Introduction
Acute respiratory distress syndrome (ARDS) a
syndrome of acute respiratory failure, which was first described in
1967 by Ashbaugh et al (1).
Affected patients present with progressive arterial hypoxemia,
dyspnea and a marked increase in the work of breathing (2), and endotracheal intubation and
positive pressure ventilation are required for most patients. ARDS
may arise from a variety of medical conditions, including sepsis,
pneumonia, major trauma or aspiration of gastric contents.
Inappropriate accumulation and activity of leukocytes and
platelets, dysregulated inflammation, altered permeability of
alveolar endothelial and epithelial barriers and uncontrolled
activation of coagulation pathways constitute the central
pathophysiologies of ARDS. In spite of substantial progress in
understanding underlying pathophysiology of ARDS, it remains a
major clinical problem with the mortality rate remaining as high as
40–46% and with no effective pharmacological therapy available
(3). However, cell-based therapy
has emerged as a promising therapeutic approach for ARDS (4).
ARDS was recently revealed to be an immune disorder
arising from an imbalance between regulatory T cells (Tregs) and
interleukin (IL)-17-producing T helper lymphocytes (Th17) (3). Th17 cells are known to have crucial
roles in the induction of autoimmune diseases, including rheumatoid
arthritis, experimental autoimmune encephalomyelitis and
allergen-specific responses. Furthermore, Th17 cells are crucial
for defense against extracellular bacteria and fungi (5,6).
Tregs are characterized by the forkhead box P3 (Foxp3).
Furthermore, transforming growth factor (TGF)-β1 promotes Treg
differentiation, which in turn suppresses adaptive T-cell responses
and prevents autoimmunity. An imbalance between Th17 and Tregs is
characteristic for ARDS (7–9).
Pharmacologically restoring the equilibrium between Th17 and Tregs
has therefore become a promising therapeutic strategy for ARDS.
Cyclic adenosine monophosphate (cAMP) signaling is a major pathway
as well as a key therapeutic target for inducing immune tolerance
and is involved in Treg function. Control of effector T-cell (Teff)
proliferation and function by cAMP signaling is well established.
Several mechanisms exist to suppress Teff cells by activation of
cAMP signaling, including the action of anti-inflammatory agents
through G protein-coupled receptors, cell-to-cell interactions and
inhibition of cyclic nucleotide phosphodiesterases (PDEs) within
cells. In addition, cAMP can also suppress TGF-β-mediated adaptive
Treg differentiation and reduce the Treg concentration (10–12).
It therefore appeared that targeting the cAMP signaling pathway to
reverse the imbalance of Treg/Th17 may represent a promising
approach for ARDS therapy.
The present study showed that activation of cAMP
partially restored the Treg/Th17 ratio in a mouse model of ARDS and
prevented the progression of the condition. Targeting of the cAMP
signaling pathway was therefore indicated to be a promising
strategy for ARDS therapy.
Materials and methods
Experimental protocols and animals
The present study was approved by the ethics
committee of Zhejiang University (Hangzhou, China). Male Kunming
mice (age, 8 weeks; weight, 20–22 g) were purchased from the Animal
Research Center of Zhejiang University (Hangzhou, China) and housed
in the laboratory animal center of Zhejiang University at 22°C with
40–60% humidity under a 12-h light/dark cycle. Mice were allowed
access to standard food and clean water ad libitum. Mice
were randomly divided into three groups (n=10): i) Sham group: Mice
were administered with normal saline, then subjected to sham
surgery; ii) ARDS group: Mice were supplemented with normal saline
and then underwent cecal ligature and puncture (CLP); iii) ARDS+PTX
group: Mice received PTX 10 mg/kg per hour for 5 h via
intraperitoneal administration, followed by undergoing CLP. CLP was
performed as follows: Subsequent to anesthesia with 8% sevoflurane
(Shanghai Hengrui Pharmaceutical Co., Ltd., Shanghai, China), a
midline laparotomy was performed. After the cecum was carefully
isolated, bowel obstruction was prevented by placing a cotton
ligature below the ileocecal valve. The cecum was then punctured
twice with an 18-gauge needle. Animals in the Sham group were
subjected to abdominal incision, while cecal ligation and
perforation were not performed. Subsequently, the two layers of the
abdominal cavity were closed with 3.0 silk sutures, followed by
fluid resuscitation by subcutaneous administration of pre-warmed
sterile saline (0.5 ml/10 g body weight). Animals in the Sham and
CLP groups received tramadol (Shanghai Hengrui Pharmaceutical Co.,
Ltd.; 0.05 mg/kg body weight subcutaneously, repeated every 8 h)
for post-operative analgesia. The animals were then returned to
their cages, where they received water and food. All of these
procedures were performed by the same investigator to ensure
consistency.
Twenty-four hours after lung injury, animals were
sacrificed with 5 mg/kg tramadol; lungs and spleens were collected
for further analysis. All animals used in the present study were
housed and cared for in accordance with the Chinese Pharmacological
Society Guidelines for Animal Use. All surgical procedures were
performed under 8% sevoflurane anesthesia, and all efforts were
made to minimize animal suffering.
