Introduction
Alzheimer's disease (AD), which is the most common
neurodegenerative disorder in the elderly population, is
characterized by deposition of amyloid-β (Aβ) and neuroinflammation
(1). Aβ deposition has been
detected in various areas of the brain, resulting in activation of
microglia and neuronal death (2).
Various mechanisms and pathways of Aβ generation and deposition
have been established, including the inflammatory response
(3). Microglia are the resident
macrophages of the brain, which have a dual role in Aβ deposition.
Microglia help eliminate Aβ deposition via phagocytosis; however,
activated microglia are capable of facilitating Aβ accumulation via
the release of inflammatory factors, including interleukin (IL)-1β,
IL-6 and tumor necrosis factor (TNF)-α, which are associated with
the Aβ cascade during the development of AD (4). Therefore, the inflammatory response
mediated by activated microglia has a key role in Aβ generation and
deposition, and thus AD pathogenesis; however, the underlying
molecular mechanisms remain to be elucidated.
Small noncoding RNA molecules, including microRNAs
(miRNAs, miRs), have an important role in cell differentiation and
activation. miRNA dysfunction may contribute to neuroinflammation
under pathological conditions (5).
Previous studies have demonstrated that miRNAs participate in the
differentiation of progenitor cells into microglia, and in the
activation process (6,7). Alterations to miRNA expression have
been associated with AD. It was previously reported that miR-155
upregulation may contribute to a microglia-mediated neurotoxic
response (8). In addition, Duan
et al (9) demonstrated that
miR-206 positively regulated the lipopolysaccharide (LPS)-induced
inflammatory response in human astrocytes.
Insulin-like growth factor 1 (IGF1) promotes
synaptogenesis, neurogenesis, neuroprotection, and exerts
anti-inflammatory effects in the brain (10). Circulating serum IGF1 can
accelerate the clearance of Aβ peptides (11), and loss of serum IGF-I input to the
brain may be an early biomarker of disease onset in a murine model
of AD (12). An IGF1 polymorphism
has been reported as a key regulator for genetic susceptibility to
late-onset AD in a Han Chinese population (13), thus indicating an essential role
for IGF1 in the development of AD.
The role of miR-206 and IGF1 in LPS-induced
microglial inflammation remains unknown; therefore, the present
study aimed to investigate the precise role of miR-206 and IGF1 in
the microglial inflammatory response induced by LPS. The expression
levels of miR-206 and IGF1 were detected in 60 peripheral blood
samples from patients with AD and matched age subjects using
quantitative polymerase chain reaction (qPCR). A dual luciferase
reporter gene assay was used to determine the association between
miR-206 and IGF1. In addition, the role of miR-206 in LPS-treated
microglia was determined by gain and loss of function experiments.
The results demonstrated that miR-206 upregulation was able to
enhance LPS-induced microglial inflammation, which was reversed
following exogenous IGF1 treatment, thus indicating that
miR-206/IGF1 signaling may be considered a novel therapeutic target
for the treatment of microglial inflammation in AD.
Materials and methods
Blood samples
The peripheral blood samples were collected from
patients with AD (30 cases; 16 male and 14 female; aged,
65-75-years-old; mean age, 68.6-years-old) and from normal
age-matched individuals (30 cases; 18 males and 12 females; age,
65–75-years-old; mean age, 67.8-years-old) at the First Affiliated
Hospital of Xinxiang Medical University (Weihui, China) between
September 2013 and October 2014, according to the legislation and
ethical boards of Xinxiang Medical University. The study was
approved by the Ethics Committee of The First Affiliated Hospital
of Xinxiang Medical University (Weihui, China). Individuals with
significant illnesses, including diabetes, heart disease, stroke or
cancer, were excluded from the present study. All individuals
provided written informed consent. The blood samples were stored at
−80°C until further use.
Cell culture and treatment
Microglial BV-2 cells were purchased from the
American Type Culture Collection (Rockville, MD, USA), and were
cultured in Dulbecco's modified Eagle's medium (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (FBS; Invitrogen; Thermo Fisher Scientific,
Inc.) at 37°C in a humidified atmosphere containing 95% air and 5%
CO2. BV-2 cells were plated in 6-well plates
(5×105 cells/well) and were grown to 80% confluence.
