Introduction
Hepatocellular carcinoma (HCC) is the most common
type of liver cancer and is the fifth most common type of cancer
worldwide (1). The rate of
HCC-associated mortality is >600,000 each year, and the 5-year
survival rate is <9% (2,3). The
majority of patients with HCC have chronic liver diseases,
including chronic hepatitis infection (4,5). HCV
infection has been the leading cause of chronic hepatitis, liver
cirrhosis and HCC for decades (6).
It is estimated that 200,000,000 individuals are chronically
infected with HCV worldwide (7,8).
Despite progress in understanding immunity against HCV infection
and HCV pathogenesis, and progress in the development of effective
direct-acting antivirals, no effective vaccine has been developed
for HCV, and the current antiviral treatments have certain
limitations.
HCV is a member of the Hepacivirus genus of
the Flaviviridae family, and is represented by seven major
genotypes. HCV contains a 9.6-kb positive strand RNA genome
comprising a 5′-noncoding region (NCR), which includes an internal
ribosome entry site, an open reading frame (ORF) that encodes
structural and non-structural proteins, and a 3′ NCR. The
structural proteins, which form the viral particle, include the
core protein, and the envelope glycoproteins, E1 and E2 (9). Among the HCV viral proteins, the
first structural protein encoded by the HCV ORF is the core
protein, which forms the viral nucleocapsid. The HCV core protein
is likely to be an important determinant in mediating the
pathological effects of HCV through several multifunctional
activities (10). Previous studies
have indicated that the HCV core protein may be a potential
oncoprotein in HCV-associated HCC, however, the HCV core regulatory
mechanisms remain to be fully elucidated (11,12).
Previously, microRNAs, a class of genes transcribing
small, highly conserved, 19–24-nucleotide noncoding RNAs, have
emerged as potent regulators of gene expression by silencing
messenger RNAs (mRNAs), which bind to the 3′-untranslated region
(3′UTR) of target genes (13).
MicroRNAs are critical in a number of normal biological processes,
including the regulation of cellular differentiation, metabolism,
proliferation, apoptosis and cancer (14). The aims of the present study were
to investigate whether microRNAs are involved in HCV core
protein-induced aberrant proliferation. The results may determine
whether HCV core protein-induced upregulation of microRNA-196a is
associated with HCC proliferation and the biological behavior of
HCC, and may assist in elucidating the underlying mechanism.
Materials and methods
Cell culture and transfection
The human HepG2 and Huh-7 HCC cell lines were
obtained from the American Type Culture Collection (Manassas, CA,
USA) and grown in Dulbecco's modified Eagle's medium (DMEM;
Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% foetal bovine serum (FBS) and 100 U/ml
penicillin/streptomycin (both purchased from Invitrogen; Thermo
Fisher Scientific, Inc.) at 37°C and 5% CO2 in a
humidified chamber. miR-196a mimics and control miR mimics were
purchased from Dharmacon (Austin, TX, USA). The cells were
transfected with either the miR mimic (50 nM) or plasmid (200 ng)
using HiPerFect transfection reagent (Qiagen, Valencia, CA, USA),
according to the manufacturer's protocol. The cells were collected
for analyses, which were performed 48 h following transfection.
Ad-HCV core adenovirus construction and
infection
The Ad-HCV core adenovirus and the control
Ad-enhanced green fluorescent protein (EGFP) adenovirus were
constructed using the Ad-easy system (Invitrogen; Thermo Fisher
Scientific, Inc.), as previously described (15). The HCC cells were seeded into a
6-well plate at a density of 3×105 cells per well and
incubated at 37°C overnight until the cells were 50–70% confluent.
The HCC cells were then infected with the Ad-MOCK, Ad-HCV core and
control Ad-EGFP adenovirus, at a multiplicity of infection (MOI) of
50. After 48 h, the infection efficiency was evaluated by observing
the expression of EGFP under a fluorescence microscope (Eclipse
90i; Nikon, Tokyo, Japan). The cells were collected for analyses,
which were performed 48 h following infection.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
The total RNA enriched with microRNAs was isolated
from the HCC cells using a mirVana™ miRNA isolation kit (Applied
Biosystems; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. The expression of miR-196a was quantified
using a Taqman MicroRNA assay (Applied Biosystems; Thermo Fisher
Scientific, Inc.). Briefly, 10 ng of total RNA was used for reverse
transcription to cDNA using specific stem-loop real-time primers
(Shanghai Jade Lee Biotechnology Co., Ltd., Shanghai, China). The
primer sequences were as follows: miR-196a forward,
5′-GTCGTATCCAGTGCAGGGTCCGAGGTA-3′ and reverse,
5′-TTCGCACTGGATACGACAGCAAAAATGTG-3′; U6 forward,
5′-GTCGTATCCAGTGCAGGGTCCGAGGT-3′ and reverse,
5′-ATTCGCACTGGATACGACAAAAATATGG-3′. The reverse transcription
primers for the specific microRNAs were used to amplify the cDNA
from the microRNA. The PCR reaction mixture was composed of 1
μl cDNA, 0.5 μl primer and 5 μl SYBR Green PCR
master mix buffer (total volume, 10 μl). qPCR was performed
with 30 cycles of the following: 30 Sec at 98°C, 90 sec at 58°C and
20 sec at 72°C; with a final extension at 72°C for 5 min using ABI
Prizm 7500 PCR machine and TaqMan Universal PCR Master mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.). All primers were
obtained from the TaqMan miRNA assays (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The relative quantification of miR-196a
was calculated using the 2−ΔΔCq method (16). The data were normalised using U6 as
an internal control, and the data were measured relative to a
calibrator sample as the external control.