Histological examination and hematoxylin
and eosin (H&E) staining
The lung tissue was fixed in 4% paraformaldehyde
(Beyotime Institute of Biotechnology, Haimen, China) and the
pulmonary lobules were cut into 3-mm thick blocks. Following
embedding in paraffin, tissues were cut into 4-µm slices.
After de-paraffinization using xylene and re-hydration using a
graded ethanol series, the sections were stained with H&E
(Bogoo, Shanghai, China) and images were captured under a
microscope (BX41TF; Olympus Corporation, Tokyo, Japan). Tissue
structure, inflammatory cell infiltration and necrosis were
observed.
Isolation of spleen lymphocytes
The spleen was harvested and gently pressed through
a 70-µm cell strainer (BD Biosciences, Franklin Lakes, NJ,
USA), followed by washing with 0.9% sodium chloride solution and
red cell lysis with ACK Lysing Buffer (Beyotime Institute of
Biotechnology). Among these splenocytes,
CD4+CD25+Foxp3+ Tregs,
CD4+IL-17+ Th17 cells and CD4+
cells were identified using fluorescence-assisted cell sorting
(FACS; Attune NxT; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). cAMP was quantified using a cAMP-specific ELISA (Direct cAMP
ELISA; cat. no. ADI900-066; Enzo Life Sciences, Farmingdale, NY,
USA).
Western blot analysis
Tissue and lymphocyte lysates were prepared and
western blotting was performed as previously described (13). In brief, the protein concentration
was determined using BCA Protein assay kit (Beyotime Institute of
Biotechnology) and 30 µg protein in each lane was separated
on 10% sodium dodecyl polyacrylamide gels and electro-transferred
onto nitrocellulose membranes (EMD Millipore, Billerica, MA, USA).
Membranes were then probed with rabbit polyclonal anti-Foxp3
(1:1,000; Abcam, Cambridge, MA, USA; cat. no. ab54501), rabbit
polyclonal anti-RAR-related orphan receptor γt (RORγt; 1:1,000;
Abcam; cat. no. 78007) or rabbit monoclonal anti-signal transducer
and activator of transcription (STAT)3 (1:1,000; Cell Signaling
Technologies, Inc., Danvers, MA, USA; cat. no. 12640) as well as
anti-glyc-eraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies
(1:1,000; Santa Cruz Biotechnology, Inc., Dallas, TX, USA; cat. no.
sc-25778) for 12 h at 4°C. Subsequently, membranes were incubated
with horseradish peroxidase-conjugated mouse anti-rabbit secondary
antibody (1:2,000; cat. no. sc-2357; Santa Cruz Biotechnology,
Inc.) for 1 h at 37°C, and SuperSignal West Pico Chemiluminescent
Substrate (Pierce Biotechnology, Inc., Rockford, IL, USA) according
to the manufacturer's instructions.
Reverse-transcription quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted with TRIzol according to the
manufacturer's instructions and was reverse-transcribed using the
Prime Script™ RT reagent kit (Takara Bio, Inc., Otsu, Japan). The
10 µl RT mixture contained 400 ng of total RNA, 2 µl
5X PrimeScript Buffer, 0.5 µl PrimeScript RT Enzyme mix I;
0.5 µl Oligo dT Primer (50 µM), 0.5 µl random
hexamers (100 µM) and RNase free distilled water to 10
µl. The complementary DNA template was amplified by
real-time PCR using the SYBR-PremixExTaq™ kit (Takara Bio, Inc.) on
a StepOne Real-Time PCR System (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The PCR reaction mixture contained 12.5
µl 2X SYBR Premix Ex Taq, 0.5 µl forward primer (10
µM), 0.5 µl reverse primer (10 µM), 2
µl cDNA and 9.5 µl distilled water. The primers were
obtained from Sangon Biotech Co., Ltd. (Shanghai, China) and the
sequences were as follows: 5′-GTGGCCCGGATGTGAGAAG-3 (forward),
5′-GGAGCCCTTG TCGGATGATG-3 (reverse) for Foxp3;
5′-TCAAGTTTGGCCGAATGTC-3′ (forward), 5′-CATCTGAGAGCCCTAAAGTGTATG-3′
(reverse) for RORγt; 5′-GCACCGTCAAGGCTG AGAAC-3′ (forward),
5′-GCCTTCTCCATGGTGGTGAA-3′ (reverse) for GAPDH. Thermal cycling was
programmed as follows: 95°C for 30 sec followed by 40 cycles of
95°C for 5 sec, 60°C for 20 sec and 72°C for 15 sec, and subsequent
final elongation at 72°C for 10 min. Gene expression was assessed
using the 2−ΔΔCq method (14) and levels of the analyte mRNAs were
normalized to those of GAPDH as an internal standard.
Cytokine analysis
IL-2, IL-6, IL-10, IL-17 and TGF-β secretion by
spleen lymphocytes was quantified using an Inflammation Kit (BD
Biosciences) according to the manufacturer's recommendations.
