Introduction
Hypoxia-ischemia (HI) is a major cause of perinatal
brain injury and results in mortality or lifelong morbidities
(1,2). Hypoxic ischemia (HI) in neonates
triggers sequential cascades of neurotoxic events within hours,
which last for days to weeks following the injury, resulting in
significant neuronal injury (2,3). The
incidence of perinatal HI reaches 2–6% in live term births each
year in Western countries (4,5).
Approximately half of the HI events result in mortality and 25% of
the survivors suffer from neurological disabilities, including
cerebral palsy, cognitive and/or sensory deficits, mental
retardation, learning disabilities and epilepsy. These impairments
significantly impact life experience and social welfare (6,7).
Additionally, the pathophysiological mechanisms are complex and
processes including apoptosis, necroptosis, mitochondrial
impairment, oxidative stress and inflammation are involved
(8). Until now, no effective
therapy to treat these neurological disorders exists. Therefore,
searching for novel and more effective therapeutic interventions is
necessary and of social value.
Apoptosis signal-regulating kinase 1 (ASK1) is one
of >20 members that make up the triple mitogen-activated protein
kinase (MAPK) family of enzymes (9). Over the past decade, numerous
previous studies have revealed that ASK1 serves a pivotal role in
the cellular response to a wide variety of environmental and
biological stresses, including reactive oxygen species such as
hydrogen peroxide, endoplasmic reticulum stress caused by protein
aggregation, influx of calcium ions, and receptor-mediated signals
transduced via lipopolysaccharides (LPS), Fas ligand, cytokines
(TNFα) and certain G protein-coupled receptor agonists (10–14).
In addition, exogenous expression of ASK1 in cells has shown that
ASK1 signaling engages the intrinsic apoptosis pathway, promoting
cytochrome c release from mitochondria and the subsequent
activation of caspase 3 and 9 (15,16).
Further notable evidence indicated that ASK1 serves
a potential role in the pathogenesis of ischemic brain injury
(17–19). Using a cerebral ischemia rat model,
as well as in an in vitro kinase assay, ASK1 exhibited
increased auto-phosphorylation and activity at various time points
following the induction of cerebral ischemia (20). Heat shock protein-27 (Hsp27) was
observed to be upregulated in cells surviving ischemic insults and
in ischemic preconditioning models (21). Additionally, Hsp27 promoted
long-term neuroprotection against cerebral ischemia by physically
interacting with ASK1 resulting in the inhibition of ASK1 activity
(22). Genetic knockdown of ASK1
or inhibition of the ASK1/MAPK kinase (MKK)4 cascade also
effectively abolished neuronal ischemia (22). Therefore, inhibition of the
pro-apoptotic ASK1 pathway may be a promising novel neuroprotective
strategy for cerebral injury.
However, the expression and distribution of ASK1 in
the brain of perinatal HI rat models remain to be elucidated. In
the present study, the 7-day-old rat was used to build the HI
model. At several time points following the insult, ASK1 expression
and the distribution were determined by western blotting and double
immunofluorescence, respectively, and indicated that ASK1 was
observed and localized within neurons and astrocytes. It was also
demonstrated that the ASK1/JNK pathway was involved in the brain
damage following HI in neonatal rats. Notably, NQDI-1, a specific
inhibitor of ASK1 was intracerebroventricularly injected following
insult of the neonatal rat brain and was demonstrated to
significantly attenuate acute hypoxic-ischemic cerebral injury by
inhibiting cell apoptosis.
Materials and methods
HI rat model and treatments
All animal procedures were approved by the Sun
Yat-Sen University Committee on Animal Use and Care. A total of 12
female Sprague-Dawley rats with litters of mixed gender pups were
acquired from the Nanjing University (Nanjing, China). The mothers
were housed at 25°C under a 12-h light/dark cycle, with ad
libitum access to food and water, until the pups were
7-days-old. The HI model was established, as described previously
(23,24). Briefly, each pup was anesthetized
with diethyl ether (100 mg/kg; Sigma-Aldrich, St. Louis, MO, USA)
during the entire procedure and the body was maintained at 37°C
using a homoisothermy bench. Following a 0.5 cm skin incision in
the midline of the neck, the right common carotid artery (CCA) was
permanently ligated with 5-0 silk. Following ligation of the CCA,
the pups were returned to their housing for 0.5 h to recover from
anesthesia. The pups were subsequently maintained in a hypoxic
chamber at 37°C, 8% O2 and 92% N2 for 6, 12,
24 or 48 h. The sham group underwent a neck dissection and the silk
was placed around the CCA, but was not ligated. The pups were
anesthetized with 2.5% halothane and were intracerebroventricularly
infused with dimethyl sulfoxide (DMSO; Sigma-Aldrich) or 250 nmol
NQDI-1 (Cayman Chemical Company, Inc., Ann. Arbor, MI, USA), a
highly specific ASK1 inhibitor, dissolved in DMSO into the right
cerebral hemisphere 30 min prior to HI using a 30-gauge needle with
a 5 μl Hamilton syringe (infusion rate, 1 μl/min).
The NQDN-1 dose (250 nmol/pup) used in the present study was
selected, according to a previous report (25).
Immunofluorescence
At 6, 12, 24 or 48 h following HI, the cortices of
the pups were harvested and cut into 5-mm sections for further
analyses. Immunofluorescence was performed using the 24-h sections
only. Sections were embedded with paraffin and incubated with
rabbit anti-ASK1 polyclonal antibody (1:200; cat. no. ab131506;
Abcam, Cambridge, UK) or rabbit anti-p-JNK monoclonal antibody
(Thr183/Tyr185; 1:200; cat. no. 9255; Cell Signaling Technology,
Inc., Beverly, MA, USA), and mouse anti-NeuN (1:150; cat. no.
MAB377; EMD Millipore, Billerica, MA, USA) or mouse anti-GFAP
(1:80; cat. no. MAB360; EMD Millipore) monoclonal antibodies.