Cell proliferation assay
To determine the cell proliferation capacity, the
HCC cells were monitored using cell growth curves. The cells were
plated into 96-well plates (4×103 cells/well), and the
cell viability was documented every day for 5 days using a Cell
Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan). The
absorbance values in each well at 450 and 510 nm were measured
using a Benchmark Microplate Reader (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Cell cycle assay
For cell cycle analysis, the cells were collected by
centrifugation (12,000 × g at 4°C for 5 min) 48 h following
infection or transfection, washed with cold phosphate-buffered
saline (PBS), fixed with 70% cold ethanol, and incubated at −20°C
overnight. The fixed cells were then collected, washed with PBS,
resuspended in Tris-HCl buffer (pH 7.4) supplemented with 100
μg/ml RNase A (Sigma-Aldrich, St. Louis, MO, USA) and
labelled for 30 min with 50 μg/ml propidium iodide
(Sigma-Aldrich). Cell cycle analysis was performed on a flow
cytometer (FACSCanto; BD Biosciences, Franklin Lakes, NJ, USA)
using ModFit LT software version 3.2 (Verity Software House, Inc.,
Topsham, ME, USA).
Western blot analysis
The cells were lysed on ice using M-PER mammalian
protein extraction reagent (Cell Signaling Technology, Inc.,
Boston, USA). Protein concentration was quantified using a Bradford
assay (Bio-Rad Laboratories, Inc.) The protein (30 mg) was
separated by 10% SDS-PAGE and transferred onto a polyvinylidene
fluoride membrane (Amersham Biosciences, Buckinghamshire, UK).
Then, the membranes were blocked with 5% skim milk at room
temperature for 1 h, and washed in 0.05% Tris-buffered saline
(Sigma-Aldrich) with Tween 20 for 5 min three times. The membranes
were probed with primary antibodies against FOXO1 (cat. no. 2880)
and GAPDH (cat. no. 5174) (both dilution, 1:1,000; both rabbit
monoclonal; both purchased from Cell Signaling Technology, Inc.)
overnight at 4°C, followed by incubation with secondary antibodies
at 37°C for 1 h. The bands were detected using enhanced
chemiluminescence (Pierce Biotechnology, Inc., Rockford, IL, USA)
and visualised using the ChemiDoc XRS system (Bio-Rad Laboratories,
Inc.).
Plasmid construction
To construct the luciferase reporter vector, the
full-length 3′UTR of FOXO1, as well as a mutant sequence of FOXO1,
were synthesised using PCR. PCR was performed under the follow
thermal cycling conditions for 30 cycles: 30 Sec at 95°C, 90 sec at
60°C and 30 sec at 72°C; with a final extension at 72°C for 5 min
using a ABI Prizm 7500 PCR machine (Applied Biosystems; Thermo
Fisher Scientific, Inc.) The primers used contained the following
restriction sites: FOXO1 3′UTR, forward
5′-ACTAGTGAAGTGCCAAACTCACTACA-3′ and reverse
5′-AAGCTTTACAAAGCTGGCATTTAATC-3′; and Mut FOXO1 3′UTR, forward
5′-CTTTTGTTTGTAAGAACTGTTTTCTGCGGA-3′ and reverse
5′-TCCGCAGAAAACAGTTCTTACAAACAAAAG-3′. The PCR product from the
FOXO1 3′UTR was cloned into the SpeI and HindIII
restriction sites downstream of the luciferase ORF in the
pMIR-report vector (Ambion, Carlsbad, CA, USA).
For re-expression, FOXO1 without the 3′UTR was
amplified using PCR with the following primers: Forward
5′-ATGGCCGAGGCGCCTCAGGTG-3′ and reverse
5′-TCAGCCTGACACCCAGCTATGT-3′. The resulting PCR amplicons of FOXO1
were cloned into the T vector (Promega Corp., Madison, WI, USA).
The correct clones were confirmed by sequencing.
Luciferase reporter assays
For the luciferase assay, the HCC cells were grown
to 70–80% confluence in 24-well plates and co-transfected with the
firefly luciferase reporter vector containing the FOXO1 3′UTR or
its mutant 3′UTR, and miRNA mimics (50 nM) using Lipofectamine™
2000 reagent (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. A luciferase activity
assay was performed 48 h following co-transfection using a
Dual-Luciferase Reporter Assay System (cat. no. E1980; Promega
Corp.), and the values were normalised with Renilla luciferase
activity.
Statistical analysis
All data were analyzed using SPSS version 17.0 (SPSS
Inc., Chicago, IL, USA). All data are shown as the mean ± standard
deviation. Significant differences were analysed using Student's
t-test for comparisons between two groups, and one-way
analysis of variance or the non-parametric Kruskal-Wallis H test
were used for multiple comparisons. P<0.05 was considered to
indicate a statistically significant difference.
