Introduction
Brugada syndrome (BS) is an electrical and
hereditary disease, including in a group of cardiac channelopathies
characterized by a right bundle branch block (RBBB), ST segment
elevation on a 12-lead surface electrocardiogram (ECG) and an
increased risk of syncope, seizures, and sudden cardiac arrest in
an otherwise healthy individual with a structurally normal heart.
In an ECG, the ST segment connects the QRS complex and the T wave
and has a duration of 0.080 to 0.120 sec. In BS, the patient may
present with 3 types of ECG (I, II or III), only type I
(coved-type) is diagnostic of BS and demonstrates an ST segment
elevation of ≥2mm and a negative T wave (1). In addition to pathological,
diagnostic ECGs, BS patients may present an alternate or normal
ECG. A pharmacological unmasking test was performed using
inhibitors of the sodium channel.
Mutations in the SCN5A gene have been identified in
patients with BS (2–5). Sequencing of the SCN5A gene revealed
that 10–20% of patients exhibited a mutation in SCN5A (6–11).
However, certain variations in SCN5A present an incomplete
penetrance in familial studies (12). Mutations in other genes, including
CACNA1c, CACNB2b, GPD1-L, KCNE3, SCN1B, SCN3B and HCN4, have been
associated with the condition to a smaller extent (~1%) (13–17).
In ~70–80% of patients with BS, direct sequencing of the associated
genes revealed no causative point mutation (18). In the present study, a novel
variant and its pathogenicity were investigated by sequencing SCN5A
and conducting a cosegregation study with the family, in order to
provide a clinical and genetic context for BS.
Materials and methods
Clinical data
The index case was a 26-year-old Latino-American
male. The patient was diagnosed following a temporary loss of
consciousness for 15 sec and a spontaneous recovery. The patient
was admitted to the intensive care unit of the Virgen de la
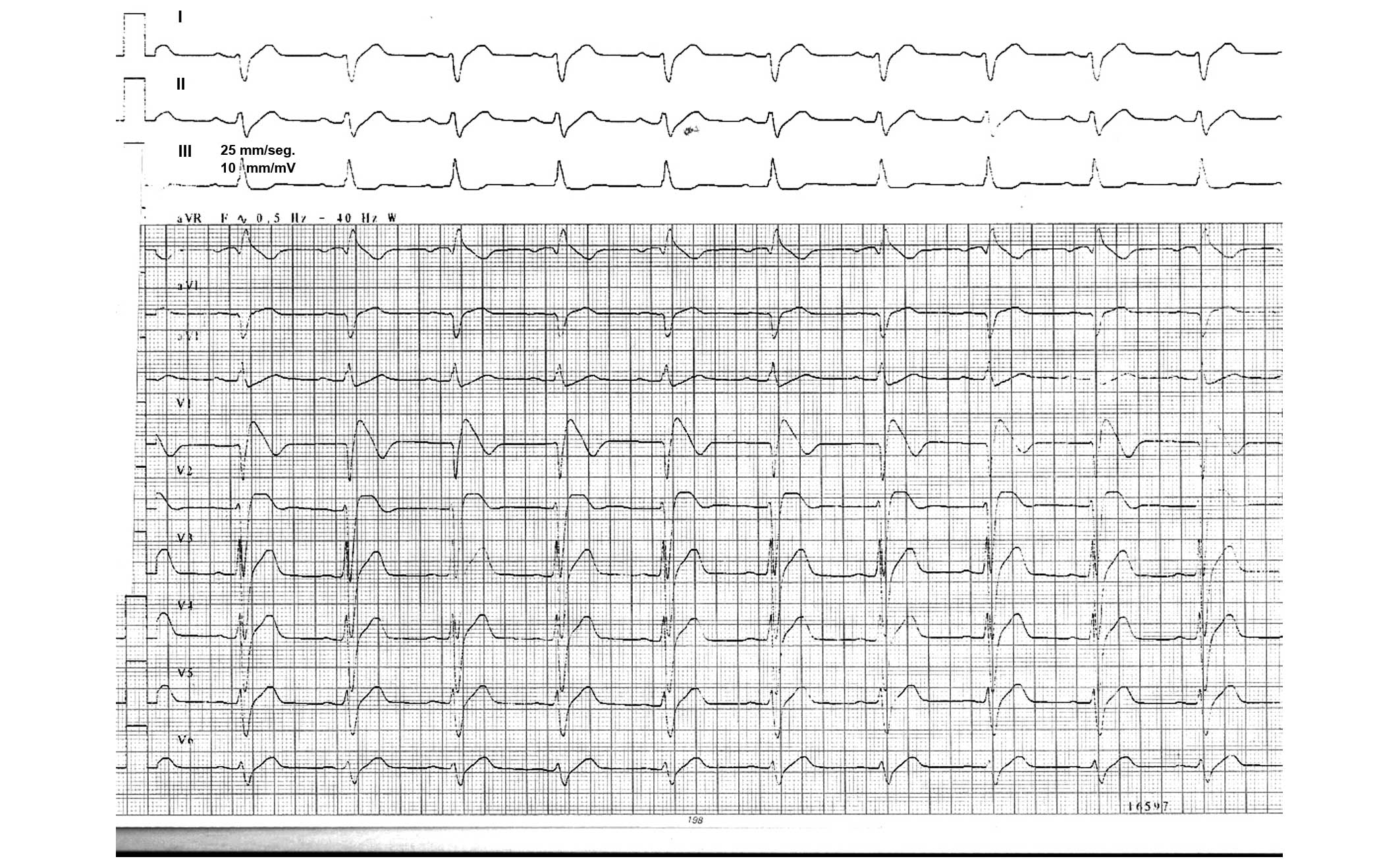