Subsequently, the sections were incubated with fluorescein
isothiocyanate-conjugated goat anti-rabbit (1:120; cat. no.
sc-2012; Santa Cruz Biotechnology, Inc.) or goat anti-mouse (1:120;
cat. no. sc-2010; Santa Cruz Biotechnology, Inc.) immunoglobulin G
(IgG), and tetramethylrhodamine-conjugated goat anti-rabbit (1:120;
cat. no. sc-2780; Santa Cruz Biotechnology, Inc.) or goat
anti-mouse (1:120; cat. no. sc-2781; Santa Cruz Biotechnology,
Inc.) IgG. The nucleus was stained with
4′,6-diamidino-2-phenylindole (DAPI; Beyotime Institute of
Biotechnology, Inc., Dalian, China; 1:100). A total of 5 sections
per rat were analyzed under a microscope (DTX500; Nikon
Corporation, Tokyo, Japan).
Western blot analysis
Isolated cortices and hippocampi from the right
hemisphere were dissected (n=4/group). Cortices were homogenized in
ice-cold lysis buffer (Beyotime Institute of Biotechnology, Inc.)
containing several protease inhibitors, including
phenylmethanesulfonyl fluoride (0.5 mmol/l; Sigma-Aldrich),
aprotinin (5 μg/ml; Sigma-Aldrich) and leupeptin (5
μg/ml; Sigma-Aldrich), and a phosphokinase inhibitor (10
μg/ml; Sigma-Aldrich). The lysates were centrifuged at
13,000 × g for 30 min at 4°C. Protein concentrations were
determined using a bicinchoninic acid protein assay kit (Pierce
Biotechnology, Inc., Rockford, IL, USA) with bovine serum albumin
as the standard. The protein samples (30 μg/lane) were
separated on 8–12% sodium dodecyl sulfate-polyacrylamide gels. The
proteins were subsequently transferred onto polyvinylidene fluoride
membranes. The membranes were blocked with 5% non-fat dry milk in
Tris-buffered saline containing 0.05% Tween-20 at room temperature
for 1 h with rotation. The membranes were incubated overnight at
4°C with rabbit anti-JNK monoclonal antibody (1:1,000; Cell
Signaling Technologies, Inc.), rabbit anti-p-JNK (Thr183/Tyr185)
monoclonal antibody (1:1,000), rabbit anti-ASK1 polyclonal antibody
(1:800), rabbit anti-c-Jun polyclonal antibody (1:800; cat. no.
9165; Cell Signaling Technologies, Inc.), rabbit anti-p-c-Jun
polyclonal antibody (1:800; cat. no. 3270; Cell Signaling
Technologies, Inc.), rabbit anti-p53 polyclonal antibody (1:200;
cat. no. ab131442; Abcam) and mouse anti-caspase 3 polyclonal
antibody (1:100; cat. no. ab3623; Abcam). A mouse anti-GAPDH
polyclonal antibody (1:2,000; cat. no. sc-365062; Santa Cruz
Biotechnologies, Inc., Santa Cruz, CA, USA) was used as an internal
loading control. The membranes were subsequently incubated with
peroxidase-conjugated goat anti-rabbit and anti-mouse
immunoglobulin G (1:10,000; cat. nos. ZB2301 and ZB2305,
respectively; ZSBIO, Beijing, China) in blocking solution for 1 h
at 37°C. The signals of the bound antibodies were visualized by
enhanced chemiluminescence (EMD Millipore). Image-Pro Plus (version
6.0; Media Cybernetics, Inc., Rockville, MD, USA) was used to
quantify the densities of the protein signals on X-ray films
following scanning. The protein levels were normalized against the
protein levels of the loading control, GAPDH.
2,3,5-triphenyltetrazolium chloride (TTC)
staining
As described previously (26), 48 h after HI induction, TTC
staining was performed to measure the infarct volume (n=4). The
animals were perfused with cold saline under deep anesthesia and
the brains were quickly removed. The brains were subsequently
embedded in brain matrix and frozen for 3 min at -80°C. Each brain
was sliced coronally at 2 mm intervals with the matrix. A total of
4 sliced sections were subsequently stained with 1% TTC (w/v) at
37°C for 12 min and were subsequently fixed in 4% (w/v)
formaldehyde in PBS for 24 h at 4°C. Finally, the brain slice
images were captured under a microscope (DTX500) and the areas of
unstained tissue (the infarct areas) were delineated manually using
Image Pro Plus (version 6.0) software by an individual in a blinded
manner.
Terminal deoxynucleotidyl transferase
deoxyuridine triphosphate nick-end labeling (TUNEL) assay
TUNEL assays were performed to assess the apoptosis
of cells in the rat brain cortex using the DeadEnd™ Fluorometric
TUNEL system (Promega Corporation, Madison, WI, USA), according to
the manufacturer's protocol. Cell nuclei with dark green
fluorescent staining, which were defined as TUNEL-positive nuclei,
were visualized using a fluorescence microscope (DTX500). The
number of TUNEL-positive cells were counted in six randomly
selected fields. The cell nuclei were then counter-stained with
4′,6-diamidino-2-phenylindole, visualized using a fluorescence
microscope (DTX500) and counted.
Statistical analysis
Statistical analysis was performed using SPSS
version 15.0 (SPSS, Inc., Chicago, IL, USA). Numerical continuous
data were presented as the mean ± standard deviation and were
analyzed using one-way analysis of variance. P<0.05 was
considered to indicate a statistically significant difference.
