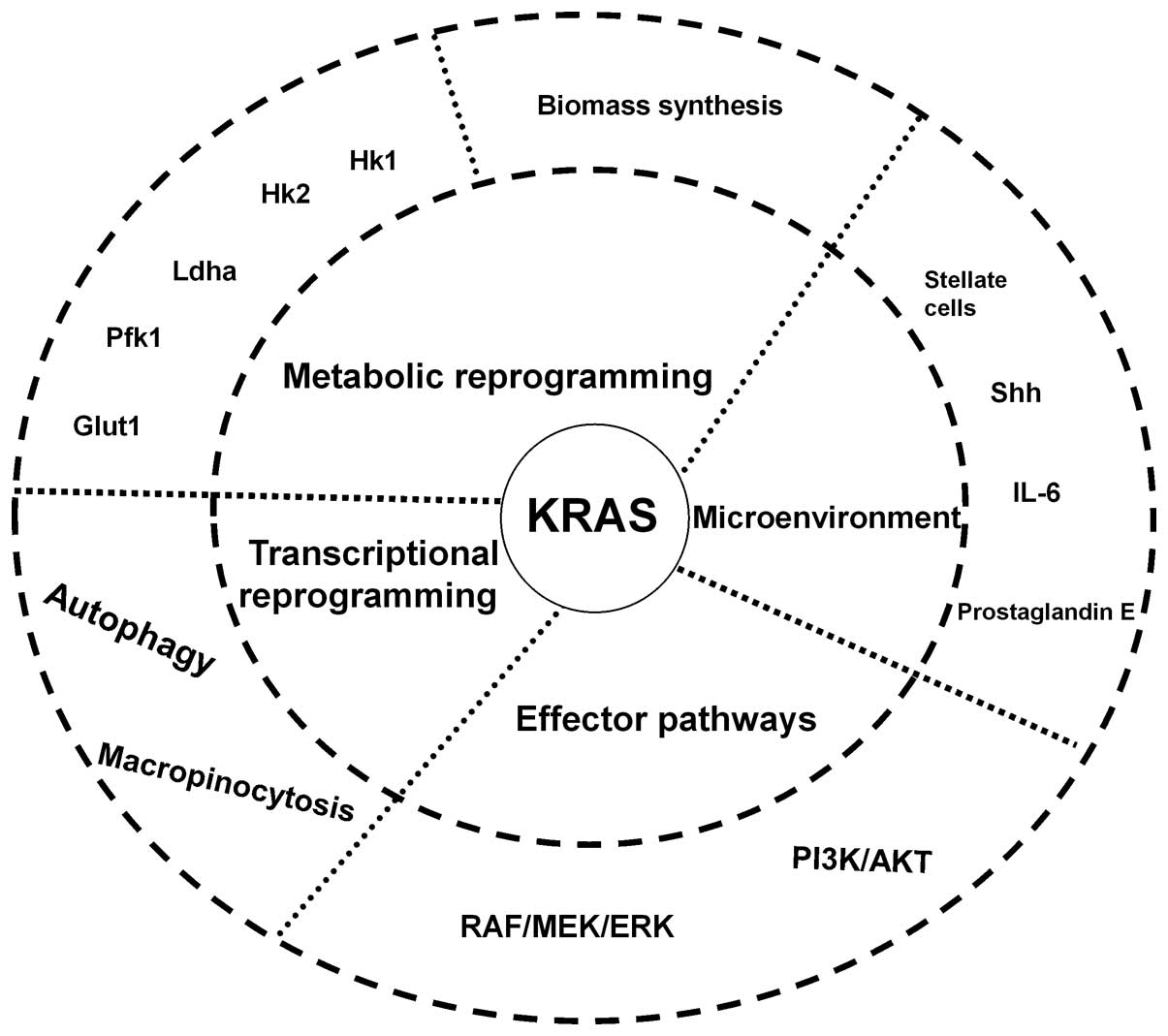
Pancreatic ductal adenocarcinoma (PDAC) is an
incurable disease that results in mortality. The number of newly
diagnosed cases almost equals the annual number of deaths despite
advances in surgery and chemoradiotherapy in the past decades.
Although the 5-year survival of pancreatic cancer patients has
almost doubled over the past decade, it remains low at ~7–8%
according to the US National Cancer Institute (1). A lack of early diagnostic strategies,
high resistance to chemoradiotherapy and early local or distant
metastatic recurrence following surgery are the three predominant
factors that contribute to poor outcomes. Further understanding of
the biological and molecular mechanisms underlying the initiation
and development of PDAC are required. Genetically, cancer
progresses as a result of the combined activation of oncogenes and
inactivation of tumor suppressors. Similarly, numerous molecular
alterations are also required for pancreatic intraepithelial
neoplasias (PanIN) lesions to develop into PDAC. Previous studies
have established that PDAC is characterized by four signature
mutations including mutations in GTPase KRas (KRAS) oncogene and in
cyclin-dependent kinase inhibitor 2A (CDKN2A), tumor protein p53
(TP53), and SMAD family member 4 (SMAD4) tumor suppressor genes
(2,3). Approximately 90% of pancreatic
neoplasms express mutant KRAS, which has been hypothesized to be
the initiator of PDAC. However, the development of therapeutic
agents targeting KRAS in PDAC remains unsuccessful. The present
review discusses recent research regarding KRAS and explores
potential therapeutic targets.