Statistical analysis
Values are expressed as the mean ± standard
deviation. Statistical analyses were conducted on Prism 6.0
(GraphPad Software, Inc., La Jolla, CA, USA) using one-way analysis
of variance or the unpaired Student's t-test as indicated.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Activation of cAMP signaling attenuates
CLP-induced ALI
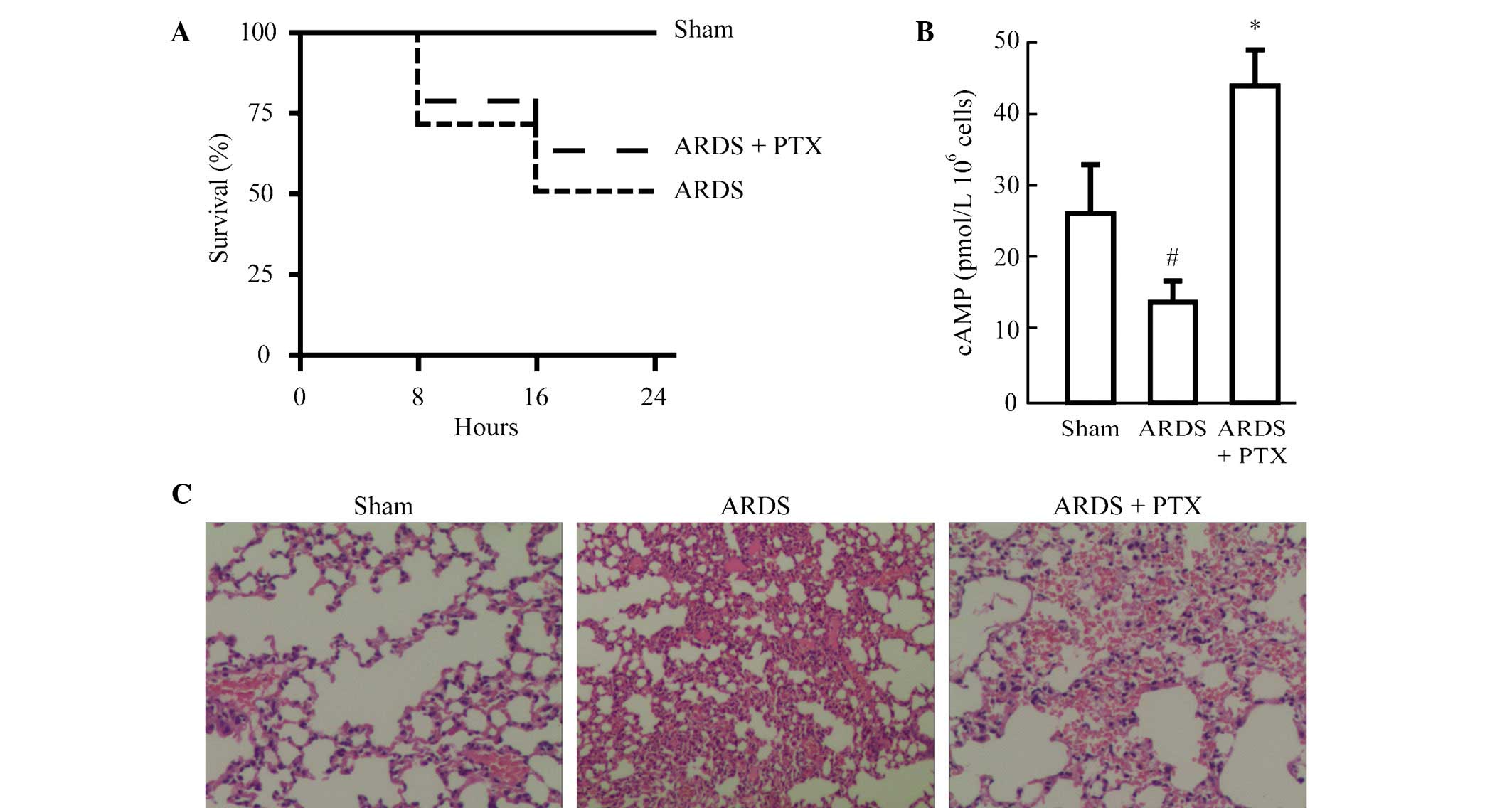
In order to identify whether activation of the cAMP
signaling pathway can ameliorate ALI and the resultant ARDS, one
group of animals was pre-treated with PDE inhibitor pentoxifylline
(PTX). As show in Fig. 1A,
although the difference was not significant, PTX pre-treatment
improved the survival rate of mice following CLP to a certain
degree (from 50 to 61%). In addition, cAMP levels were
significantly reduced in the splenocytes of mice following CLP,
while they were significantly increased in splenocytes from animals
pre-treated with PTX compared with those in the Sham group
(Fig. 1B). Furthermore, the degree
of lung injury was evaluated in mice treated with or without PTX
prior to CLP. Histological analysis revealed that CLP induced
alveolar septal thickening, accumulation of inflammatory cells in
the interstitium and alveoli and an influx of protein-rich fluid
into the alveolar space. However, mice pre-treated with PTX
demonstrated reduced structural changes following CLP (Fig. 1C).