Results
HI induces the expression and
distribution of ASK1
The expression levels of ASK1 were determined by
western blotting in the rat HI model. A significant increase in the
expression of ASK1 was observed in each hemisphere 6 h after
insult, followed by a >4-fold increase in the protein expression
of ASK1 in the brain of HI model compared with the sham group at 6,
12 and 24 h (Fig. 1A and B). The
expression of ASK1 at 48 h after insult was increased 2.86-fold
compared with the sham group (Fig. 1A
and B). To further determine the distribution of ASK1 in the
rat cortex following HI, double immunofluorescence was performed
using paraffin-embedded sections from sham controls, as well as
from control rats at 24 h after HI (n=5/group). The
neuronal-specific marker, NeuN, and the astrocyte-specific marker,
glial fibrillary acidic protein (GFAP), was used to indicate the
neurons. As shown in Fig. 1C,
increased expression of ASK1 was observed and ASK1 was localized in
the astrocytes at 24 h after insult, compared with the sham group.
NeuN and ASK1 double immunofluorescence indicated increased
expression of ASK1 and that it was localized to the neurons at 24 h
after insult, compared with sham group (Fig. 1D). These results indicated that
ASK1 was localized to the astrocytes and neurons following HI in
developing brain.
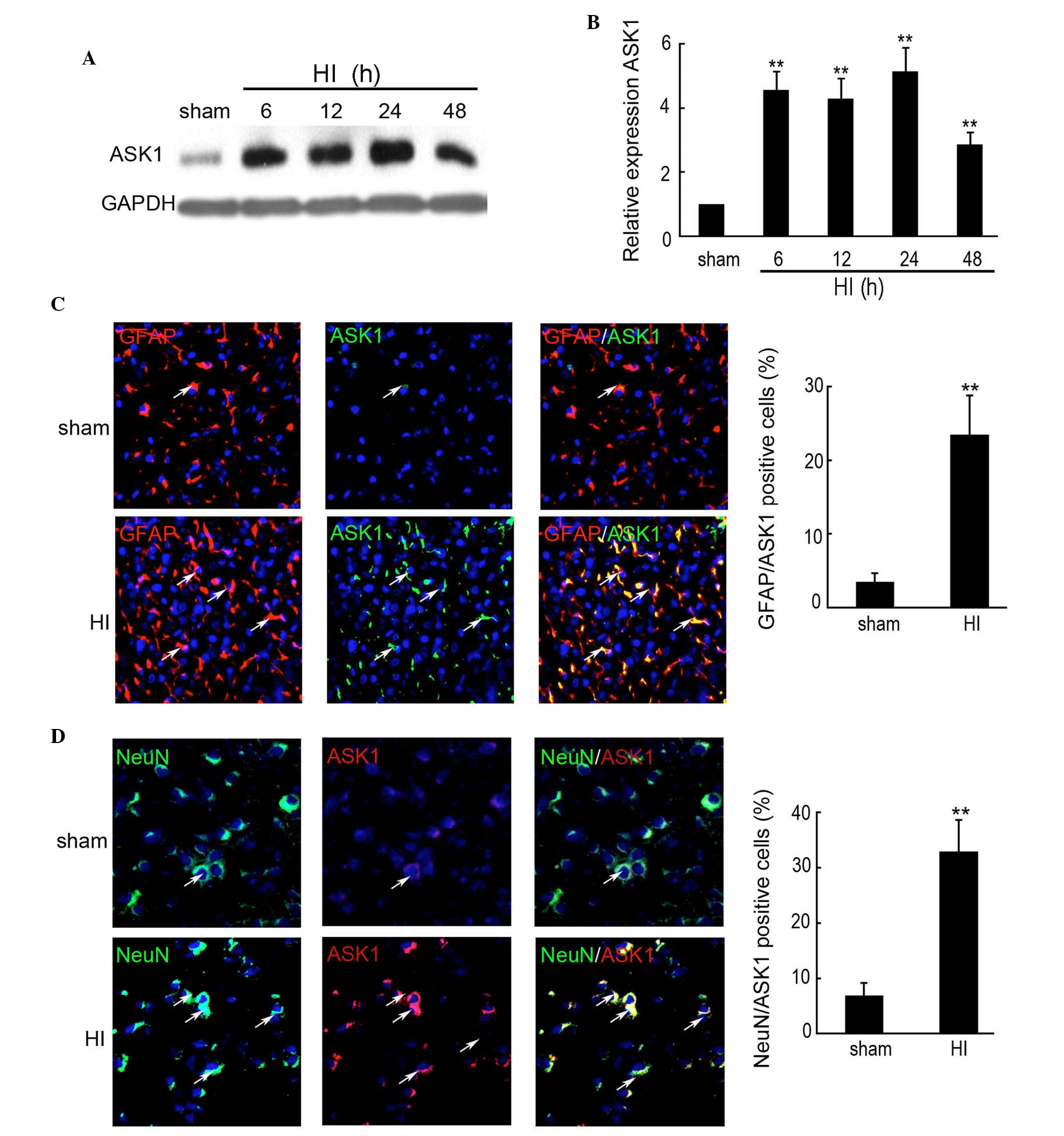 | Figure 1Expression of ASK1 in the brain cortex
of the rat HI model was determined by western blotting and
immunofluorescence. The brain cortex of the rat was perfused and
collected at 6, 12, 24 and 48 h after brain insult. No brain insult
was used as a sham control. (A and B) Western blotting detection of
the protein expression of ASK1 in the sham and HI model rats at 6,
12, 24 and 48 h after brain insult (n=4; **P<0.01).
GAPDH was used as a loading control. (C) Double immunofluorescence
with GFAP (red) and ASK1 (green) was used in paraffin-embedded
sections from sham controls, as well as from control rats at 24 h
after HI (n=6/group; magnification, ×200). DAPI was used to
indicate the cell nucleus. (**P<0.01). (D) Double
immunofluorescence of NeuN (green) and ASK1 (red) was used in
paraffin-embedded sections from sham controls, as well as from
control rats at 24 h after HI (n=6/group). DAPI was used to
indicate the cell nucleus. (**P<0.01; magnification,
×200). ASK, apoptosis signal-regulating kinase 1; HI,
hypoxia-ischemia; DAPI, 4′,6-diamidino-2-phenylindole; GFAP, glial
fibrillary acidic protein. |
HI increases the expression levels of
p-JNK, p53, p-c-Jun and caspase 3
A previous study indicated that p-JNK is the direct
target of ASK1 (27). Therefore,
western blotting and immunofluorescence were used to determine the
expression of p-JNK in the rat cortex following HI at different
time points. As shown in Fig. 2A and
B, the expression of p-JNK was significantly increased between
6 and 48 h after insult, compared with the sham controls.