Arrixaca Clinical University Hospital (Murcia, Spain). An ECG was
performed and revealed an elevation in ST (V1–V3), with a negative
t wave and RBBB. The flecainide drug test was positive and an
implantable cardioverter defibrillator was implanted. The ST
elevation in V1 was up to 2 mm and 1 mm in V2, so the patient
demonstrated a diagnostic ECG (coved-type) in V1 and
undifferentiated in V2 (Fig.
1).
To date, no family history of sudden death was
observed. The patient's brother (33-year-old) and sister
(18-year-old) underwent physical examination, ECG and Doppler ECG
investigations, similar to the index patient. The present study was
approved by the ethics committee of Virgen de la Arrixaca Clinical
University Hospital and signed informed consent was obtained from
the proband.
Genetic study
The index case was included in a genetic study of
BS. The genomic DNA was extracted from peripheral blood samples
using Maxwell® 16 Blood DNA purification kit (Promega
Corporation, Madison, WI, USA). All known exons in the SCN5A gene
were amplified with intronic primers (Bonsai Technologies Group,
SA, Alcobendas, Spain) and sequenced in both directions using
BigDye v1.1 (Applied Biosystems, Foster City, CA, USA) chemistry in
an ABI3130 analyzer (Applied Biosystems). The intronic primers were
forward, 5′-CCCTGCTGAGCACTTTCCATTTG-3′ and reverse,
5′-TACAAGTCAGCTGGACGGAGAAGC-3′. The genomic sequence from the
samples obtained from the index patient was compared with the SCN5A
sequence in the NCBI sequence database (NM_198056). In
silico analysis was performed using pathogenicity prediction
software: PMUT (http://mmb.irbbarcelona.org/PMut/), MutationTaster
(http://www.mutationtaster.org/) and
Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/). The siblings
of the index patient also underwent genomic sequencing from
obtained blood samples and a cosegregation study was conducted to
elucidate the pathogenicity of the variant detected.