PTX partly restores the Treg/Th17
imbalance in mice with CLP-induced ARDS
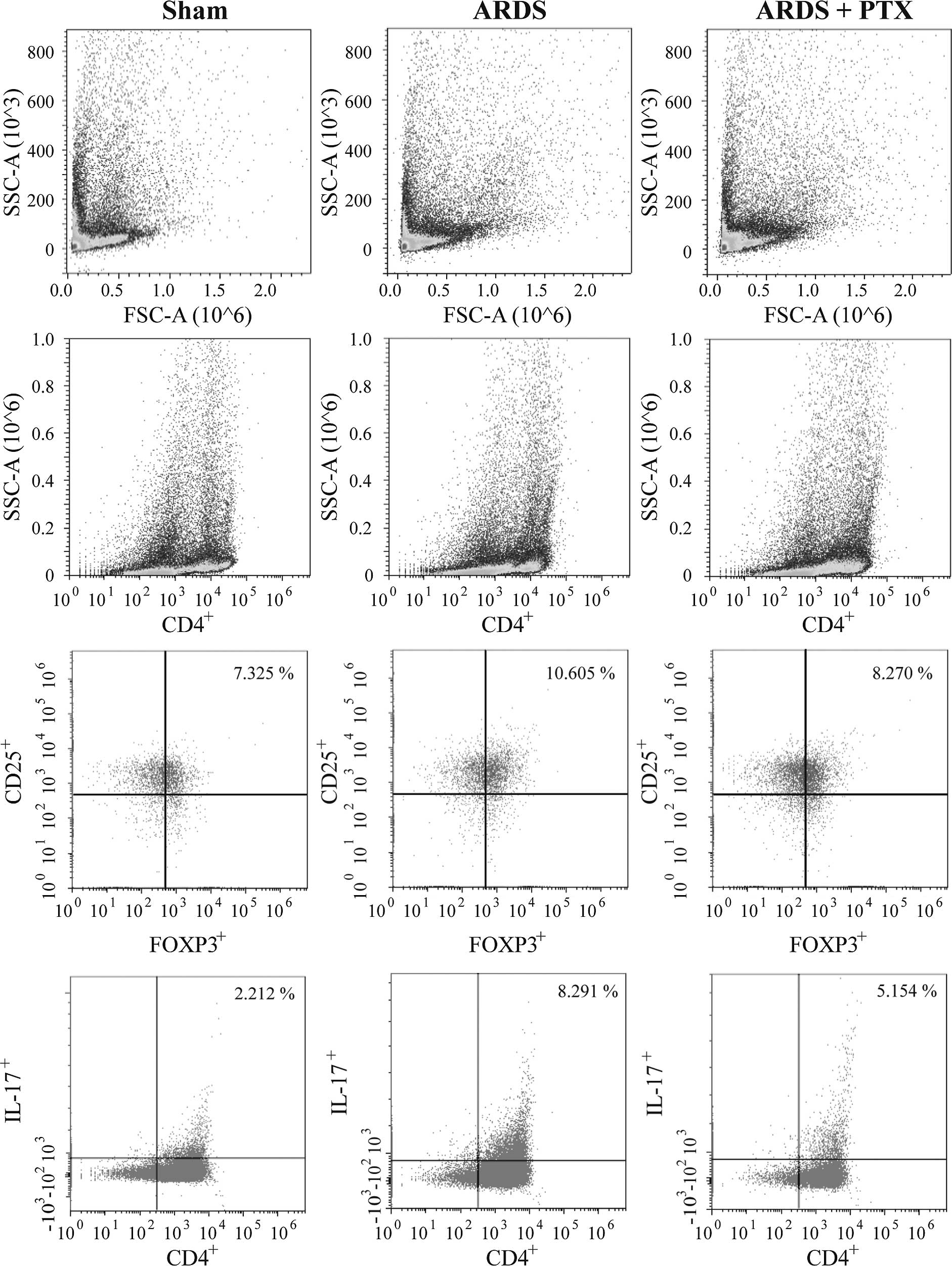
FACS analysis revealed that the proportion of
CD4+CD25+Foxp3+ Tregs and
CD4+IL-17+ Th17 cells in spleen lymphocytes
were significantly increased after CLP (Fig. 2), which was consistent with the
results of previous studies (15,16).