Additionally, no significant change in the expression of JNK was
observed at different time points following insult. Furthermore,
immunofluorescence also indicated an increased expression of ASK1
in the rat cortex following HI at 6, 12, 24 and 48 h (Fig. 2C). Western blotting was used to
determine the expression of ASK1-associated downstream targets. As
shown in Fig. 2D and E, the
expression levels of p-c-Jun, p53 and caspase 3 were significantly
upregulated between 6 and 48 h after insult, compared with the sham
controls.
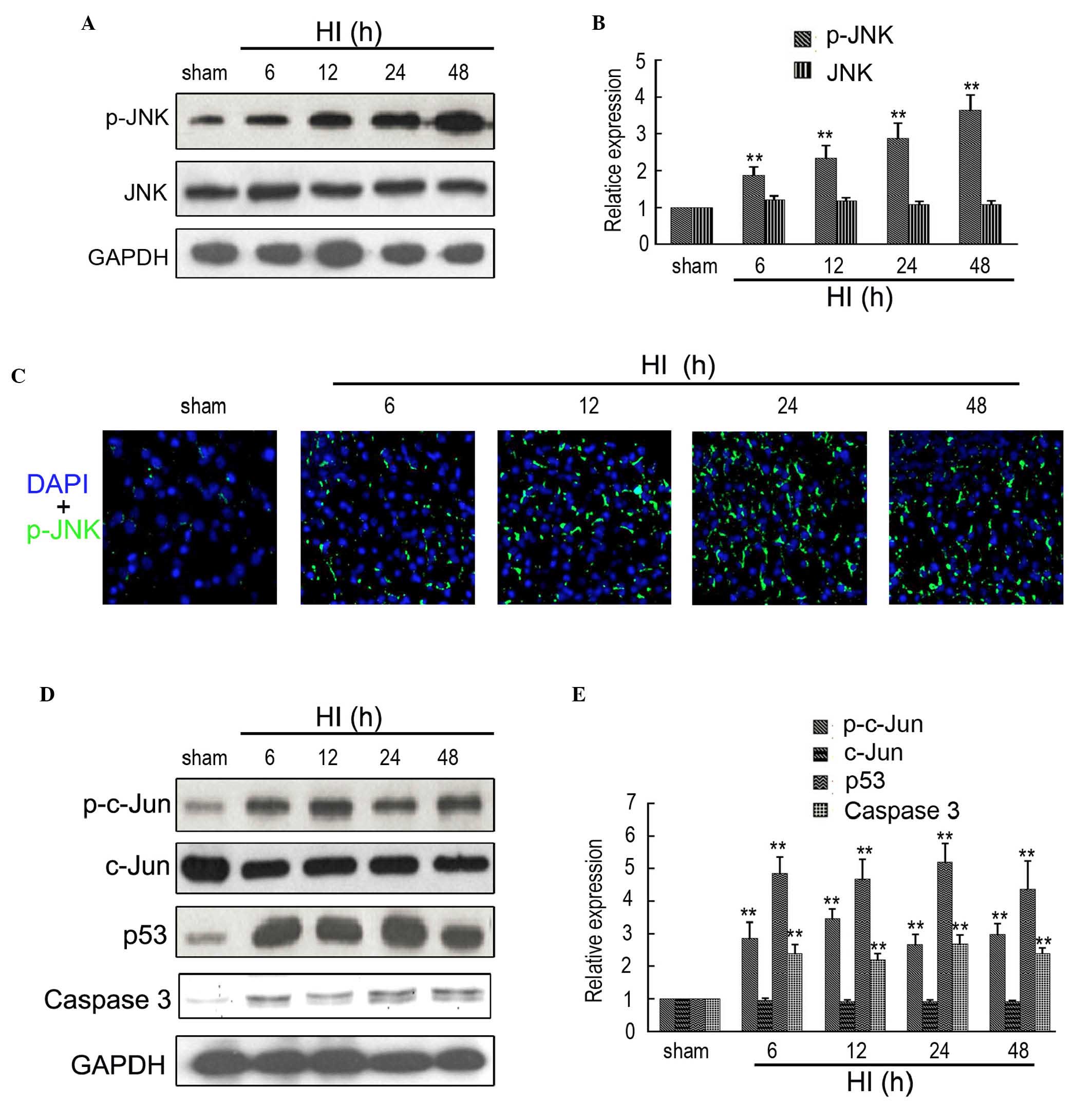 | Figure 2Expression of ASK1-associated
downstream targets in the brain of the rat HI model. (A and B)
Western blotting detection of p-JNK and JNK expression in the sham
and HI model at 6, 12, 24 and 48 h after brain insult. GAPDH was
used as a loading control (n=4; **P<0.01). (C)
Immunofluorescence detection of p-JNK in the brain of the sham and
HI model at 6, 12, 24 and 48 h after brain insult. DAPI was used to
indicate the cell nucleus (magnification, ×200). (D and E) Western
blotting detection of p-c-Jun, c-Jun, p53 and Caspase 3 in the sham
and HI model at 6, 12, 24 and 48 h after brain insult. GAPDH was
used as a loading control (n=4; **P<0.01). ASK,
apoptosis signal-regulating kinase 1; HI, hypoxia-ischemia; DAPI,
4′,6-diamidino-2-phenylindole; GFAP, glial fibrillary acidic
protein. |
NQDI-1 inhibits the expression of ASK1
and downstream targets in the HI model
NQDI-1 has been approved as a inhibitor of ASK1 and
is widely used in vitro and in vivo. In the present
study, 250 nmol NQDI-1 in DMSO was intracerebroventricularly
injected following brain insult. Western blotting was performed to
determine the expression of ASK1 in the sham, HI, DMSO and NQDI-1
groups and indicated that NQDI-1 markedly inhibited the expression
of ASK1 in the brain cortex, compared with the HI and DMSO group
(Fig. 3A and B). Furthermore,
immunofluorescence staining also indicated that the expression of
ASK1 was inhibited by NQDI-1 in the brain cortex (Fig. 3C). The expression of downstream
targets of ASK1 were also determined in the present study. As shown
in Fig. 3D and E, the expression
levels of p-JNK, p-c-Jun, p53 and caspase 3 were significantly
decreased by NQDI-1, compared with the HI and DMSO groups. Low
expression of p-JNK in the brain cortex was also observed by
immunofluorescence in the NQDI-1-treated group (Fig. 3F).