PDAC develops with progressive cellular,
morphological and architectural changes from normal ductal
epithelium to preneoplastic lesions, and then PanINs and PDAC. The
majority of PDAC and early PanIN lesions involve mutations in the
KRAS oncogene. Almoguera et al (4) and Smit et al (5) first established the association
between the mutant KRAS gene and PDAC in 1988. To investigate the
role of the KRAS oncogene in the onset of PDAC, multiple
genetically engineered mouse (GEM) models were established. The
first model was the endogenous KRAS-based model, Ptf1a-Cre
(6), followed by the Pdx1-Cre
model (7).
Pdx1-Cre;LSL-KRASG12D and
Ptf1a-Cre;LSL-KRASG12D mice were also generated, these
are generally referred to as KC mice (8) and express oncogenic KRAS from the
earliest stage of pancreatic development. KC mice demonstrate that
mutant KRAS is sufficient for the initiation of PDAC. The use of KC
mice is a useful tool for pancreatic cancer research, as other
signaling pathways and genetic events resulting in pancreatic
carcinogenesis may be investigated (9). As tumor suppressor genes are usually
lost or inactivated in human PDAC, KC mice have been crossed with
mice with non-functional or mutant alleles of CDKN2A or p53
(10,11). The latter model, usually referred
to as KPC mice, is currently the most promising preclinical model
of PDAC. When treated with standard therapeutic strategies for
PDAC, KPC mice are observed to react in same way as human patients
(12). PanIN with oncogenic KRAS
is able to rapidly progress to PDAC when subjected to inflammatory
insult (13).
PDAC is a highly aggressive neoplasm that has a
marked fibro-inflammatory microenvironment, promoting cancer
induction and growth. GEM models have been used to investigate the
role of KRAS on the PDAC microenvironment, which contains large
quantities of inflammatory stroma. Immune cells infiltrate around
the lowest grade preinvasive lesions, but immunosuppressive cells,
including macrophages, regulatory T cells and myeloid-derived
suppressor cells (MDSC), predominate in the early response and
persist through invasive cancer (14). Phenotype changes of the stellate
cells occur earlier than noticeable changes in other pancreatic
components (15). Thus, even low
levels of KRAS activity generate signals that influence the
microenvironment.
By contrast to the majority of other solid tumors,
pancreatic tumors are considered to be hypovascularized, although
blood vessels are present within the tumor microenvironment as
stellate cells produce angiogenic factors (16). In addition to the cellular
components, the stroma comprises components of the extracellular
matrix, including collagen fibers and hyaluronic acid (17,18).
Inactivation of KRAS also results in resolution of the chronic
inflammation associated with pancreatic cancer. Thus, KRAS is
hypothesized to regulate the production of factors that maintain an
active stroma. These factors and their activities remain to be
further elucidated, however, Sonic Hedgehog, interleukin-6 (IL-6),
and prostaglandin E are considered factors, each of which is
expressed in a KRAS-dependent manner (19). Sonic hedgehog (SHH) is one of the
ligands of the hedgehog signaling pathway, it is expressed by
pancreatic tumor cells (20,21)
and functions in a paracrine manner (22), activating hedgehog signaling in the
stroma and potentially mediating its maintenance (23). The inflammatory cytokine IL-6 is
overexpressed in pancreatic tumors and it important in the
development of PanINs in mice (24). Prostaglandin E and prostaglandin E
receptor 4 exert a direct effect on stellate cells to stimulate the
production of stroma (19). All
these factors are generated by sustained high-level KRAS
activity.
The immune cells that infiltrate the pancreas also
appear to be regulated by KRAS. In mouse models of PDAC, PanINs are
infiltrated by immune cells, including those that suppress the
immune response, including regulatory T cells, MDSC, and mast cells
(25). Tumor cells secrete
cytokines, such as granulocyte-macrophage colony-stimulating factor
(26,27), which further promotes infiltration
of MDSC that inhibit anti-tumor immune responses. KRAS inactivation
results in an overall reduction in the number of infiltrating
immune cells. Thus, the inflammatory environment of pancreatic
tumors also appears to be regulated by KRAS in a paracrine manner,
forming part of a KRAS-associated positive-feedback loop of
inflammation that requires further elucidation in the future.
