcl-2 in vitro, but it also reduced the
expression levels of ER stress moderators, Grp78, ATF6 and PERK,
and CHOP in particular. I/R induces a prominent increase in CHOP
activation, a common effect of the three main signaling pathways of
ER stress, in short time, which triggers JNK- regulated apoptosis.
The phosphorylation of PERK in response to ER stress is considered
to be a protective response, as it attenuates protein synthesis and
provides cells with more time to manage correct folding or degrade
accumulated misfolded proteins (33). Thus, the lack of PERK- kinase
activity and lack of eIF2α phosphorylation make the cells more
vulnerable to apoptosis in response to ER stress. The ATF6 pathway
is considered to control a rapid response to stimulus-induced ER
stress (25). Western blot
analysis identified 92 and 125 kDa processed fragments, confirming
the activation of ATF6 and PERK in I/R cardiomyocytes (Fig. 5C and D). Members of the Bcl-2
family are also important in the transduction of an internal
apoptotic signal.
In conclusion, the present study reported for the
first time, to the best of our knowledge, that I/R induced the ER
stress-associated pathways and their downstream apoptotic targets.
The mechanism by which I/R affects the homeostasis of ER and
induces ER stress remains to be fully elucidated, and only indirect
evidence is available to suggest that I/R in the heart induces ER
stress. In the present study, the results showed the inhibitory
effect of 4-PBA on decreasing myocardium apoptosis and preventing
cell death induced by I/R, suggesting the possibility of the use of
4-PBA as a therapeutic agent for cardiac ischemia/reperfusion
diseases. However, the mechanism underlying the cardioprotective
action of 4-PBA remains to be fully elucidated. Further
investigations are required to elucidate other mechanisms involved
in the effect of 4-PBA in ER stress, and the distinct effects of
cardiac injury and heart failure. Improved understanding may lead
to the design of improved ER stress-targeted therapies against
life-threatening cardiac diseases.
References
|
1
|
Di Diego JM and Antzelevitch C: Acute
myocardial ischemia: Cellular mechanisms underlying ST segment
elevation. J Electrocardiol. 47:486–490. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Logue SE, Gustafsson AB, Samali A and
Gottlieb RA: Ischemia/reperfusion injury at the intersection with
cell death. J Mol Cell Cardiol. 38:21–33. 2005. View Article : Google Scholar
|
|
3
|
Minamino T and Kitakaze M: ER stress in
cardiovascular disease. J Mol Cell Cardiol. 48:1105–1110. 2010.
View Article : Google Scholar
|
|
4
|
Scarabelli TM and Gottlieb RA: Functional
and clinical repercussions of myocyte apoptosis in the multifaceted
damage by ischemia/reperfusion injury: Old and new concepts after
10 years of contributions. Cell Death Differ. 11(Suppl 2):
S144–S152. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jennings RB, Sommers HM, Smyth GA, Flack
HA and Linn H: Myocardial necrosis induced by temporary occlusion
of a coronary artery in the dog. Arch Pathol. 70:68–78.
1960.PubMed/NCBI
|
|
6
|
Duan SR, Wang JX, Wang J, Xu R, Zhao JK
and Wang DS: Ischemia induces endoplasmic reticulum stress and cell
apoptosis in human brain. Neurosci Lett. 475:132–135. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim I, Xu W and Reed JC: Cell death and
endoplasmic reticulum stress: Disease relevance and therapeutic
opportunities. Nat Rev Drug Discov. 7:101–1030. 2008. View Article : Google Scholar
|
|
8
|
Shen X, Zhang K and Kaufman RJ: The
unfolded protein response-a stress signaling pathway of the
endoplasmic reticulum. J Chem Neuroanat. 28:79–92. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nishitoh H: Life and death under the ER
stress condition. Journal of Oral Biosciences. 46:259–269. 2004.
View Article : Google Scholar
|
|
11
|
Sozen E, Karademir B and Ozer NK: Basic
mechanisms in endoplasmic reticulum stress and relation to
cardiovascular diseases. Free Radic Biol Med. 78:30–41. 2015.
View Article : Google Scholar
|
|
12
|
Verfaillie T, Garg AD and Agostinis P:
Targeting ER stress induced apoptosis and inflammation in cancer.
Cancer Lett. 332:249–264. 2013. View Article : Google Scholar
|
|
13
|
Brusilow SW and Maestri NE: Urea cycle
disorders: Diagnosis, pathophysiology and therapy. Adv Pediatr.
43:127–170. 1996.
|
|
14
|
Qi X, Hosoi T, Okuma Y, Kaneko M and
Nomura Y: Sodium 4-phenylbutyrate protects against cerebral
ischemic injury. Mol Pharmacol. 66:899–908. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vilatoba M, Eckstein C, Bilbao G, Smyth
CA, Jenkins S, Thompson JA, Eckhoff DE and Contreras JL: Sodium
4-phenylbutyrate protects against liver ischemia reperfusion injury
by inhibition of endoplasmic reticulum-stress mediated apoptosis.