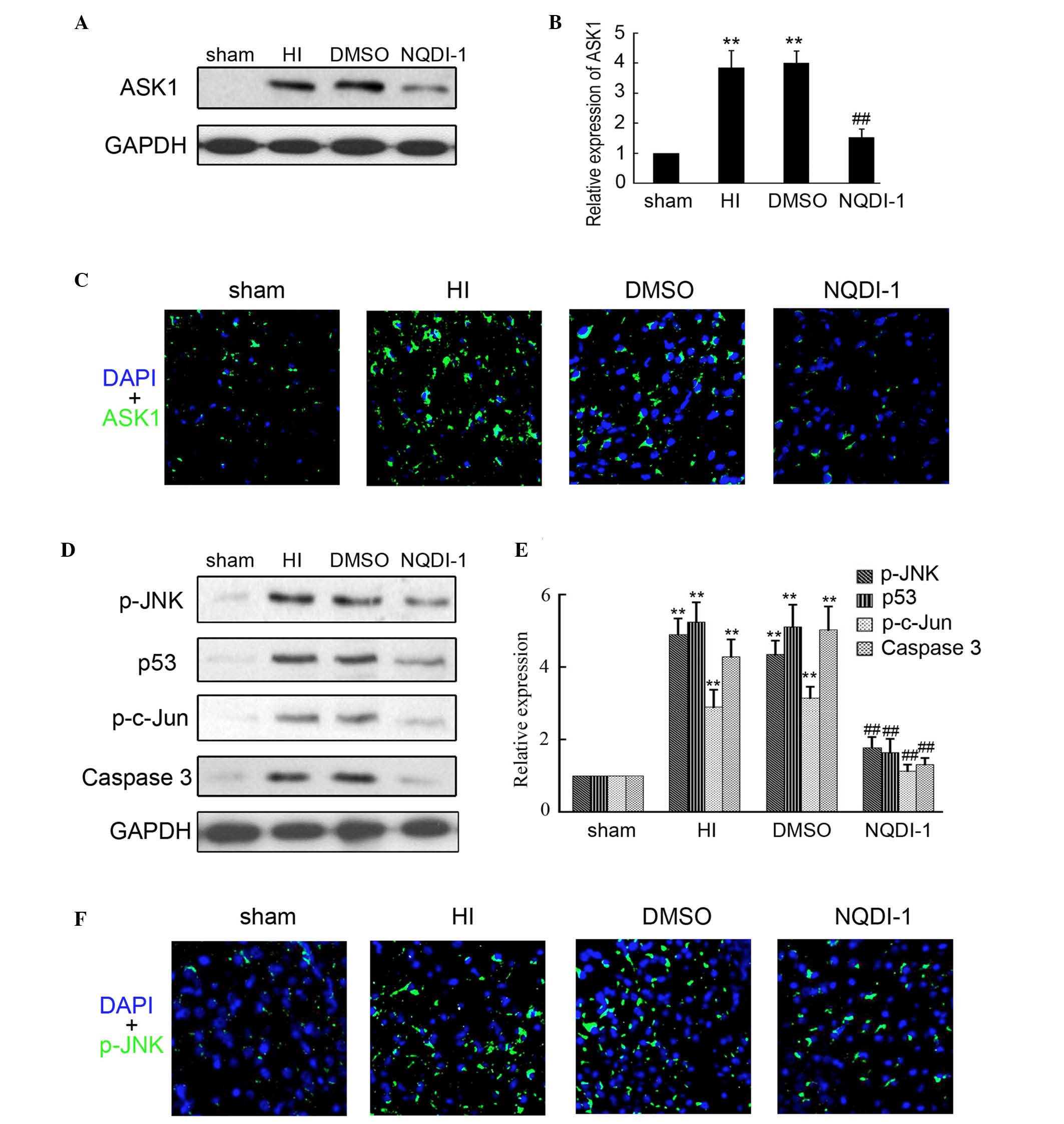 | Figure 3Expression of ASK1 and downstream
targets following NQDI-1 treatment in vivo. After 250 nmol
NQDI-1 treatment for 48 h, the brain cortex was perfused and
collected. (A and B) Western blotting detection of ASK1 expression
in the sham, HI, DMSO- and NQDI-1-treated brain cortex (n=4;
**P<0.01 compared with the sham controls;
##P<0.01 compared with the HI group). (C)
Immunofluorescence detection of ASK1 in the sham, HI, DMSO- and
NQDI-1-treated brain cortex (n=3; magnification, ×200). DAPI was
used to indicate the cell nucleus. (D and E) Western blotting
detection of p-JNK, p-c-Jun, p53 and Caspase 3 expression in sham,
HI, DMSO- and NQDI-1-treated brain cortex (n=4;
**P<0.01 compared with the sham controls;
##P<0.01 compared with the HI group). (F)
Immunofluorescence detection of p-JNK in sham, HI, DMSO-and
NQDI-1-treated brain cortex (n=3; magnification, ×200). DAPI was
used to indicate the cell nucleus. p-, phosphorylated; ASK,
apoptosis signal-regulating kinase 1; HI, hypoxia-ischemia; DMSO,
dimethyl sulfoxide; DAPI, 4′,6-diamidino-2-phenylindole; JNK, c-Jun
N-terminal kinase. |
NODI-1 inhibits injury and apoptosis in
the brain cortex
TTC staining was used to evaluate the brain infract
volume in the different groups. As shown in Fig. 4A, injection of NQDI-1 markedly
decreased the infract volume of the brain at 48 h after HI. It is
known that apoptosis is the major cause of brain injury following
HI. Additionally, ASK1, as an inductor of apoptosis, has been
widely researched in tumors. Therefore, in the present study, the
apoptosis was determined by TUNEL. An increased number of apoptotic
cells were observed in the brain cortex of the HI and DMSO-treated
rats, compared with the sham controls (Fig. 4B). Notably, the apoptosis of neuron
cells in the brain cortex was significantly inhibited following
intracerebroventricularly injection of NQDI-1, compared with the HI
and DMSO-treated groups. These results indicated that NQDI-1
markedly inhibited apoptosis in the brain cortex.