Results and Discussion
A total of three variants in the SCN5A gene were
identified. Two were known variants, which are expressed in the
normal population (p.A29A and p.E1061E). Sequence analysis
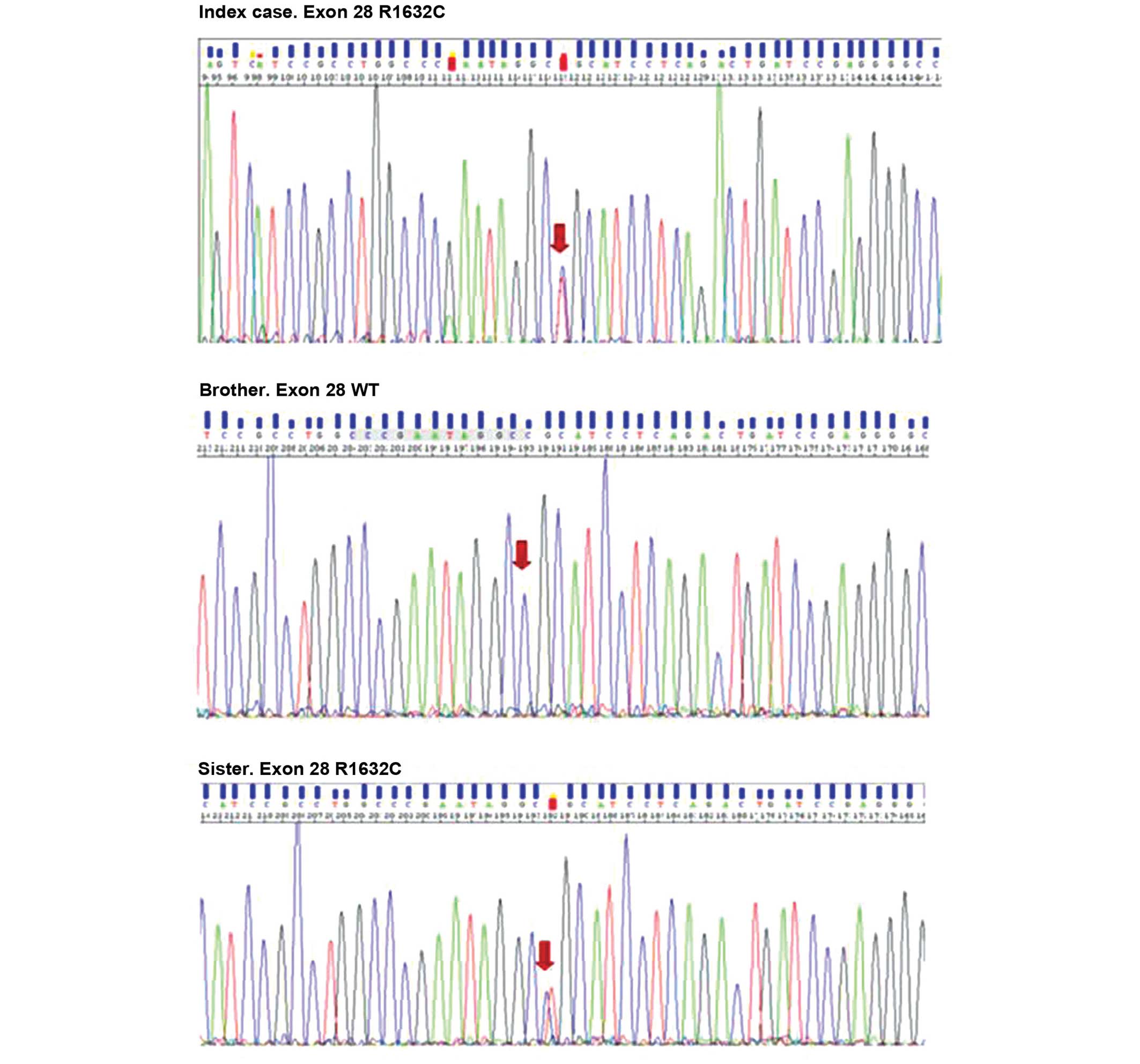
identified a novel missense variation in heterozygotes (the index
case and the patient's sister), c.4894C>T within exon 28,
resulting in the replacement of arginine 1632 by cysteine (Fig. 2). The Grantham score for this
replacement was 180 (scores between 0–250). The variant, p.R1632C,
affected a highly conserved region in the protein, the fourth
transmembrane loop (S4) of the IV domain. S4 acts as the 'voltage
sensor' and is activated by changes in membrane potential, and is
also involved in channel gating.