Results
Ad-HCV core infection upregulates the
expression of miR-196a in HCC cells
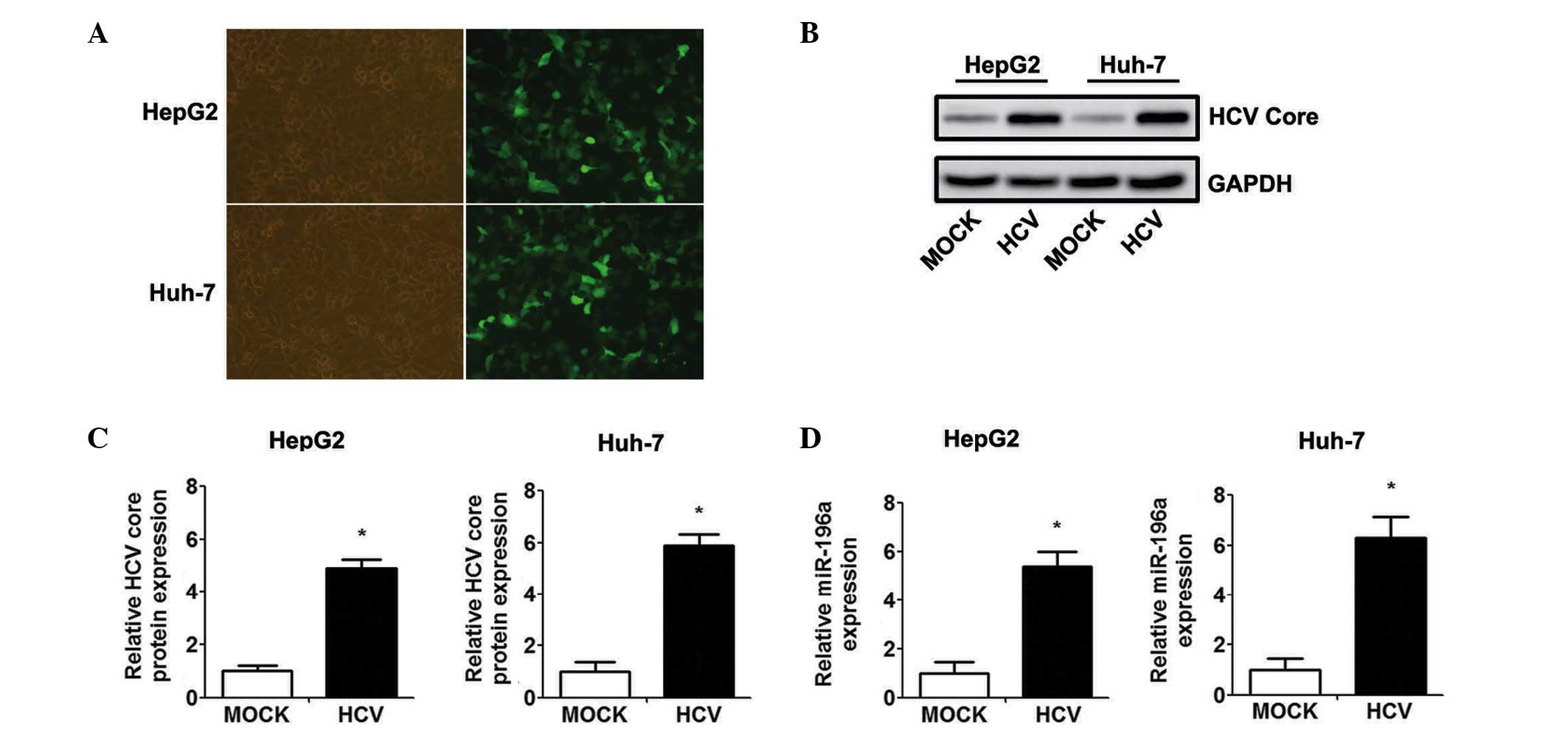
The HepG2 and Huh-7 cells were infected with Ad-HCV
core or Ad-GFP at an MOI of 50, and the infection efficiency was
evaluated by observing the expression of EGFP under a fluorescence
microscope (Fig. 1A). Western blot
analysis confirmed the efficient transient expression of the HCV
core protein at 48 h-post Ad-HCV core infection (Fig. 1B and C). The present study
subsequently examined whether the level of miR-196a is affected by
HCV core overexpression. As shown in Fig. 1D, the level of miR-196a was
significantly higher in the Ad-HCV core-infected cells, compared
with the expression level in the Ad-MOCK control group.