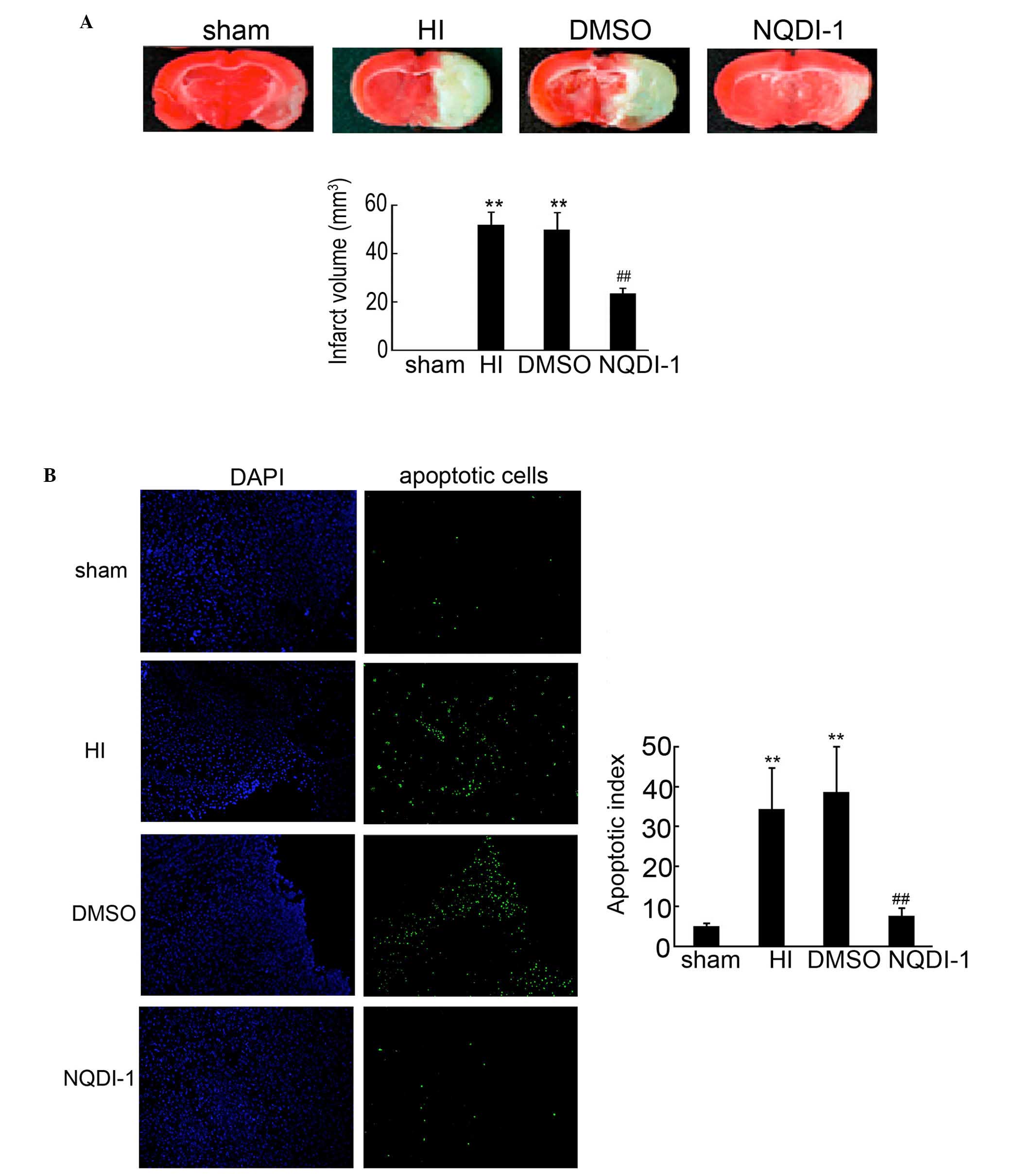 | Figure 4Apoptosis detection by TUNEL in the
brain cortex. (A) Infarct volume in the neonatal brain induced by
HI was assessed by TTC staining (n=4; **P<0.01
compared with the sham controls; ##P<0.01 compared
with the HI group). (B) Paraffin-embedded sections from the sham,
HI, DMSO- and NQDI-1-treated brain cortex was used for apoptosis
detection by TUNEL. DAPI was used to indicate the cell nucleus.