The bioinformatics analysis of the novel variant was
performed using three online tools, MutationTaster, PolyPhen-2 and
PMUT, to predict the effect of an amino acid substitution on the
structure and function of the protein (Table 1). These three software programmes
predicted that R1632C is pathogenic. Furthermore, the variant is
localized in a key domain for channel formation, and amino acid
substitution results in severe physiochemical changes. The scores
in the table are the probability of the prediction, a score close
to 1 indicates a high likelihood that the prediction is
correct.
 | Table IIn silico analysis using
prediction software. |
Table I
In silico analysis using
prediction software.
| Parameter | MutationTaster | Polyphen-2 | PMUT |
|---|
| Results | Prediction disease
causing | Probably
damaging | Pathological |
| Score | 0.9999 | 1.0 | 0.8846 |
The genetic analysis was performed on his siblings.
A healthy carrier, the patient's sister, exhibited a normal
baseline and drug challenged ECG.
This variant was not present in public databases,
including HGMD, ClinVar and Exome Variant Server. However, in the
identical position, there was a R1632H (c.4895G>A) disease
causing variation (rs199473286). This variant was investigated by
Gui et al (19) using
whole-cell patch clamp experiments. The expression of this variant
causes inactivated channels at physiological membrane potentials
and normal heart rates in patients with Sick Sinus Syndrome.
The present case report, identified a novel amino
acid change located in a known replacement position. Although the
replacement is not identical and in the majority of cases, distinct
changes in the identical position causes similar pathological
result, this variation may cause a different phenotypic expression.
The present study revealed no evidence that this variant
co-segregated with the disease, therefore, more extensive familial
analysis is required. Also, it is necessary to perform a more
frequent clinical follow up with the novel carriers to assess their
risk of developing the disease.
When a novel variation is detected, in silico
analysis can assist with determining the pathogenicity of the novel
alteration, however, a genetic and clinical familial study is
crucial to elucidate the causality of the variation. Genotypic
investigation of family members is required when a potential
pathogenic variant in a proband is identified, to assess the risk
of developing the characteristic phenotype.
Acknowledgments
The authors would like to thank Mr. David Lopez
(Hereditary Cardiopathies Unit, Virgen de la Arrixaca University
Hospital) and Dr Roberto Barriales (Hospital Marítimo de Oza,
Coruña, Spain) for their assistance with the clinical work during
the present study. The current study was supported, in part, by a
national grant from the Sociedad Española de Cardiología and by the
Cardiovascular Research Network (RECAVA) from the Carlos III Health
Institute (grant nos. C03/01, RD06/0014/0017 and
RD06/0014/0018).
References
|
1
|
Brugada R, Campuzano O, Sarquella-Brugada
G, Brugada J and Brugada P: Brugada syndrome. Methodist Debakey
Cardiovasc J. 10:25–28. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Potet F, Mabo P, Le Coq G, Probst V,
Schott JJ, Airaud F, Guihard G, Daubert JC, Escande D and Le Marec
H: Novel brugada SCN5A mutation leading to ST segment elevation in
the inferior or the right precordial leads. J Cardiovasc
Electrophysiol. 14:200–203. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen Q, Kirsch GE, Zhang D, Brugada R,
Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G,
et al: Genetic basis and molecular mechanism for idiopathic
ventricular fibrillation. Nature. 392:293–296. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Antzelevitch C: Ion channels and
ventricular arrhythmias: Cellular and ionic mechanisms underlying
the Brugada syndrome. Curr Opin Cardiol. 14:274–279. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Antzelevitch C: Genetic basis of brugada
syndrome. Heart Rhythm. 4:756–757. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moric E, Herbert E, Trusz-Gluza M,
Filipecki A, Mazurek U and Wilczok T: The implications of genetic
mutations in the sodium channel gene (SCN5A). Europace. 5:325–334.