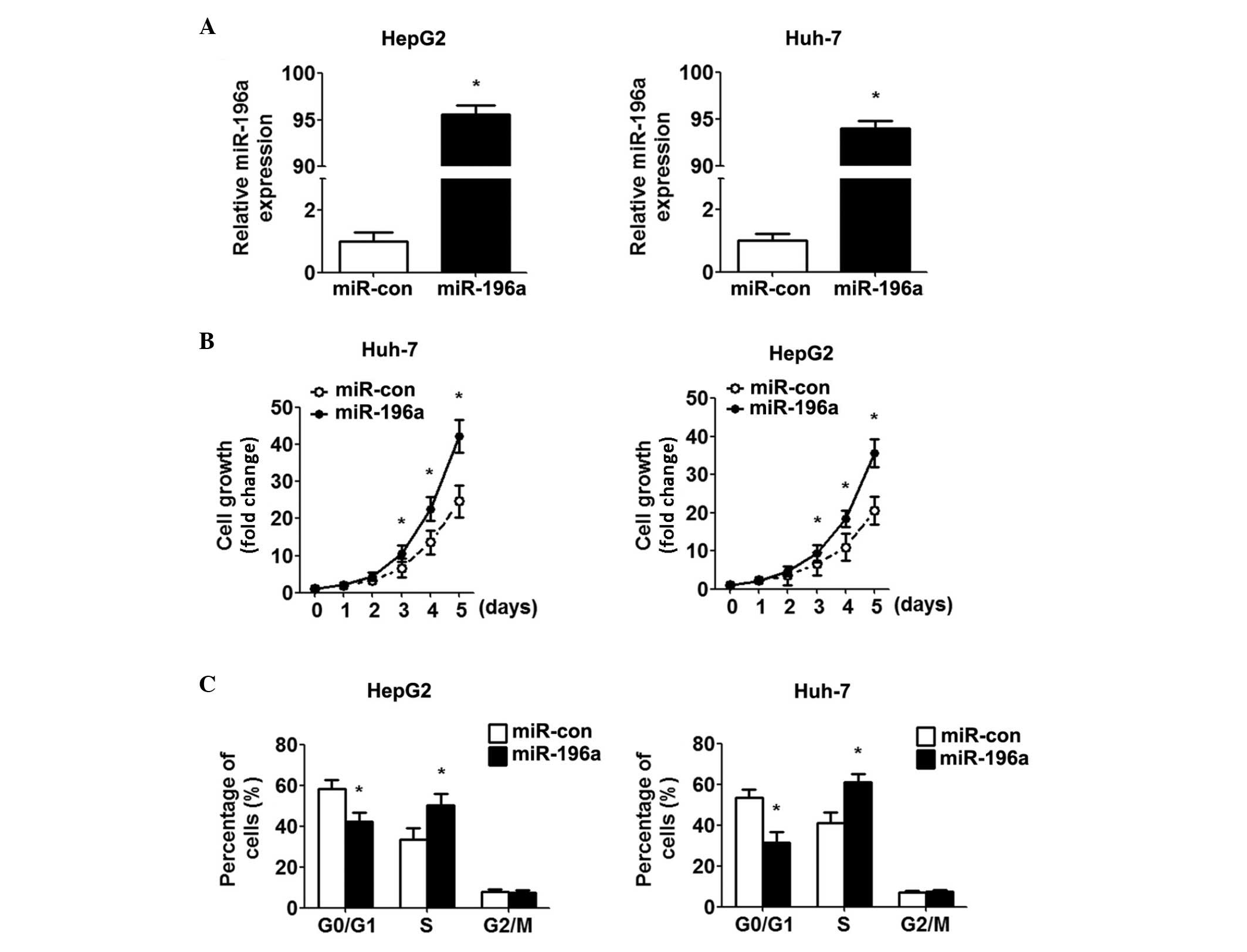
Overexpression of miR-196a promotes cell
growth and the G1-S transition
A previous study demonstrated that Ad-HCV core
infection promotes cell growth and the G1-S transition (15). To confirm the hypothesis that the
HCV core protein affects the proliferation of HCC cells via the
upregulation of miR-196a, miR-196a was overexpressed in the HepG2
and Huh-7 cells by transfection with miR-196a mimics. Increased
expression of miR-196a was confirmed using RT-qPCR (Fig. 2A). Using cell growth curves, it was
found that the miR-196a mimic promoted the growth of the HCC cells,
compared with the control cells (Fig.
2B). In addition, marked promotion of the G1/S transition was
observed by the miR-196a mimics (Fig.
2C). Overall, these data suggested that miR-196a promoted HCC
cell proliferation by inducing the G1-S transition.
FOXO1 is a direct target of miR-196a
As microRNAs regulate the expression of mRNAs by
targeting the 3′UTR of relative mRNAs, the present study detected
the target of miR-196a in HCC cells. A previous study reported that
FOXO1 serves as a target of miR-196a post-transcriptional
repression in cervical cancer (17). To identify whether FOXO1 is
regulated by miR-196a in HCC, HepG2 and Huh-7 cells were
transfected with miR-196a mimics. Western blot analysis showed that
the miR-196a mimics markedly inhibited the protein expression of
FOXO1 in HCC cells (Fig. 3A). To
verify whether FOXO1 is a direct target of miR-196a in HCC cells,
the potential seed sequence for miR-196a in the 3′UTR of FOXO1 was
analysed, and the wild-type and mutant FOXO1 3′UTR fragments were
cloned into a luciferase reporter gene system (Fig. 3B). The wild-type or mutant FOXO1
3′UTR constructs were co-transfected with the miR-196a or control
mimics into the HepG2 and Huh-7 cells, and luciferase activity was
measured. The luciferase reporter assay indicated that transfection
with miR-196a led to inhibition of the wild-type 3′UTR, however,
miR-196a had no effect on the luciferase intensity controlled by
the mutant 3′UTR (Fig. 3C).