Surgery. 138:342–351. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu CX, Liu R, Gao M, Zhao G, Wu S, Wu CF
and Du GH: Pinocembrin protects brain against ischemia/reperfusion
injury by attenuating endoplasmic reticulum stress induced
apoptosis. Neurosci Lett. 546:57–62. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mimori S, Ohtaka H, Koshikawa Y, Kawada K,
Kaneko M, Okuma Y, Nomura Y, Murakami Y and Hamana H:
4-Phenylbutyric acid protects against neuronal cell death by
primarily acting as a chemical chaperone rather than histone
deacetylase inhibitor. Bioorg Med Chem Lett. 23:6015–6018. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kuang X, Hu W, Yan M and Wong PK:
Phenylbutyric acid suppresses protein accumulation-mediated ER
stress in retrovirus-infected astrocytes and delays onset of
paralysis in infected mice. Neurochem Int. 57:738–748. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun J, Sun G, Meng X, Wang H, Wang M, Qin
M, Ma B, Luo Y, Yu Y, Chen R, et al: Ginsenoside RK3 prevents
hypoxia-reoxygenation induced apoptosis in H9c2 cardiomyocytes via
AKT and MAPK pathway. Evid Based Complement Alternat Med.
2013:6901902013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun L, Isaak CK, Zhou Y, Petkau JC, O K,
Liu Y and Siow YL: Salidroside and tyrosol from Rhodiola protect
H9c2 cells from ischemia/reperfusion-induced apoptosis. Life Sci.
91:151–158. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yuan Y, Guo Q, Ye Z, Pingping X, Wang N
and Song Z: Ischemic postconditioning protects brain from
ischemia/reperfusion injury by attenuating endoplasmic reticulum
stress-induced apoptosis through PI3K-Akt pathway. Brain Res.
1367:85–93. 2011. View Article : Google Scholar
|
|
22
|
Kim HD, Jang CY, Choe JM, Sohn J and Kim
J: Phenylbutyric acid induces the cellular senescence through an
Akt/p21(WAF1) signaling pathway. Biochem Biophys Res Commun.
422:213–218. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Park CS, Cha H, Kwon EJ, Sreenivasaiah PK
and Kim do H: The chemical chaperone 4-phenylbutyric acid
attenuates pressure-overload cardiac hypertrophy by alleviating
endoplasmic reticulum stress. Biochem Biophys Res Commun.
421:578–584. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ayala P, Montenegro J, Vivar R, Letelier
A, Urroz PA, Copaja M, Pivet D, Humeres C, Troncoso R, Vicencio JM,
et al: Attenuation of endoplasmic reticulum stress using the
chemical chaperone 4-phenylbutyric acid prevents cardiac fibrosis
induced by isoproterenol. Exp Mol Pathol. 92:97–104. 2012.
View Article : Google Scholar
|
|
25
|
Yamamoto K, Yoshida H, Kokame K, Kaufman
RJ and Mori K: Differential contributions of ATF6 and XBP1 to the
activation of endoplasmic reticulum stress-responsive cis-acting
elements ERSE, UPRE and ERSE-II. J Biochem. 136:343–350. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang WA, Groenendyk J and Michalak M:
Endoplasmic reticulum stress associated responses in cancer.
Biochim Biophys Acta. 1843:2143–2149. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Powell KS and Latterich M: The making and
breaking of the endoplasmic reticulum. Traffic. 1:689–694. 2000.
View Article : Google Scholar
|
|
28
|
Cullinan SB and Diehl JA: Coordination of
ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway.
Int J Biochem Cell Biol. 38:317–332. 2006. View Article : Google Scholar
|
|
29
|
Ye J, Rawson RB, Komuro R, Chen X, Davé
UP, Prywes R, Brown MS and Goldstein JL: ER stress induces cleavage
of membrane-bound ATF6 by the same proteases that process SREBPs.
Mol Cell. 6:1355–1364. 2000. View Article : Google Scholar
|
|
30
|
Owen CR, Kumar R, Zhang P, McGrath BC,
Cavener DR and Krause GS: PERK is responsible for the increased
phosphorylation of eIF2alpha and the severe inhibition of protein
synthesis after transient global brain ischemia. J Neurochem.
94:1235–1242. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhu S, Jin J, Wang Y, Ouyang Z, Xi C, Li
J, Qiu Y, Wan J, Huang M and Huang Z: The endoplasmic reticulum
stress response is involved in apoptosis induced by aloe-emodin in
HK-2 cells. Food Chem Toxicol. 50:1149–1158. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li H, Zhu X, Fang F, Jiang D and Tang L:
Down-regulation of GRP78 enhances apoptosis via CHOP pathway in
retinal ischemia-reperfusion injury. Neurosci Lett. 575:68–73.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vaughn LS, Snee B and Patel RC: Inhibition
of PKR protects against tunicamycin-induced apoptosis in
neuroblastoma cells. Gene. 536:90–96. 2014. View Article : Google Scholar
|