In addition to intracellular factors, the
interactions between the tumor cells and their microenvironment are
also controlled by KRAS, although the mechanisms require further
elucidation. In iKRAS mice, inactivation of oncogenic KRAS at any
stage of carcinogenesis results in reduced proliferation and smooth
muscle actin expression in the stroma (32). SHH, secreted by tumor cells, is one
of the signals mediating the interaction between the tumor cells
and the surrounding fibroblasts within the stroma (20,21),
and it also activates paracrine signaling in fibroblasts (22). However, it is probable that
additional signals are involved in the regulation of the
interactions between KRAS-expressing epithelial cells and the
surrounding microenvironment. Oncogenic KRAS mutations and the
immune microenvironment may act synergistically to promote the
development and progression of PDAC.
Transcriptional reprogramming of key metabolic
enzymes (for example glutamate dehydrogenase-1 and
glutamic-oxaloacetic transaminase 1) in the glutamine pathway,
which is involved in the utilization of autophagy in PDAC, is
driven by KRAS (39,40). Inhibition of autophagy in mouse
models blocked KRAS tumorigenicity in a wild-type TP53 background,
but resulted in PanIN transformation into invasive PDAC in the
presence of an oncogenic KRAS mutation and a TP53 deletion
(41). KRAS has an additional role
in absorbing and degrading the extracellular components of cancer
cells, referred to as macropinocytosis. Upregulation of
macropinocytosis by KRAS contributes to the metabolic requirements
of PDAC cell lines, however, inhibition of macropinocytosis results
in slowing of KRAS-transformed cell growth (42,43).
Thus, it may be possible to design therapeutic agents to target
KRAS, or its effectors, that alter pancreatic cancer metabolism and
impair the ability of the cancer cells to maintain high levels of
glycolysis (44).
Cancer-associated mortality is predominantly due to
a lack of early diagnosis and effective therapeutic strategies.
However, the underlying molecular mechanisms of PDAC development
and progression are little known, thus, the development of mouse
models is required, particularly GEM models, to investigate the
mechanisms of pancreatic tumorigenesis and reproduce the
pathogenesis of PDAC to aid development of diagnostic and
therapeutic strategies.
Notch signaling pathways are important in the
progression of pancreatic cancer, which has been investigated in
GEM models. Deletion of Notch-1 resulted in an increased tumor
incidence and progression in Pdx-1-Cre;LSL-KRASG12D
mice, indicating that Notch-1 may be a tumor suppressor gene in
pancreatic cancer development (50). However, another previous study
demonstrated that deficiency of Notch-2, but not Notch-1, blocked
PanIN progression and prolonged survival (51). These findings suggest further
investigation into the exact physiological role of Notch-1 in
pancreatic cancer initiation and progression is required.
Though numerous similarities are observed between
the PanIN lesions and PDAC in GEM models and those of human
patients, the etiology is distinct. PDAC is not a pediatric
disease, which indicates PDAC tumors are more likely to arise as a
result of sporadic mutations in adult individuals. In addition,
KRAS mutations are not exhibited by the entire pancreas but in
certain PDAC cell types. To begin to address these issues, a second
generation of GEM models was generated by crossing mice with a
resident KRASLSLG12Vgeo allele with double transgenic mice
(Elas-tTA;Tet-O-Cre). During late embryonic development, these
composite mice express the knocked-in KRASG12 V oncogene in ~20–30%
of acinar cells (31). Notably,
the latent period and penetrance of PanIN lesion development are
similar to those expressing the KRASG12D oncogene. The
above model enables expression of KRAS oncogene to be activated in
a controlled temporal manner by feeding the mice with doxycycline
(52).
In PDAC, serine/threonine-protein kinase B-raf
(BRAF) or phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic
subunit α (PIK3CA) activation is uncommon (53). However, GEM models with BRAF or
PIK3CA mutations may provide key information to aid understanding
of PDAC development. For example, the expression of the BRAF V600E
mutation in early pancreatic precursors results in embryonic
lethality. However, in P14 mice that express the same BRAF V600E
mutation, activation of the Pdx1-CreERT2 (estrogen receptor 2)
transgene by exposure to tamoxifen results in widespread PanIN
development (54). Notably, these
PanINs did not progress to PDAC tumors within one year. However,
with the same Pdx1-CreERT2 transgene, activation of the PIK3CA
H1047R oncogene did not induce any PanIN lesions. This suggests
that it is the RAF/mitogen-activated protein kinase kinase
(MEK)/extracellular signal-regulated kinase (ERK) signaling pathway
activated when KRAS oncogenes initiate PanIN lesions rather than
the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/AKT
signaling pathway. This indicates the importance of the RAF/MEK/ERK
signaling pathway in tumor initiation and maintenance. Furthermore,
future therapeutic strategies may be developed to target it.
Among patients with pancreatic cancer, ~10% have a
family history of the disease. There is a 2 times greater risk of
pancreatic cancer when a first-degree relative is diagnosed.