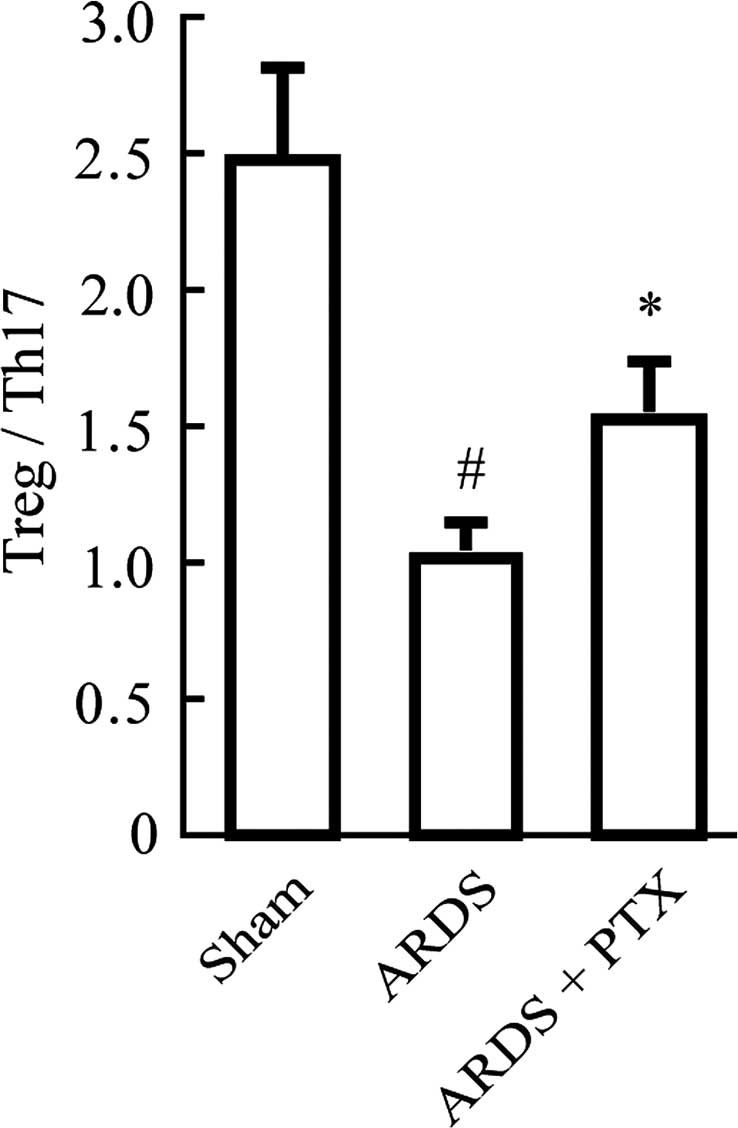
However, in CLP-induced mouse models of ARDS, the Treg/Th17 ratio
was obviously decreased compared with that in the Sham group, which
suggested that the Treg/Th17 balance was shifted towards Th17
(Fig. 3). Of note, pre-treatment
with PTX significantly attenuated CLP-induced increases in the
number of Tregs and Th17 as well as the decrease of the Treg/Th17
ratio.
Activation of cAMP signaling decreases
CLP-induced cytokine secretion
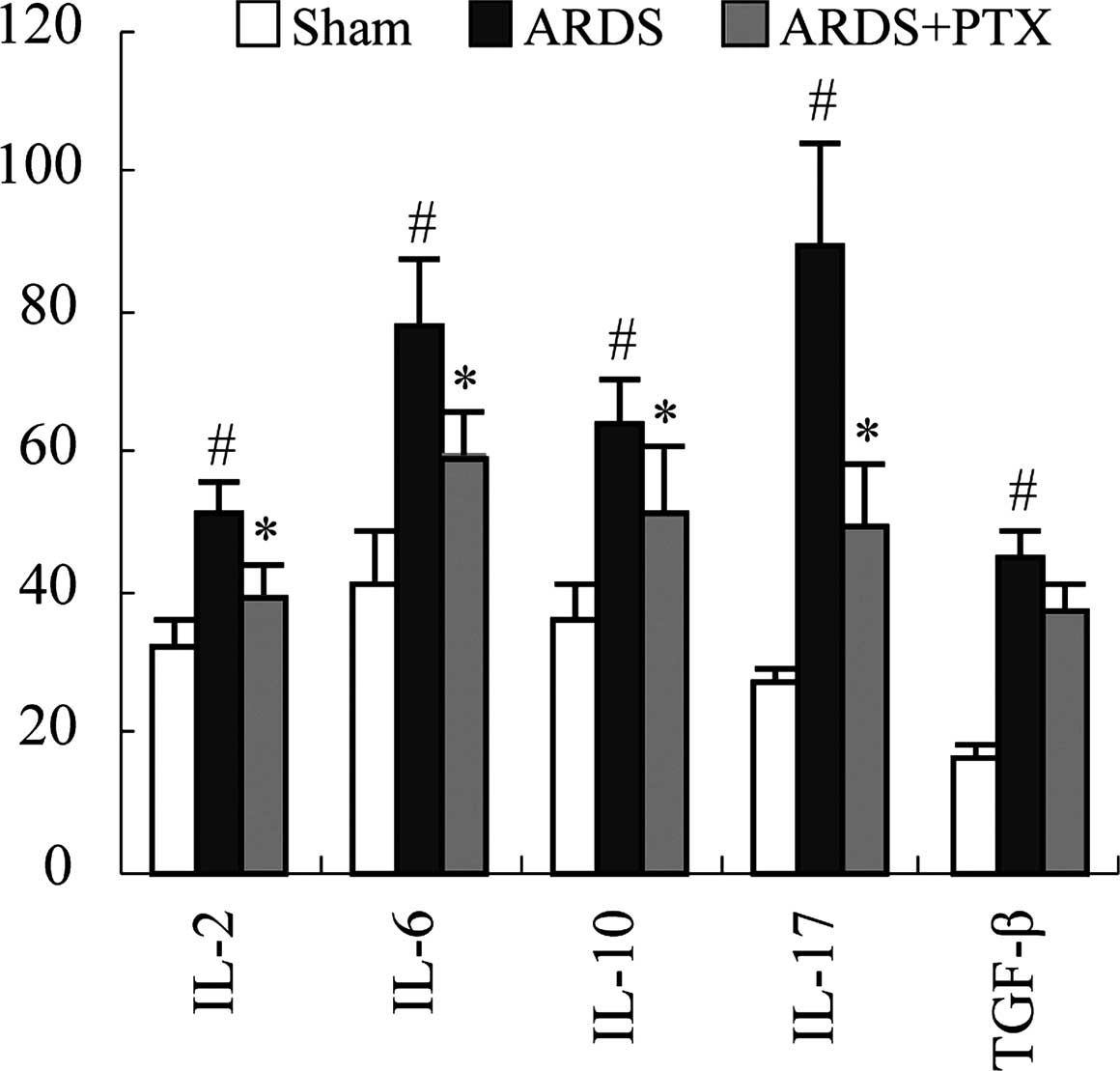
Inflammation is the main pathological consequence of
CLP-induced sepsis, which is characterized by the release of
inflammatory factors and contributes to ARDS. Thus, the ability of
PTX to prevent cytokine production of spleen lymphocytes following