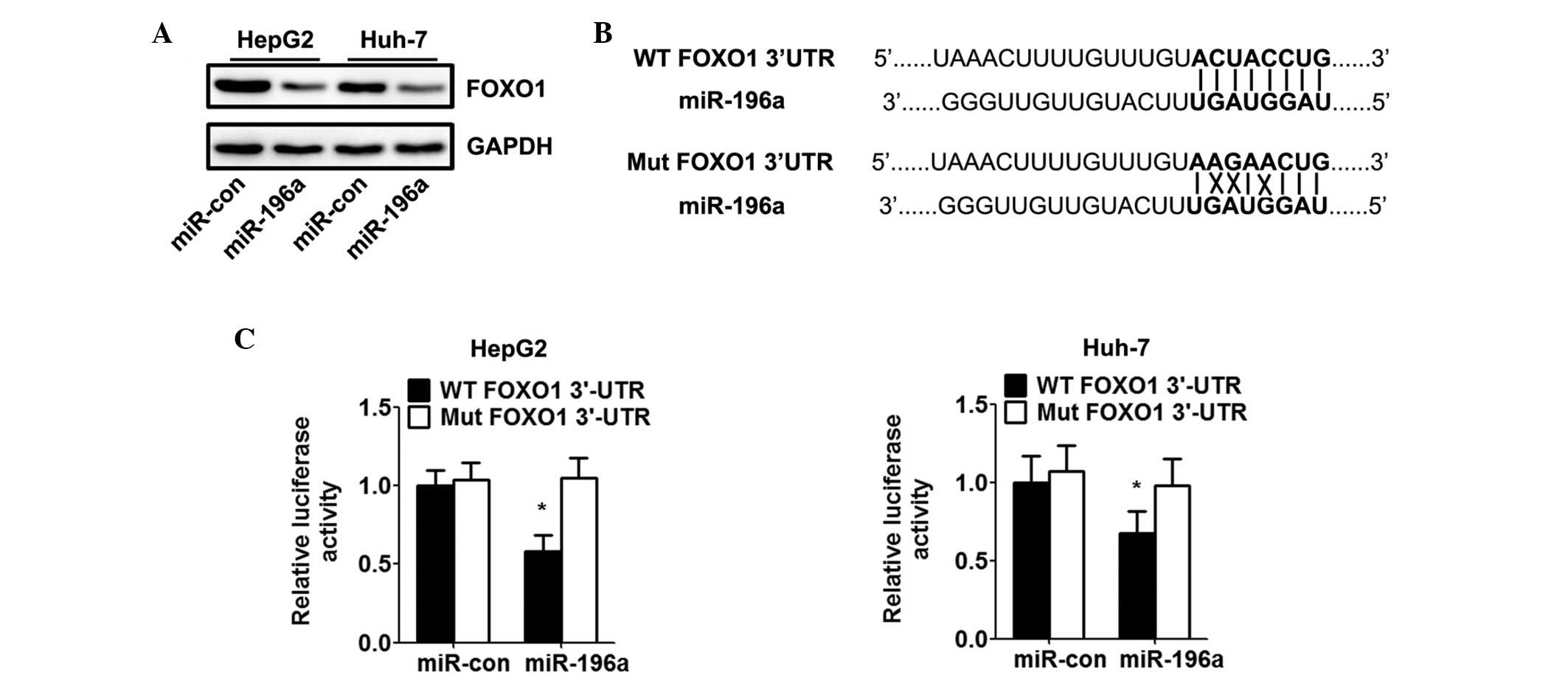 | Figure 3miR-196a directly targets FOXO1 and
inhibits the expression of FOXO1. HepG2 and Huh-7 cells were
transfected with miR-con or miR-196 mimics for 48 h. (A) Western
blot analysis of the effect of miR-196a on protein levels of FOXO1
in hepatocellular carcinoma cells. Relative expression of FOXO1 was
calculated based on densitometric analysis of band intensities. (B)
Full-length 3′-UTR sequences of WT FOXO1 and Mut FOXO1 were cloned
into the pMIR-report vector. Predicted base pairing between
miR-196a and the 3′-UTR sequence of FOXO1 is shown (solid lines
indicate matching base pairs; crosses indicate non-matching base
pairs). (C) Effects of miR-196a on luciferase intensity, controlled
by the WT or Mut 3′UTR of FOXO1, were determined using a luciferase
assay. Data are expressed as the mean ± standard deviation (n=5).