Families carrying germline mutations in certain genes, including
breast cancer 2, early onset (BRCA2), CDKN2A, LKB1, protease serine
1 and partner and localizer of BRCA2 also have an increased risk
(55). Through mixing the KC
strain of mice with mice with a truncated BRCA2 gene, two GEM
models for hereditary pancreatic cancer have been produced. PDAC
formed in these mice with high penetrance and a shorter latency
period compared with those carrying wild-type BRCA2 alleles
(56). However, in a similar
study, a KRASG12D-background mouse with BRCA2
homozygously inactivated developed acinar carcinoma, not PDAC
(57). The KC mouse model with a
conditional floxed LKB1 allele also resulted in an increased number
of PanINs, and complete penetrance and shorter latency of PDAC
tumor formation (58). Notably,
where the KRAS oncogene is not observed, homozygous loss of BRCA2
or LKB1 has different results in early pancreatic precursors.
Pdx1-Cre;LKB1lox/lox mice have very short latency periods prior to
development of pancreatic mucinous cystadenomas (58), though knockout of BRCA2 does not
induce histological alterations (57). These observations suggest that
there are a variety of methods to control the malignant
transformation of pancreatic cells.
The KRAS gene has been demonstrated to be important
in the development of PDAC, demonstrating the development of KRAS
inhibitors is required. Blocking the KRAS GTP binding site directly
prevents KRAS signaling. However, effective therapies that directly
target mutated KRAS remain unavailable, thus, research has focused
on targeting KRAS indirectly. Farnesylated KRAS following
translation is trans-located to the membrane and the Ras-activating
proteins located there. It is then activated by guanine nucleotide
exchange factors (Ras-GEFs). Farnesyltransferase inhibitors (FTIs)
are important in the post-translational modification of KRAS
activation. Certain FTIs, including lonafarnib and tipifarnib, have
been tested clinically tested, though the results are not yet
satisfactory in treating KRAS-driven tumors (64). This failure may be due to the three
different types of Ras proteins.
The majority of successful results of FTIs in
preclinical studies have focused on GTPase HRas (HRAS)-dependent
tumors (65). Compared with HRAS,
KRAS may be gera-nylgeranylated by inhibiting farnesyltransferase
(66). Via the alternate
post-translational modification of farnesylation, KRAS may be
localized to the membrane and so be activated. This has led to the
development of potential therapeutic strategies to prevent KRAS
reaching the membrane. Deltarasin is an inhibitor that binds to the
farnesyl-binding pocket of phosphodiesterase (PDE) (67). Following farnesylation, KRAS
interacts with PDE and is translocated to the membrane (68). Salirasib is another inhibitor that
limit KRAS activity in the membrane. Unlike PDE, Salirasib removes
the farnesylated protein from the membrane, thus blocking KRAS
activity (69). Salirasib has
shown potential as a KRAS inhibitor in preclinical and clinical
trials against PDAC (70).
When KRAS cannot be blocked from reaching the
membrane, other therapeutic strategies are required to prevent
activation of KRAS on the membrane. Patgiri et al (71) designed a small-molecule α-helix
mimic, using the hydrogen bond surrogate, to block the exchange of
GDP for GTP, and thus inhibit the interaction between KRAS and its
Ras-GEF SOS. Post-translational acetylation of KRAS alters the
ability of SOS to exchange GDP for GTP, however, further research
is required to elucidate the role acetylation has in the activity
of mutant KRAS.
The RAF/MEK/ERK and PI3K/AKT signaling pathways are
the targets of an increasing amount of research and numerous
inhibitors targeting these signaling pathways are already being
tested in clinical trials. In KRASG12D-driven GEM
models, inhibition of PI3K signaling has been demonstrated to be
efficient at inhibiting growth in vivo (72). Inhibition of MEK1/2 has
demonstrated suppression of cell growth in cell lines of
orthotopically transplanted human and mouse PDAC. Preclinical
studies of non-small cell lung cancer (NSCLC) have also
demonstrated successful results for this potential therapy
(73).
Dual-pathway inhibition has demonstrated promising
results, however, its toxicity is markedly higher than single-agent
therapy (74). To ameliorate this,
tissue-specific effectors required for the activation of the two
signaling pathways should be targeted. In preclinical studies of
NSCLC, Molina-Arcas et al (75) demonstrated dual-pathway inhibition
via inhibiting IGF1R and MEK. However, further investigation is
required to determine the efficacy of dual signaling pathway
inhibition against KRAS-driven PDAC.
Although oncogenic KRAS has been associated with
PDAC for over 20 years, pharmacological attempts to target KRAS
directly have been unsuccessful. Single downstream effector
inhibition may only be modestly effective as oncogenic KRAS
activates multiple downstream signaling pathways (Fig. 1). More studies are required to
further elucidate the underlying mechanisms of PDAC initiation and
progression. Comprehensive investigation into PDAC may provide
potential therapeutic strategies against pancreatic cancer for the
future.