CLP was assessed. An ELISA revealed that the secretion of the
classic inflammatory factors IL-2, IL-6, IL-10, IL-17 and TGF-β was
significantly upregulated following CLP. However, PTX treatment
significantly attenuated CLP-induced upregulation of IL-2, IL-6,
IL-10, IL-17 (P<0.01). While PTX also decreased TGF-β, levels
compared to those in the ARDS group, differences were not
significant (Fig. 4).
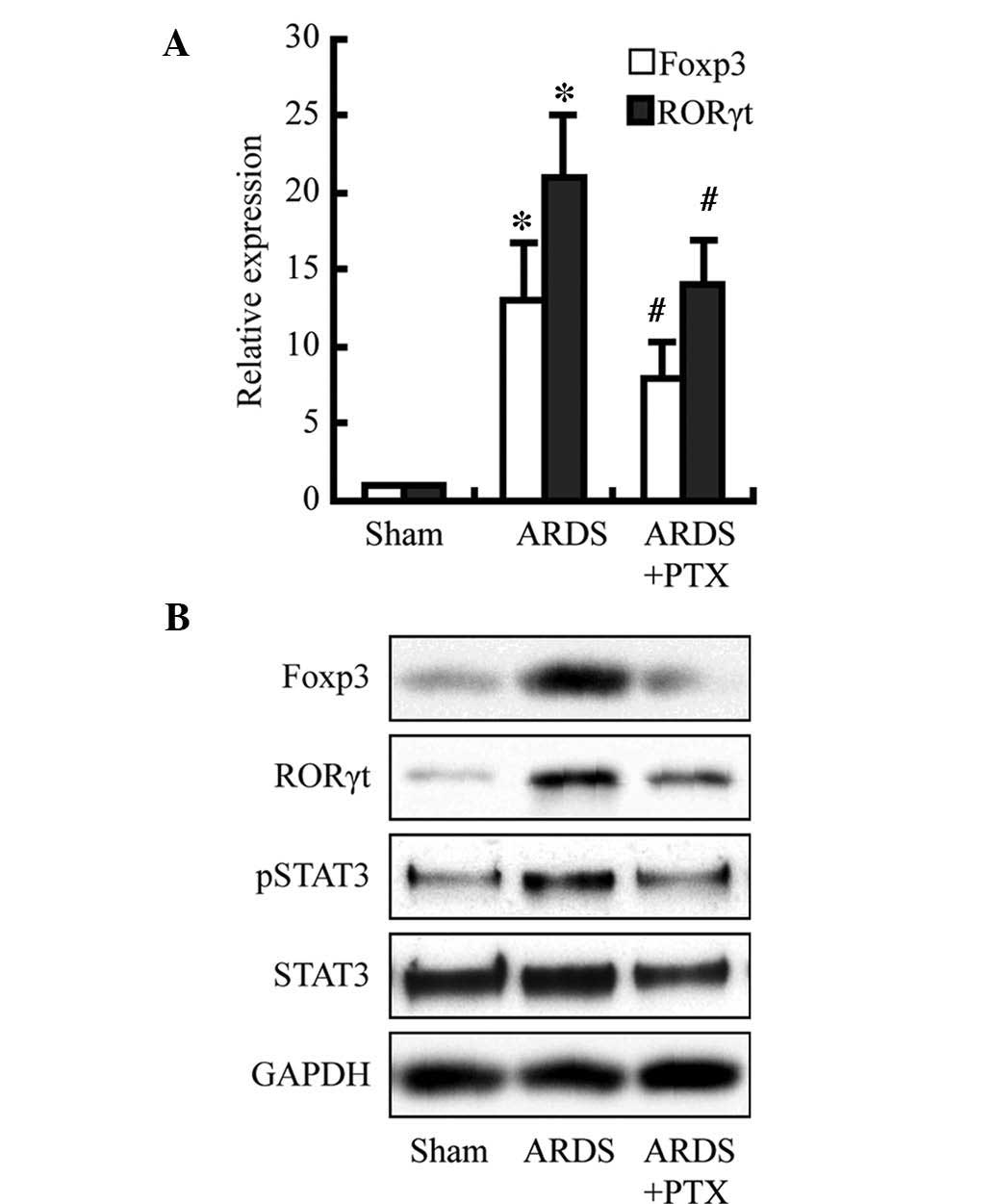
PTX attenuates CLP-induced expression of
Foxp3 and RORγt as well as activation of STAT3
To investigate the underlying mechanisms of the
PTX-induced reduction of cytokine production and partial
restoration of the Treg/Th17 balance following CLP, Foxp3, RORγt
and STAT3 were assessed as members of the cAMP signaling pathway.