*P<0.05, vs. miR-con group. HCV, hepatitis C virus;
HCC, hepatocellular carcinoma; miR, microRNA; FOXO1, forkhead box
O1; UTR, untranslated region; Mut, mutant; WT, wild-type; con,
control. |
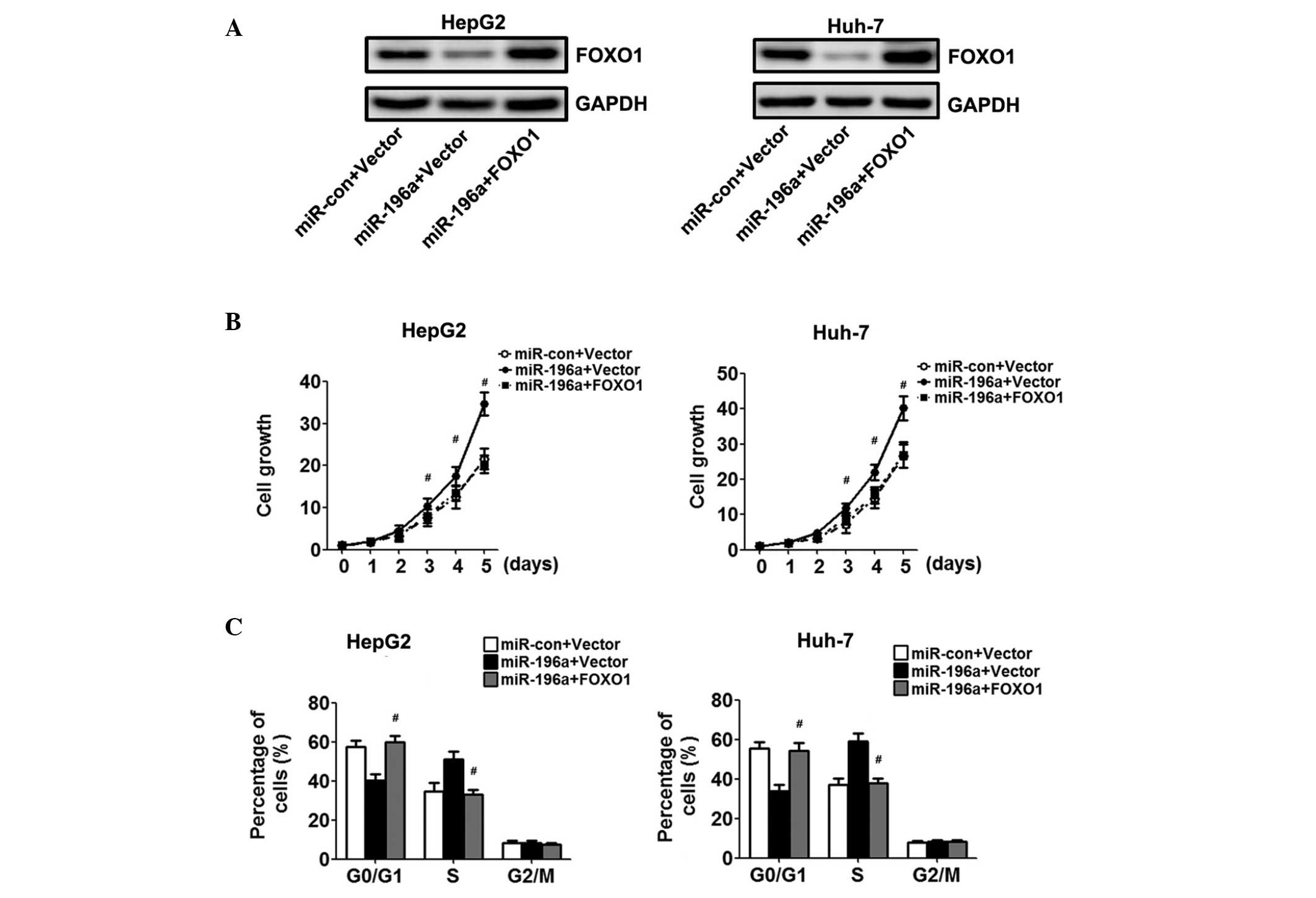
Overexpression of FOXO1 reverses
miR-196a-mediated proliferation
To confirm the hypothesis that miR-196a affects the
proliferation of HCC cells via the downregulation of FOXO1, the
HepG2 and Huh-7 cells overexpressing miR-196a were transfected with
a FOXO1 construct lacking the respective 3′UTR, or an empty vector.
Western blot analysis demonstrated increased protein expression
levels of FOXO1 in the cells co-transfected with the FOXO1
recombinant plasmid, compared with the cells transfected with the
empty vector (Fig. 4A).
Overexpression of FOXO1 resulted in growth inhibition of the HCC
cells, as assessed using the CCK-8 assay (Fig. 4B). Cell cycle assays showed that
the overexpression of FOXO1 restored the percentage of cells in the
G0/G1 phase and reversed the enhanced
proliferation effect of miR-196a (Fig.
4C).
Discussion
HCV-associated infection, which is present
worldwide, increasingly contributes to health care expenditures,
morbidity rates and mortality rates (18). Although substantial progress has
been made in previous years in determining the structure and
function of HCV proteins, the HCV viral load does not necessarily
correlate with the severity and progression of the disease.
Therefore, the investigation of a novel regulatory mechanism for
the diagnosis and treatment of HCV-associated infections is
required. The HCV core protein is likely to be an important
determinant in mediating the pathological effects of HCV (11,12).
There is accumulating evidence that the HCV core protein can exert
a regulatory function through the expression of microNAs (19).
miR-196a has been shown to be crucial in normal cell
differentiation and proliferation, and in the tumorigenesis of
various types of cancer (20–23).