2003. View Article : Google Scholar
|
|
7
|
Priori SG, Napolitano C, Gasparini M,
Pappone C, Della Bella P, Giordano U, Bloise R, Giustetto C, De
Nardis R, Grillo M, et al: Natural history of Brugada syndrome:
Insights for risk stratification and management. Circulation.
105:1342–1347. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schulze-Bahr E, Eckardt L, Breithardt G,
Seidl K, Wichter T, Wolpert C, Borggrefe M and Haverkamp W: Sodium
channel gene (SCN5A) mutations in 44 index patients with Brugada
syndrome: Different incidences in familial and sporadic disease.
Hum Mutat. 21:651–652. 2003. View Article : Google Scholar
|
|
9
|
Priori SG, Napolitano C, Gasparini M,
Pappone C, Della Bella P, Brignole M, Giordano U, Giovannini T,
Menozzi C, Bloise R, et al: Clinical and genetic heterogeneity of
right bundle branch block and ST-segment elevation syndrome: A
prospective evaluation of 52 families. Circulation. 102:2509–2515.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bai R, Napolitano C, Bloise R, Monteforte
N and Priori SG: Yield of genetic screening in inherited cardiac
channelopathies: How to prioritize access to genetic testing. Circ
Arrhythm Electrophysiol. 2:6–15. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kapplinger JD, Tester DJ, Alders M, Benito
B, Berthet M, Brugada J, Brugada P, Fressart V, Guerchicoff A,
Harris-Kerr C, et al: An international compendium of mutations in
the SCN5A-encoded cardiac sodium channel in patients referred for
Brugada syndrome genetic testing. Heart Rhythm. 7:33–46. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Campuzano O, Allegue C, Iglesias A and
Brugada R: Genetic basis of Brugada syndrome. J Genet Syndr Gene
Ther. 4:72013.
|
|
13
|
Delpón E, Cordeiro JM, Núñez L, Thomsen
PE, Guerchicoff A, Pollevick GD, Wu Y, Kanters JK, Larsen CT,
Hofman-Bang J, et al: Functional effects of KCNE3 mutation and its
role in the development of Brugada syndrome. Circ Arrhythm
Electrophysiol. 1:209–218. 2008. View Article : Google Scholar
|
|
14
|
London B, Michalec M, Mehdi H, Zhu X,
Kerchner L, Sanyal S, Viswanathan PC, Pfahnl AE, Shang LL,
Madhusudanan M, et al: Mutation in glycerol-3-phosphate
dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+
current and causes inherited arrhythmias. Circulation.
116:2260–2268. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Watanabe H, Koopmann TT, Le Scouarnec S,
Yang T, Ingram CR, Schott JJ, Demolombe S, Probst V, Anselme F,
Escande D, et al: Sodium channel β1 subunit mutations
associated with Brugada syndrome and cardiac conduction disease in
humans. J Clin Invest. 118:2260–2268. 2008.PubMed/NCBI
|
|
16
|
Antzelevitch C, Pollevick GD, Cordeiro JM,
Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva
A, Wollnik B, et al: Loss-of-function mutations in the cardiac
calcium channel underlie a new clinical entity characterized by
ST-segment elevation, short QT intervals, and sudden cardiac death.
Circulation. 115:442–449. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weiss R, Barmada MM, Nguyen T, Seibel JS,
Cavlovich D, Kornblit CA, Angelilli A, Villanueva F, McNamara DM
and London B: Clinical and molecular heterogeneity in the Brugada
syndrome: A novel gene locus on chromosome 3. Circulation.
105:707–713. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hedley PL, Jørgensen P, Schlamowitz S,
Moolman-Smook J, Kanters JK, Corfield VA and Christiansen M: The
genetic basis of Brugada syndrome: A mutation update. Hum Mutat.
30:1256–1266. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gui J, Wang T, Jones RPO, Trump D, Zimmer
T and Lei M: Multiple loss-of-function mechanisms contribute to
SCN5A-related familial Sick Sinus Syndrome. PLoS ONE. 5:e109852010.
View Article : Google Scholar : PubMed/NCBI
|