As shown in the Fig. 5, Foxp3 and
RORγt expression were significantly increased at the protein and
mRNA level following CLP; furthermore, the protein levels of
activated STAT3 were obviously increased. However, pre-treatment
with PTX obviously attenuated these increases. These results may
indicate that the observed PTX-induced decreases in cytokine levels
and restoration of the Treg/Th17 balance following CLP may be due
to decreasing Foxp3 and RORγt expression as well as the levels of
activated STAT3, which is consistent with the findings of a
previous study (17).
Discussion
The present study provided a novel strategy for the
treatment of ARDS by specifically interfering with cAMP-associated
signaling. It was revealed that pre-treatment with the PDE
antagonist PTX obviously attenuated lung injury and mortality, and
reduced the production of IL-2, IL-6 IL-10, IL-17 and TGF-β in a
mouse model of CLP-induced ARDS. To investigate the underlying
mechanism, the lymphocytes from the spleens of the mice were
isolated and analyzed, revealing that the number of Tregs as well
as that of Th17 cells was increased after CLP, while the Treg/Th17
ratio was shifted towards Th17. Of note, pre-treatment with PTX
decreased the number of Tregs and Th17 cells and partially restored
the Treg/Th17 ratio. Furthermore, while cAMP levels were
significantly decreased in the splenocytes of mice following CLP,
pre-treatment with PTX led to a marked increase of cAMP.
Furthermore, PTX decreased CLP-induced increases in Foxp3 and RORγt
expression as well as STAT3 activation.
ARDS is a fatal but potentially reversible clinical
syndrome of lung inflammation caused by numerous direct and
indirect lung insults. Pulmonary and extra-pulmonary infection,
aspiration and trauma are common causes of ARDS. ARDS is also
increasingly recognized as a Th17- and Treg-associated immune
disease. The present study found that the proportion of
CD4+CD25+Foxp3+ Tregs amongst all
CD4+ splenocytes increased in mice with ARDS induced by
CLP, which was consistent with the results of previous studies.
Adamzik et al (15)
reported that the ratio of Tregs amongst all CD4+
lymphocytes in patients succumbed to ARDS (16.5%; P=0.025) was
almost two-fold greater than that in ARDS survivors (9.0%; P=0.015)
compared to that in control subjects (5.9%). Furthermore, D'Alessio
et al (16) showed that the
number of CD4+CD25+Foxp3+ Tregs
was increased in mice with lipopolysaccharide-induced lung injury
and remained elevated until day 10. Of note, administration of
CD4+CD25+Foxp3+ Tregs was able to
remedy experimental lung injury (16). These observations may indicate that
CD4+CD25+Foxp3+ Tregs are a
double-edged sword in ARDS: While a modest increase of Tregs may
have anti-inflammatory action, excessive increases of Tregs may
lead to tissue injury. While the roles of Th17 cells in ARDS have
remained largely elusive, they have been investigated in several
types of lung disease (18–20).
Recently, Yu et al (21)
showed that ARDS patients exhibited a significant increase in the
frequency of Th17 cells and their signature cytokine, IL-17. The
present study revealed that PTX decreased the percentage of Tregs
and Th17 cells in spleen lymphocytes, and partially restored the
Treg/Th17 ratio. The transcriptional regulators Foxp3 and RORγt are
important for regulating the differentiation and function of
Treg/Th17 (22,23), and the present study found that PTX
significantly inhibited the expression of Foxp3 and RORγt.
In conclusion, the present study provided a novel
treatment strategy for ARDS by restoring the Treg/Th17 balance and
the subsequent immune response via cAMP signaling, which requires
pre-clinical and clinical validation.
Acknowledgments
The present study was supported by Natural Science
Foundation of Zhejiang Province (grant no. LQ12H01002) and the
Zhejiang Provincial Medical and Health Platform Key Program (grant
no. 2012ZDA002).
References
|
1
|
Ashbaugh DG, Bigelow DB, Petty TL and
Levine BE: Acute respiratory distress in adults. Lancet. 2:319–323.
1967. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
ARDS Definition Task Force; Ranieri VM,
Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E,
Camporota L and Slutsky AS: Acute respiratory distress syndrome:
The Berlin Definition. JAMA. 307:2526–2533. 2012.PubMed/NCBI
|
|
3
|
Pierrakos C, Karanikolas M, Scolletta S,
Karamouzos V and Velissaris D: Acute respiratory distress syndrome:
Pathophysiology and therapeutic options. J Clin Med Res. 4:7–16.