The upregulation of miR-196a correlates with aggressive progression
and unfavourable prognoses in colorectal cancer, gastric cancer,
non-small cell lung cancer, osteosarcoma and other types of cancer
(24–27). A previous report suggested that
circulating miR-196a is a specific and non-invasive candidate
biomarker for the diagnosis of chronic HCV infection (28). A subsequent study demonstrated that
HCV infection markedly enhances the expression of miR-196a in
hepatocytes, and that HCV replication is required for the
upregulation of miR-196a, which suggests that miR-196a is critical
in HCV-associated infection (29).
The data obtained in the present study indicated that ectopic
expression of the HCV core protein enhanced the expression of
miR-196a. The overexpression of miR-196a significantly inhibited
HCC cell growth due to S phase accumulation. These results were
consistent with previous studies demonstrating that miR-196a may
have oncogenic role in tumourigenesis and tumour progression.
As for the mechanism of miRNA regulation in cancer,
it is crucial to validate target genes. FOXO1 has been identified
as one of the high-scoring candidate genes for miR-196a targets by
several in silico methods for target gene prediction,
including PicTar, TargetScan and microRNA databases (30). A previous study provided direct
evidence that FOXO1 is a direct target of miR-196a in cervical
cancer cells (16). FOXO1 belongs
to the FOXO subfamily of forkhead transcription factors, which
contain evolutionarily conserved transcriptional activators that
are characterised by a highly conserved forkhead domain with a
DNA-binding motif (31). A
previous study provided direct evidence that FOXO1 is a potent
transcriptional activator, which triggers the expression of a
program of genes involved in cell cycle arrest, apoptosis, DNA
repair and hypoxia responsiveness (32–34).
FOXO1 is also known to be a tumour suppressor, and the upregulation
of FOXO1 induces cell cycle arrest in HCC (35,36).
In the present study, through the prediction of molecular
information, FOXO1 was confirmed as a direct target gene for
miR-196a, and it was shown that miR-196a negatively regulated the
expression of FOXO1 expression by directly targeting the 3′UTR of
FOXO1 mRNA in the cells. Additionally, the present study
demonstrated that the inhibitory effect of miR-196a on cell growth
was reversed by overexpressing FOXO1, which suggested that miR-196a
promoted cell growth by suppressing the expression of FOXO1.
In conclusion, the present study was the first, to
the best of our knowledge, to identify the potential role of
miR-196a in HCV core-mediated HCC proliferation and its underlying
mechanisms. The findings of the present study revealed that the HCV
core promoted the upregulation of miR-196a, and was crucial in HCC
cell proliferation, possibly through the indirect targeting of
FOXO1 by miR-196a. Understanding the precise role of miR-196a in
the induction of tumour cell proliferation is likely to increase
our understanding of the biology of HCV-associated HCC, and the
inhibition of miR-196a may represent a novel therapeutic strategy
for the treatment of HCV-associated HCC.
References
|
1
|
Berasain C, Perugorria MJ, Latasa MU,
Castillo J, Goñi S, Santamaría M, Prieto J and Avila MA: The
epidermal growth factor receptor: A link between inflammation and
liver cancer. Exp Biol Med (Maywood). 234:713–725. 2009. View Article : Google Scholar
|
|
2
|
Sherman M: Hepatocellular carcinoma:
Epidemiology, risk factors and screening. Semin Liver Dis.
25:143–154. 2005. View Article : Google Scholar
|
|
3
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kremsdorf D, Soussan P, Paterlini-Brechot
P and Brechot C: Hepatitis B virus-related hepatocellular
carcinoma: Paradigms for viral-related human carcinogenesis.
Oncogene. 25:3823–3833. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lupberger J and Hildt E: Hepatitis B
virus-induced oncogenesis. World J Gastroenterol. 13:74–81. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brody H: Hepatitis C. Nature. 474:S12011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gravitz L: Introduction: A smouldering
public-health crisis. Nature. 474:S2–S4. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thomas DL: Global control of hepatitis C:
Where challenge meets opportunity. Nat Med. 19:850–858. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moradpour D and Penin F: Hepatitis C virus
proteins: from structure to function. Curr Top Microbiol Immunol.
369:113–142. 2013.PubMed/NCBI
|
|
10
|
Zhang Y, Li RQ, Feng XD, Zhang YH and Wang
L: Down-regulation of PTEN by HCV core protein through activating
nuclear factor-κB. Int J Clin Exp Pathol. 7:7351–7359. 2014.
|
|
11
|
Watashi K and Shimotohno K: The roles of
hepatitis C virus proteins in modulation of cellular functions: A
novel action mechanism of the HCV core protein on gene regulation
by nuclear hormone receptors. Cancer Sci. 94:937–943. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Siavoshian S, Abraham JD, Kieny MP and
Schuster C: HCV core, NS3, NS5A and NS5B proteins modulate cell
proliferation independently from p53 expression in hepatocarcinoma
cell lines. Arch Virol. 149:323–336. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Farh KK, Grimson A, Jan C, Lewis BP,
Johnston WK, Lim LP, Burge CB and Bartel DP: The widespread impact
of mammalian MicroRNAs on mRNA repression and evolution. Science.
310:1817–1821. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ell B and Kang Y: MicroRNAs as regulators
of bone homeostasis and bone metastasis. Bonekey Rep. 3:5492014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang S, Xie Y, Yang P, Chen P and Zhang
L: HCV core protein-induced down-regulation of microRNA-152
promoted aberrant proliferation by regulating Wnt1 in HepG2 cells.