The present review was supported by the National
Natural Science Foundation (grant nos. 81372651, 81201900, 81172276
and 81101565), and the Sino-German Center (grant. no. GZ857).
|
1
|
National Cancer Institute: SEER stat fact
sheets, pancreas. http://seer.cancer.gov/statfacts/html/pancreas.html.
Accessed April 15, 2016.
|
|
2
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wood LD and Hruban RH: Pathology and
molecular genetics of pancreatic neoplasms. Cancer J. 18:492–501.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Almoguera C, Shibata D, Forrester K,
Martin J, Arnheim N and Perucho M: Most human carcinomas of the
exocrine pancreas contain mutant c-K-ras genes. Cell. 53:549–554.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Smit VT, Boot AJ, Smits AM, Fleuren GJ,
Cornelisse CJ and Bos JL: KRAS codon 12 mutations occur very
frequently in pancreatic adenocarcinomas. Nucleic Acids Res.
16:7773–7782. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kawaguchi Y, Cooper B, Gannon M, Ray M,
MacDonald RJ and Wright CV: The role of the transcriptional
regulator Ptf1a in converting intestinal to pancreatic progenitors.
Nat Genet. 32:128–134. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hingorani SR, Petricoin EF, Maitra A,
Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD,
Hitt BA, et al: Preinvasive and invasive ductal pancreatic cancer
and its early detection in the mouse. Cancer Cell. 4:437–450. 2003.
View Article : Google Scholar
|
|
8
|
Olive KP and Tuveson DA: The use of
targeted mouse models for preclinical testing of novel cancer
therapeutics. Clin Cancer Res. 12:5277–5287. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Morris JP IV, Wang SC and Hebrok M: KRAS,
Hedgehog, Wnt and the twisted developmental biology of pancreatic
ductal adenocarcinoma. Nat Rev Cancer. 10:683–695. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Aguirre AJ, Bardeesy N, Sinha M, Lopez L,
Tuveson DA, Horner J, Redston MS and DePinho RA: Activated KRAS and
Ink4a/Arf deficiency cooperate to produce metastatic pancreatic
ductal adenocarcinoma. Genes Dev. 17:3112–3126. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hingorani SR, Wang L, Multani AS, Combs C,
Deramaudt TB, Hruban RH, Rustgi AK, Chang S and Tuveson DA:
Trp53R172H and KRASG12D cooperate to promote chromosomal
instability and widely metastatic pancreatic ductal adenocarcinoma
in mice. Cancer Cell. 7:469–483. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Singh M, Lima A, Molina R, Hamilton P,
Clermont AC, Devasthali V, Thompson JD, Cheng JH, Bou Reslan H, Ho
CC, et al: Assessing therapeutic responses in KRAS mutant cancers
using genetically engineered mouse models. Nat Biotechnol.
28:585–593. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Carrière C, Young AL, Gunn JR, Longnecker
DS and Korc M: Acute pancreatitis accelerates initiation and
progression to pancreatic cancer in mice expressing oncogenic KRAS
in the nestin cell lineage. PLoS One. 6:e277252011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Clark CE, Hingorani SR, Mick R, Combs C,
Tuveson DA and Vonderheide RH: Dynamics of the immune reaction to
pancreatic cancer from inception to invasion. Cancer Res.
67:9518–9527. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Won JH, Zhang Y, Ji B, Logsdon CD and Yule
DI: Phenotypic changes in mouse pancreatic stellate cell Ca2+
signaling events following activation in culture and in a disease
model of pancreatitis. Mol Biol Cell. 22:421–436. 2011. View Article : Google Scholar :
|
|
16
|
Erkan M, Adler G, Apte MV, Bachem MG,
Buchholz M, Detlefsen S, Esposito I, Friess H, Gress TM, Habisch
HJ, et al: StellaTUM: Current consensus and discussion on
pancreatic stellate cell research. Gut. 61:172–178. 2012.
View Article : Google Scholar
|
|
17
|
Jacobetz MA, Chan DS, Neesse A, Bapiro TE,
Cook N, Frese KK, Feig C, Nakagawa T, Caldwell ME, Zecchini HI, et
al: Hyaluronan impairs vascular function and drug delivery in a
mouse model of pancreatic cancer. Gut. 62:112–120. 2013. View Article : Google Scholar :
|
|
18
|
Provenzano PP, Cuevas C, Chang AE, Goel
VK, Von Hoff DD and Hingorani SR: Enzymatic targeting of the stroma
ablates physical barriers to treatment of pancreatic ductal
adenocarcinoma. Cancer Cell. 21:418–429. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Charo C, Hwang RF, Arumugam T, Hwang R,
Yang P, Dubois RN, Menter DG, Logsdon CD and Ramachandran V:
Prostaglandin E2 regulates pancreatic stellate cell activity via
the EP4 receptor. Pancreas. 42:467–474. 2013. View Article : Google Scholar :
|
|
20
|
Thayer SP, di Magliano MP, Heiser PW,
Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernández-del
Castillo C, Yajnik V, et al: Hedgehog is an early and late mediator
of pancreatic cancer tumorigenesis. Nature. 425:851–856. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Berman DM, Karhadkar SS, Maitra A, Montes
De Oca R, Gerstenblith MR, Briggs K, Parker AR, Shimada Y, Eshleman
JR, Watkins DN and Beachy PA: Widespread requirement for hedgehog
ligand stimulation in growth of digestive tract tumours. Nature.