2012.PubMed/NCBI
|
|
4
|
Curley GF and Laffey JG: Future therapies
for ARDS. Intensive Care Med. 41:322–326. 2015. View Article : Google Scholar
|
|
5
|
Besnard AG, Togbe D, Couillin I, Tan Z,
Zheng SG, Erard F, Le Bert M, Quesniaux V and Ryffel B:
Inflammasome-IL-1-Th17 response in allergic lung inflammation. J
Mol Cell Biol. 4:3–10. 2012. View Article : Google Scholar
|
|
6
|
Tsai HC, Velichko S, Hung LY and Wu R:
IL-17A and Th17 cells in lung inflammation: An update on the role
of Th17 cell differentiation and IL-17R signaling in host defense
against infection. Clin Dev Immunol. 2013:2679712013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ji L, Zhan Y, Hua F, Li F, Zou S, Wang W,
Song D, Min Z, Chen H and Cheng Y: The ratio of Treg/Th17 cells
correlates with the disease activity of primary immune
thrombocytopenia. PLoS One. 7:e509092012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kimura A and Kishimoto T: IL-6: Regulator
of Treg/Th17 balance. Eur J Immunol. 40:1830–1835. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma L, Xue HB, Guan XH, et al: The
Imbalance of Th17 cells and CD4(+) CD25(high) Foxp3(+) Treg cells
in patients with atopic dermatitis. J Eur Acad Dermatol Venereol.
28:1079–1086. 2014. View Article : Google Scholar
|
|
10
|
Bourne HR, Lichtenstein LM, Melmon KL,
Henney CS, Weinstein Y and Shearer GM: Modulation of inflammation
and immunity by cyclic AMP. Science. 184:19–28. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cao J, Zhang X, Wang Q, Wang X, Jin J, Zhu
T, Zhang D, Wang W, Li X, Li Y, et al: Cyclic AMP suppresses
TGF-β-mediated adaptive Tregs differentiation through inhibiting
the activation of ERK and JNK. Cell Immunol. 285:42–48. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee J, Kim TH, Murray F, et al: Cyclic AMP
concentrations in dendritic cells induce and regulate Th2 immunity
and allergic asthma. Proc Natl Acad Sci USA. 112:1529–1534. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ding XF, Zhou J, Hu QY, Liu SC and Chen G:
The tumor suppressor pVHL down-regulates never-in-mitosis A-related
kinase 8 via hypoxia-inducible factors to maintain cilia in human
renal cancer cells. J Biol Chem. 290:1389–1394. 2015. View Article : Google Scholar :
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
15
|
Adamzik M, Broll J, Steinmann J,
Westendorf AM, Rehfeld I, Kreissig C and Peters J: An increased
alveolar CD4+CD25+Foxp3+T-regulatory cell ratio in acute
respiratory distress syndrome is associated with increased 30-day
mortality. Intensive Care Med. 39:1743–1751. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
D'Alessio FR, Tsushima K, Aggarwal NR,
West EE, Willett MH, Britos MF, Pipeling MR, Brower RG, Tuder RM,
McDyer JF and King LS: CD4+CD25+Foxp3+ Tregs resolve experimental
lung injury in mice and are present in humans with acute lung
injury. J Clin Invest. 119:2898–2913. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang F, Fuss IJ, Yang Z and Strober W:
Transcription of RORγt in developing Th17 cells is regulated by
E-proteins. Mucosal Immunol. 7:521–532. 2014. View Article : Google Scholar
|
|
18
|
Newcomb DC and Peebles RS Jr:
Th17-mediated inflammation in asthma. Curr Opin Immunol.
25:755–760. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kononova TE, Urazova OI, Novitskii VV,
Churina EG, Kolobovnikova YV, Ignatov MV, Zakharova PA and
Pechenova OV: Functional activity of Th-17 lymphocytes in pulmonary
tuberculosis. Bull Exp Biol Med. 156:743–745. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vargas-Rojas MI, Ramírez-Venegas A,
Limón-Camacho L, Ochoa L, Hernández-Zenteno R and Sansores RH:
Increase of Th17 cells in peripheral blood of patients with chronic
obstructive pulmonary disease. Respir Med. 105:1648–1654. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu ZX, Ji MS, Yan J, Cai Y, Liu J, Yang
HF, Li Y, Jin ZC and Zheng JX: The ratio of Th17/Treg cells as a
risk indicator in early acute respiratory distress syndrome. Crit
Care. 19:822015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen Z, Lin F, Gao Y, Li Z, Zhang J, Xing
Y, Deng Z, Yao Z, Tsun A and Li B: FOXP3 and RORγt: Transcriptional
regulation of Treg and Th17. Int Immunopharmacol. 11:536–542. 2011.
View Article : Google Scholar
|
|
23
|
Chu S, Zhong X, Zhang J, Lao Q, He Z and
Bai J: The expression of Foxp3 and ROR gamma t in lung tissues from
normal smokers and chronic obstructive pulmonary disease patients.
Int Immunopharmacol. 11:1780–1788. 2011. View Article : Google Scholar : PubMed/NCBI
|