PLoS One. 9:e817302014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCt method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
17
|
Hou T, Ou J, Zhao X, Huang X, Huang Y and
Zhang Y: MicroRNA-196a promotes cervical cancer proliferation
through the regulation of FOXO1 and p27Kip1. Br J Cancer.
110:1260–1268. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Eckman MH, Talal AH, Gordon SC, Schiff E
and Sherman KE: Cost-effectiveness of screening for chronic
hepatitis C infection in the United States. Clin Infect Dis.
56:1382–1393. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Akamatsu S, Hayes CN, Tsuge M, Miki D,
Akiyama R, Abe H, Ochi H, Hiraga N, Imamura M, Takahashi S, et al:
Differences in serum microRNA profiles in hepatitis B and C virus
infection. J Infect. 70:273–287. 2015. View Article : Google Scholar
|
|
20
|
Liu CJ, Tsai MM, Tu HF, Lui MT, Cheng HW
and Lin SC: miR-196a overexpression and miR-196a2 gene polymorphism
are prognostic predictors of oral carcinomas. Ann Surg Oncol.
20(Suppl 3): S406–S414. 2013. View Article : Google Scholar
|
|
21
|
Liu P, Xin F and Ma CF: Clinical
significance of serum miR-196a in cervical intraepithelial
neoplasia and cervical cancer. Genetics and molecular research:
GMR. 14:17995–18002. 2015. View Article : Google Scholar
|
|
22
|
Li JH, Luo N, Zhong MZ, Xiao ZQ, Wang JX,
Yao XY, Peng Y and Cao J: Inhibition of microRNA-196a might reverse
cisplatin resistance of A549/DDP non-small-cell lung cancer cell
line. Tumour Biol. Sept 16–2015.Epub ahead of print.
|
|
23
|
Li SC, Shi H, Khan M, Caplin M, Meyer T,
Öberg K and Giandomenico V: Roles of miR-196a on gene regulation of
neuroendocrine tumor cells. Mol Cell Endocrinol. 412:131–139. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ge J, Chen Z, Li R, Lu T and Xiao G:
Upregulation of microRNA-196a and microRNA-196b cooperatively
correlate with aggressive progression and unfavorable prognosis in
patients with colorectal cancer. Cancer Cell Int. 14:1282014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu XH, Lu KH, Wang KM, Sun M, Zhang EB,
Yang JS, Yin DD, Liu ZL, Zhou J, Liu ZJ, et al: MicroRNA-196a
promotes non-small cell lung cancer cell proliferation and invasion
through targeting HOXA5. BMC Cancer. 12:3482012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tsai MM, Wang CS, Tsai CY, Chen CY, Chi
HC, Tseng YH, Chung PJ, Lin YH, Chung IH, Chen CY and Lin KH:
MicroRNA-196a/-196b promote cell metastasis via negative regulation
of radixin in human gastric cancer. Cancer Lett. 351:222–231. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang C, Yao C, Li H, Wang G and He X:
Combined elevation of microRNA-196a and microRNA-196b in sera
predicts unfavorable prognosis in patients with osteosarcomas. Int
J Mol Sci. 15:6544–6555. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu B, Xiang Y and Zhang HS: Circulating
microRNA-196a as a candidate diagnostic biomarker for chronic
hepatitis C. Mol Med Rep. 12:105–110. 2015.PubMed/NCBI
|
|
29
|
Mukherjee A, Di Bisceglie AM and Ray RB:
Hepatitis C virus-mediated enhancement of microRNA miR-373 impairs
the JAK/STAT signaling pathway. J Virol. 89:3356–3365. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shang Y, Wang LQ, Guo QY and Shi TL:
MicroRNA-196a overexpression promotes cell proliferation and
inhibits cell apoptosis through PTEN/Akt/FOXO1 pathway. Int J Clin
Exp Pathol. 8:2461–2472. 2015.PubMed/NCBI
|
|
31
|
Greer EL and Brunet A: FOXO transcription
factors at the interface between longevity and tumor suppression.
Oncogene. 24:7410–7425. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Calnan DR and Brunet A: The FoxO code.
Oncogene. 27:2276–2288. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Myatt SS and Lam EW: The emerging roles of
forkhead box (Fox) proteins in cancer. Nat Rev Cancer. 7:847–859.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Paik JH, Kollipara R, Chu G, Ji H, Xiao Y,
Ding Z, Miao L, Tothova Z, Horner JW, Carrasco DR, et al: FoxOs are
lineage-restricted redundant tumor suppressors and regulate
endothelial cell homeostasis. Cell. 128:309–323. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang XW, Shen GZ, Cao LQ, Jiang XF, Peng
HP, Shen G, Chen D and Xue P: MicroRNA-1269 promotes proliferation
in human hepatocellular carcinoma via downregulation of FOXO1. BMC
Cancer. 14:9092014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee SY, Lee GR, Woo DH, Park NH, Cha HJ,
Moon YH and Han IS: Depletion of Aurora A leads to upregulation of
FoxO1 to induce cell cycle arrest in hepatocellular carcinoma
cells. Cell Cycle. 12:67–75. 2013. View
Article : Google Scholar :
|