Apoptotic cells in each frame were counted and used for apoptosis
index analysis (n=5; **P<0.01 compared with the sham
controls; ##P<0.01 compared with the HI group;
magnification, ×100). TUNEL, terminal deoxynucleotidyl transferase
dUTP nick end labeling; TTC; 2,3,5-triphenyltetrazolium chloride;
HI, hypoxia-ischemia; DMSO, dimethyl sulfoxide; DAPI,
4′,6-diamidino-2-phenylindole. |
Discussion
Numerous previous studies have shown that ASK1
serves a potential role in the pathogenesis of ischemic brain
injury (17–19). However, the expression and
distribution of ASK1 in the brain of a perinatal HI rat model
remains to be elucidated. In the present study, it was shown for
the first time, to the best of our knowledge, that the ASK1/JNK
pathway is involved in neuronal apoptosis in developing rat brain
following HI. Additionally, the present study indicated that
intracerebroventricularly injection of NQDI-1, a specific inhibitor
of ASK1, after neonatal rats brain HI significantly attenuated
acute HI cerebral injury by inhibiting cell apoptosis.
ASK1 is a ubiquitously expressed kinase protein,
which regulates cell fate in numerous injury conditions and disease
models (17,28,29).
In the ischemic perfused heart, the activation of ASK1 was observed
to have a similar time course compared with that of MKK6 (28). In ischemic brain injury, slight
upregulation of ASK1 by LPS promoted the differentiation of
endogenous neuronal stem cells into the neurons, however, highly
increased ASK1 levels following cerebral ischemic damage led to
high levels of cell death (17).
The expression and distribution of ASK1 following HI in the brain
of the perinatal HI rat model remains to be elucidated. To
determine this, 7-day-old rat pups were insulted to establish the
HI model, and, at 6, 12, 24, 48 and 72 h after insult, ASK1
expression and distribution was determined by western blotting and
double immunofluorescence. These results indicated that ASK1 was
significantly increased at 6 h after brain insult. It was also
demonstrated that ASK1 localized with neurons and astrocytes. These
results clarified the expression and distribution of ASK1 in the
brain of perinatal HI rat models.
It was previously reported that ASK1 is a
serine/threonine protein kinase, which can activate both the
SEK1-JNK and MKK3/6-p38 MAPK pathways and constitute a pivotal
signaling pathway in cytokine- and stress-induced apoptosis
(27). Additionally, JNK
activation was observed in several injury diseases, accompanied
with changes in the expression of downstream targets, including
p-c-Jun, p53, Bim and caspase 3 upregulation and B-cell lymphoma
(Bcl)-2 downregulation (30,31).
In the present study, the expression of p-JNK was determined by
western blotting and immunofluorescence. This showed that the
expression levels of p-JNK and downstream targets were increased in
developing rat brain following HI. These data indicated that the
ASK1/JNK pathway is involved in HI-induced cell apoptosis in the
perinatal rat brain.
ASK1 is widely used as a therapeutic target in
injury diseases. Following ischemia/reperfusion (I/R) in mice,
treatment with ASK1-small interfering RNA significantly attenuates
the upregulation of ASK1, which was followed by the reduction of
infarction in the ischemic brain (18). Administration of NQDI-1 attenuated
renal dysfunction and histological changes characteristic of renal
I/R injury (IRI), accompanied by the upregulation of superoxide
dismutase and Bcl-2 in the kidney (25). Based on the upregulation of ASK1 in
the perinatal rat brain following HI, NQDI-1 was
intracerebroventricularly injected to determine whether inhibition
of ASK1 prevents apoptotic neuronal cell death and brain damage
following HI in the perinatal rat. The results indicated that
injection of NQDI-1 markedly inhibited the expression of ASK1
following HI in the perinatal rat, accompanied by a decrease of
p-JNK, p-c-Jun, p53 and caspase 3, which were the downstream
targets of ASK1. TTC staining and the TUNEL assay demonstrated that
inhibition of ASK1 by NQDI-1 markedly suppressed infract volume of
the brain, according to apoptosis inhibition.
In conclusion, the present study showed for the
first time, to the best of our knowledge, that the ASK1/JNK pathway
is involved in neuronal apoptosis in developing rat brain following
HI. Additionally, the present study indicated that
intracerebroventricularly injection of NQDI-1, a specific inhibitor
of ASK1, following neonatal rats brain HI significantly attenuated
acute HI cerebral injury by inhibiting cell apoptosis.
References
|
1
|
du Plessis AJ and Volpe JJ: Perinatal
brain injury in the preterm and term newborn. Curr Opin Neurol.
15:151–157. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferriero DM: Neonatal brain injury. N Engl
J Med. 351:1985–1995. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Johnston MV, Fatemi A, Wilson MA and
Northington F: Treatment advances in neonatal neuroprotection and
neurointensive care. Lancet Neurol. 10:372–382. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Selway LD: State of the science: Hypoxic
ischemic encephalopathy and hypothermic intervention for neonates.
Adv Neonatal Care. 10:60–66; quiz 67–68. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wachtel EV and Hendricks-Munoz KD: Current
management of the infant who presents with neonatal encephalopathy.
Curr Probl Pediatr Adolesc Health Care. 41:132–153. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bass JL, Corwin M, Gozal D, Moore C,
Nishida H, Parker S, Schonwald A, Wilker RE, Stehle S and Kinane
TB: The effect of chronic or intermittent hypoxia on cognition in
childhood: A review of the evidence. Pediatrics. 114:805–816. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nelson KB and Lynch JK: Stroke in newborn
infants. Lancet Neurol. 3:150–158. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thornton C, Rousset CI, Kichev A, Miyakuni
Y, Vontell R, Baburamani AA, Fleiss B, Gressens P and Hagberg H:
Molecular mechanisms of neonatal brain injury. Neurol Res Int.
2012:5063202012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Adekeye A, Haeri M, Solessio E and Knox
BE: Ablation of the proapoptotic genes CHOP or Ask1 does not
prevent or delay loss of visual function in a P23H transgenic mouse
model of retinitis pigmentosa. PloS One. 9:e838712014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ichijo H, Nishida E, Irie K, ten Dijke P,
Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K and Gotoh
Y: Induction of apoptosis by ASK1, a mammalian MAPKKK that
activates SAPK/JNK and p38 signaling pathways. Science. 275:90–94.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen Z, Seimiya H, Naito M, Mashima T,
Kizaki A, Dan S, Imaizumi M, Ichijo H, Miyazono K and Tsuruo T:
ASK1 mediates apoptotic cell death induced by genotoxic stress.