425:846–851. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yauch RL, Gould SE, Scales SJ, Tang T,
Tian H, Ahn CP, Marshall D, Fu L, Januario T, Kallop D, et al: A
paracrine requirement for hedgehog signalling in cancer. Nature.
455:406–410. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Olive KP, Jacobetz MA, Davidson CJ,
Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA,
Caldwell ME, Allard D, et al: Inhibition of hedgehog signaling
enhances delivery of chemotherapy in a mouse model of pancreatic
cancer. Science. 324:1457–1461. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lesina M, Kurkowski MU, Ludes K, Rose-John
S, Treiber M, Klöppel G, Yoshimura A, Reindl W, Sipos B, Akira S,
et al: Stat3/Socs3 activation by IL-6 transsignaling promotes
progression of pancreatic intraepithelial neoplasia and development
of pancreatic cancer. Cancer Cell. 19:456–469. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chang DZ, Ma Y, Ji B, Wang H, Deng D, Liu
Y, Abbruzzese JL, Liu YJ, Logsdon CD and Hwu P: Mast cells in tumor
micro-environment promotes the in vivo growth of pancreatic ductal
adenocarcinoma. Clin Cancer Res. 17:7015–7023. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller
G and Bar-Sagi D: Oncogenic KRAS-induced GM-CSF production promotes
the development of pancreatic neoplasia. Cancer Cell. 21:836–847.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bayne LJ, Beatty GL, Jhala N, Clark CE,
Rhim AD, Stanger BZ and Vonderheide RH: Tumor-derived
granulocyte-macrophage colony-stimulating factor regulates myeloid
inflammation and T cell immunity in pancreatic cancer. Cancer Cell.
21:822–835. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yadav D and Lowenfels AB: The epidemiology
of pancreatitis and pancreatic cancer. Gastroenterology.
144:1252–1261. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pylayeva-Gupta Y, Grabocka E and Bar-Sagi
D: RAS oncogenes: Weaving a tumorigenic web. Nat Rev Cancer.
11:761–774. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Steele CW, Jamieson NB, Evans TR, McKay
CJ, Sansom OJ, Morton JP and Carter CR: Exploiting inflammation for
therapeutic gain in pancreatic cancer. Br J Cancer. 108:997–1003.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guerra C, Schuhmacher AJ, Cañamero M,
Grippo PJ, Verdaguer L, Pérez-Gallego L, Dubus P, Sandgren EP and
Barbacid M: Chronic pancreatitis is essential for induction of
pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice.
Cancer Cell. 11:291–302. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Collins MA, Bednar F, Zhang Y, Brisset JC,
Galbán S, Galbán CJ, Rakshit S, Flannagan KS, Adsay NV and Pasca di
Magliano M: Oncogenic Kras is required for both the initiation and
maintenance of pancreatic cancer in mice. J Clin Invest.
122:639–653. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Feldmann G, Beaty R, Hruban RH and Maitra
A: Molecular genetics of pancreatic intraepithelial neoplasia. J
Hepatobiliary Pancreat Surg. 14:224–232. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gaglio D, Metallo CM, Gameiro PA, Hiller
K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G and
Chiaradonna F: Oncogenic K-Ras decouples glucose and glutamine
metabolism to support cancer cell growth. Mol Syst Biol. 7:5232011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dell'Antone P: Energy metabolism in cancer
cells: How to explain the Warburg and Crabtree effects? Med
Hypotheses. 79:388–392. 2012. View Article : Google Scholar
|
|
37
|
Bryant KL, Mancias JD, Kimmelman AC and
Der CJ: KRAS: Feeding pancreatic cancer proliferation. Trends
Biochem Sci. 39:91–100. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ying H, Kimmelman AC, Lyssiotis CA, Hua S,
Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff
JL, et al: Oncogenic Kras maintains pancreatic tumors through
regulation of anabolic glucose metabolism. Cell. 149:656–670. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wise DR, DeBerardinis RJ, Mancuso A, Sayed
N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon
SB and Thompson CB: Myc regulates a transcriptional program that
stimulates mitochondrial glutaminolysis and leads to glutamine
addiction. Proc Natl Acad Sci USA. 105:18782–18787. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Son J, Lyssiotis CA, Ying H, Wang X, Hua
S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et
al: Glutamine supports pancreatic cancer growth through a
KRAS-regulated metabolic pathway. Nature. 496:101–105. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rosenfeldt MT, O'Prey J, Morton JP, Nixon
C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, et al:
p53 status determines the role of autophagy in pancreatic tumour
development. Nature. 504:296–300. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kamphorst JJ, Cross JR, Fan J, de
Stanchina E, Mathew R, White EP, Thompson CB and Rabinowitz JD:
Hypoxic and Ras-transformed cells support growth by scavenging
unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci
USA. 110:8882–8887. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Commisso C, Davidson SM, Soydaner-Azeloglu
RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin
JA, Thompson CB, et al: Macropinocytosis of protein is an amino
acid supply route in Ras-transformed cells. Nature. 497:633–637.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Neesse A, Michl P, Frese KK, Feig C, Cook
N, Jacobetz MA, Lolkema MP, Buchholz M, Olive KP, Gress TM and
Tuveson DA: Stromal biology and therapy in pancreatic cancer. Gut.