Oncogene. 18:173–180. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McDonald PH, Chow CW, Miller WE, Laporte
SA, Field ME, Lin FT, Davis RJ and Lefkowitz RJ: Beta-arrestin 2: A
receptor-regulated MAPK scaffold for the activation of JNK3.
Science. 290:1574–1577. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Matsuzawa A, Nishitoh H, Tobiume K, Takeda
K and Ichijo H: Physiological roles of ASK1-mediated signal
transduction in oxidative stress- and endoplasmic reticulum
stress-induced apoptosis: Advanced findings from ASK1 knockout
mice. Antioxid Redox Signal. 4:415–425. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Matsukawa J, Matsuzawa A, Takeda K and
Ichijo H: The ASK1-MAP kinase cascades in mammalian stress
response. J Biochem. 136:261–265. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hatai T, Matsuzawa A, Inoshita S, Mochida
Y, Kuroda T, Sakamaki K, Kuida K, Yonehara S, Ichijo H and Takeda
K: Execution of apoptosis signal-regulating kinase 1 (ASK1)-induced
apoptosis by the mitochondria-dependent caspase activation. J Biol
Chem. 275:26576–26581. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hao W, Takano T, Guillemette J, Papillon
J, Ren G and Cybulsky AV: Induction of apoptosis by the Ste20-like
kinase SLK, a germinal center kinase that activates apoptosis
signal-regulating kinase and p38. J Biol Chem. 281:3075–3084. 2006.
View Article : Google Scholar
|
|
17
|
Song J, Cho KJ, Cheon SY, Kim SH, Park KA,
Lee WT and Lee JE: Apoptosis signal-regulating kinase 1 (ASK1) is
linked to neural stem cell differentiation after ischemic brain
injury. Exp Mol Med. 45:e692013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim HW, Cho KJ, Lee SK and Kim GW:
Apoptosis signal-regulating kinase 1 (Ask1) targeted small
interfering RNA on ischemic neuronal cell death. Brain Res.
1412:73–78. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu DN, Pei DS, Wang Q and Zhang GY:
Down-regulation of PTEN by sodium orthovanadate inhibits ASK1
activation via PI3-K/Akt during cerebral ischemia in rat
hippocampus. Neurosci Lett. 404:98–102. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang Q and Zhang G: Activation and
autophosphorylation of apoptosis signal-regulating kinase 1 (ASK1)
following cerebral ischemia in rat hippocampus. Neurosci Lett.
329:232–236. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Currie RW, Ellison JA, White RF,
Feuerstein GZ, Wang X and Barone FC: Benign focal ischemic
preconditioning induces neuronal Hsp70 and prolonged astrogliosis
with expression of Hsp27. Brain Res. 863:169–181. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Stetler RA, Cao G, Gao Y, Zhang F, Wang S,
Weng Z, Vosler P, Zhang L, Signore A, Graham SH and Chen J: Hsp27
protects against ischemic brain injury via attenuation of a novel
stress-response cascade upstream of mitochondrial cell death
signaling. J Neurosci. 28:13038–13055. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kichev A, Rousset CI, Baburamani AA,
Levison SW, Wood TL, Gressens P, Thornton C and Hagberg H: Tumor
necrosis factor-related apoptosis-inducing ligand (TRAIL) signaling
and cell death in the immature central nervous system after
hypoxia-ischemia and inflammation. J Biol Chem. 289:9430–9439.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li D, Qu Y, Mao M, Zhang X, Li J, Ferriero
D and Mu D: Involvement of the PTEN-AKT-FOXO3a pathway in neuronal
apoptosis in developing rat brain after hypoxia-ischemia. J Cereb
Blood Flow Metab. 29:1903–1913. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
El Eter E: NQDI 1, an inhibitor of ASK1
attenuates acute ischemic renal injury by modulating oxidative
stress and cell death. Cardiovasc Hematol Agents Med Chem.
11:179–186. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tian SF, Yang HH, Xiao DP, Huang YJ, He
GY, Ma HR, Xia F and Shi XC: Mechanisms of neuroprotection from
hypoxia-ischemia (HI) brain injury by up-regulation of cytoglobin
(CYGB) in a neonatal rat model. J Biol Chem. 288:15988–16003. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Matsuzawa A and Ichijo H: Molecular
mechanisms of the decision between life and death: Regulation of
apoptosis by apoptosis signal-regulating kinase 1. J Biochem.
130:1–8. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Harding SJ, Browne GJ, Miller BW, Prigent
SA and Dickens M: Activation of ASK1, downstream MAPKK and MAPK
isoforms during cardiac ischaemia. Biochim Biophys Acta.
1802:733–740. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nako H, Kataoka K, Koibuchi N, Dong YF,
Toyama K, Yamamoto E, Yasuda O, Ichijo H, Ogawa H and Kim-Mitsuyama
S: Novel mechanism of angiotensin II-induced cardiac injury in
hypertensive rats: The critical role of ASK1 and VEGF. Hypertens
Res. 35:194–200. 2012. View Article : Google Scholar
|
|
30
|
Zhu Y, Mao XO, Sun Y, Xia Z and Greenberg
DA: p38 Mitogen-activated protein kinase mediates hypoxic
regulation of Mdm2 and p53 in neurons. J Biol Chem.
277:22909–22914. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu G, Zhao J, Chang Z and Guo G: CaMKII
activates ASK1 to induce apoptosis of spinal astrocytes under
oxygen-glucose deprivation. Cell Mol Neurobiol. 33:543–549. 2013.
View Article : Google Scholar : PubMed/NCBI
|