60:861–868. 2011. View Article : Google Scholar
|
|
45
|
Zhu L, Shi G, Schmidt CM, Hruban RH and
Konieczny SF: Acinar cells contribute to the molecular
heterogeneity of pancreatic intraepithelial neoplasia. Am J Pathol.
171:263–273. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mazur PK and Siveke JT: Genetically
engineered mouse models of pancreatic cancer: Unravelling tumour
biology and progressin translational oncology. Gut. 61:1488–1500.
2012. View Article : Google Scholar
|
|
47
|
Pérez-Mancera PA, Guerra C, Barbacid M and
Tuveson DA: What we have learned about pancreatic cancer from mouse
models. Gastroenterology. 142:1079–1092. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Qiu W and Su GH: Challenges and advances
in mouse modeling for human pancreatic tumorigenesis and
metastasis. Cancer Metastasis Rev. 32:83–107. 2013. View Article : Google Scholar
|
|
49
|
Westphalen CB and Olive KP: Genetically
engineered mouse models of pancreatic cancer. Cancer J. 18:502–510.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hanlon L, Avila JL, Demarest RM, Troutman
S, Allen M, Ratti F, Rustgi AK, Stanger BZ, Radtke F, Adsay V, et
al: Notch1 functions as a tumor suppressor in a model of
K-ras-induced pancreatic ductal adenocarcinoma. Cancer Res.
70:4280–4286. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Mazur PK, Einwächter H, Lee M, Sipos B,
Nakhai H, Rad R, Zimber-Strobl U, Strobl LJ, Radtke F, Klöppel G,
et al: Notch2 is required for progression of pancreatic
intraepithelial neoplasia and development of pancreatic ductal
adenocarcinoma. Proc Natl Acad Sci USA. 107:13438–13443. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Guerra C, Collado M, Navas C, Schuhmacher
AJ, Hernández-Porras I, Cañamero M, Rodriguez-Justo M, Serrano M
and Barbacid M: Pancreatitis-induced inflammation contributes to
pancreatic cancer by inhibiting oncogene induced senescence. Cancer
Cell. 19:728–739. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Jones S, Zhang X, Parsons DW, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et
al: Core signaling pathways in human pancreatic cancers revealed by
global genomic analyses. Science. 321:1801–1806. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Collisson EA, Trejo CL, Silva JM, Gu S,
Korkola JE, Heiser LM, Charles RP, Rabinovich BA, Hann B, Dankort
D, et al: A central role for RAF-MEK-ERK signaling in the genesis
of pancreatic ductal adenocarcinoma. Cancer Discov. 2:685–693.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hruban RH, Canto M, Goggins M, Schulick R
and Klein AP: Update on familial pancreatic cancer. Adv Surg.
44:293–311. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Skoulidis F, Cassidy LD, Pisupati V,
Jonasson JG, Bjarnason H, Eyfjord JE, Karreth FA, Lim M, Barber LM,
Clatworthy SA, et al: Germline Brca2 heterozygosity promotes Kras
(G12D)-driven carcinogenesis in a murine model of familial
pancreatic cancer. Cancer Cell. 18:499–509. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Rowley M, Ohashi A, Mondal G, Mills L,
Yang L, Zhang L, Sundsbak R, Shapiro V, Muders MH, Smyrk T and
Couch FJ: Inactivation of Brca2 promotes Trp53-associated but
inhibits KrasG12D-dependent pancreatic cancer development in mice.
Gastroenterology. 140:1303–1313. e1–e3. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Morton JP, Jamieson NB, Karim SA, Athineos
D, Ridgway RA, Nixon C, McKay CJ, Carter R, Brunton VG, Frame MC,
et al: LKB1 haploinsufficiency cooperates with Kras to promote
pancreatic cancer through suppression of p21-dependent growth
arrest. Gastroenterology. 139:586–597. e1–e6. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hanahan D: Heritable formation of
pancreatic beta-cell tumours in transgenic mice expressing
recombinant insulin/simian virus 40 oncogenes. Nature. 315:115–122.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Bardeesy N, Cheng KH, Berger JH, Chu GC,
Pahler J, Olson P, Hezel AF, Horner J, Lauwers GY, Hanahan D and
DePinho RA: Smad4 is dispensable for normal pancreas development
yet critical in progression and tumor biology of pancreas cancer.
Genes Dev. 20:3130–3146. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Izeradjene K, Combs C, Best M, Gopinathan
A, Wagner A, Grady WM, Deng CX, Hruban RH, Adsay NV, Tuveson DA and
Hingorani SR: Kras (G12D) and Smad4/Dpc4 haploinsufficiency
cooperate to induce mucinous cystic neoplasms and invasive
adenocarcinoma of the pancreas. Cancer Cell. 11:229–243. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Siveke JT, Lubeseder-Martellato C, Lee M,
Mazur PK, Nakhai H, Radtke F and Schmid RM: Notch signaling is
required for exocrine regeneration after acute pancreatitis.
Gastroenterology. 134:544–555. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Vincent DF, Yan KP, Treilleux I, Gay F,
Arfi V, Kaniewski B, Marie JC, Lepinasse F, Martel S, Goddard-Leon
S, et al: Inactivation of TIF1gamma cooperates with Kras to induce
cystic tumors of the pancreas. PLoS Genet. 5:e10005752009.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Appels NM, Beijnen JH and Schellens JH:
Development of farnesyl transferase inhibitors: A review.
Oncologist. 10:565–578. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kohl NE, Omer CA, Conner MW, Anthony NJ,
Davide JP, deSolms SJ, Giuliani EA, Gomez RP, Graham SL, Hamilton
K, et al: Inhibition of farnesyltransferase induces regression of
mammary and salivary carcinomas in ras transgenic mice. Nat Med.
1:792–797. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Whyte DB, Kirschmeier P, Hockenberry TN,
Nunez-Oliva I, James L, Catino JJ, Bishop WR and Pai JK: K- and
N-Ras are geranylgeranylated in cells treated with farnesyl protein
transferase inhibitors. J Biol Chem. 272:14459–14464. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zimmermann G, Papke B, Ismail S, Vartak N,
Chandra A, Hoffmann M, Hahn SA, Triola G, Wittinghofer A, Bastiaens
PI and Waldmann H: Small molecule inhibition of the KRAS-PDEδ
interaction impairs oncogenic KRAS signalling. Nature. 497:638–642.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Chandra A, Grecco HE, Pisupati V, Perera
D, Cassidy L, Skoulidis F, Ismail SA, Hedberg C, Hanzal-Bayer M,
Venkitaraman AR, et al: The GDI-like solubilizing factor PDEδ
sustains the spatial organization and signalling of Ras family
proteins. Nat Cell Biol. 14:148–158. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Weisz B, Giehl K, Gana-Weisz M, Egozi Y,
Ben-Baruch G, Marciano D, Gierschik P and Kloog Y: A new functional
Ras antagonist inhibits human pancreatic tumor growth in nude mice.
Oncogene. 18:2579–2588. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Laheru D, Shah P, Rajeshkumar NV,
McAllister F, Taylor G, Goldsweig H, Le DT, Donehower R, Jimeno A,
Linden S, et al: Integrated preclinical and clinical development of
S-trans, trans-Farnesylthiosalicylic Acid (FTS, Salirasib) in
pancreatic cancer. Invest New Drugs. 30:2391–2399. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Patgiri A, Yadav KK, Arora PS and Bar-Sagi
D: An orthosteric inhibitor of the Ras-Sos interaction. Nat Chem
Biol. 7:585–587. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Eser S, Reiff N, Messer M, Seidler B,
Gottschalk K, Dobler M, Hieber M, Arbeiter A, Klein S, Kong B, et
al: Selective requirement of PI3K/PDK1 signaling for Kras
oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell.
23:406–420. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Engelman JA: Targeting PI3K signalling in
cancer: Opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Shimizu T, Tolcher AW, Papadopoulos KP,
Beeram M, Rasco DW, Smith LS, Gunn S, Smetzer L, Mays TA, Kaiser B,
et al: The clinical effect of the dual-targeting strategy involving
PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced
cancer. Clin Cancer Res. 18:2316–2325. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Molina-Arcas M, Hancock DC, Sheridan C,
Kumar MS and Downward J: Coordinate direct input of both KRAS and
IGF1 receptor to activation of PI3 kinase in KRAS-mutant lung
cancer. Cancer Discov. 3:548–563. 2013. View Article : Google Scholar : PubMed/NCBI
|