Ectopic miR-206 expression was introduced to the cells by
transfection with miR-206 mimics, and miR-206 expression was
inhibited by transfection with a miR-206 inhibitor (both Shanghai
GenePharma Co., Ltd., Shanghai, China) using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions. Negative control mimics and negative
control inhibitor were used as negative control (Shanghai
GenePharma Co., Ltd., Shanghai, China). Following incubation of the
BV-2 cells in 6-well plates for 24 h, the cell medium was removed,
and the cells were treated with or without LPS (1 µg/ml;
Sigma-Aldrich, St. Louis, MO, USA) for a further 24 h, or the cells
were transfected with miR-206 mimics (50 nM) or a miR-206 inhibitor
(100 mM), alongside IGF1 (5 µg/ml; Sigma-Aldrich) and/or LPS
(1 µg/ml) for a further 12 h.
RNA extraction and reverse
transcription-qPCR
TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.) was used to extract total RNA from the cells and
blood samples. A total of 0.5 µg RNA was used to synthesize
cDNA using a RevertAid First Strand cDNA Synthesis kit (Thermo
Fisher Scientific, Inc.) at 42°C for 60 min and 72°C for 10 min.
qPCR analysis was performed using a CFX96 Touch Real-Time PCR
Detection system and SsoFast EvaGreen Supermix (both Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Data were normalized to the
expression levels of β-actin in each sample. β-actin was used as an
endogenous control. The specific primers used were as follows:
IGF1, forward 5′-AGAGCCTGCGCAATGGAATA-3′, reverse
5′-ACCCTGTGGGCTTGTTGAAA-3′; β-actin, forward
5′-CATTAAGGAGAAGCTGTGCT-3′ and reverse 5′-GTTGAAGGTAGTTTCGTGGA-3′;
miR-206, forward 5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCCACACAC-3′
and reverse 5′-ACACTCCAGCTGGGTGGAATGTAAGGAAGT-3; U6, forward
5′-CTCGCTTCGGCAGCACA-3′ and reverse 5′-AACGCTTCACGAATTTGCGT-3′.
To quantify mature miR-206 expression, a MiScript
SYBR-Green PCR kit (Guangzhou Ribobio, Co., Ltd., Guangzhou, China)
was used. The specific primer sets for miRNA-206 and U6 were
purchased from Genecopoeia (Rockville, MD, USA). The relative
expression levels of miR-206 were normalized to U6. All PCR
analyses were performed at the following conditions: 2 min at 55°C
and 7 min at 95°C, followed by 40 cycles at 95°C for 10 sec, 60°C
for 10 sec and 72°C for 30 sec, and a final extension at 65–95°C
for 5 min. The 2−ΔΔCq method was used to analyze the
data (14).
Western blot analysis
Cells were collected and resuspended in cold
radioimmunoprecipitation lysis buffer (Beijing CWBio Co., Ltd.,
Beijing, China) for 1 h on ice. The lysates were then centrifuged
at 12,000 × g for 20 min at 4°C. Protein samples (50 µg)
were separated by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and were transferred onto a nitrocellulose membrane
(Wuhan Boster Biological Technology, Ltd., Wuhan, China). The
membrane was blocked in 5% nonfat dried milk in Tris-buffered
saline containing 0.1% Tween-20 (TBST) for 2 h, and was incubated
with the primary antibody against anti-IGF1 (cat. no. 250710;
1:200; Abbiotec LLC, San Diego, CA, USA) and β-actin (cat. no.
BA2305; 1:5,000; Wuhan Boster Biotechnology, Ltd.) overnight at
4°C. The membrane was then washed three times in TBST (15
min/wash), and was incubated with the corresponding horseradish
peroxidase-conjugated anti-rabbit immunoglobulin G secondary
antibody (cat. no. BA1055; 1:3,000; Wuhan Boster Biological
Technology, Ltd.) for 1 h at 37°C. Immunoreactive proteins were
detected using enhanced chemiluminescence (Wuhan Boster Biological
Technology, Ltd., Wuhan, China). Bands were quantified using Image
J software (NIH, Bethesda, MD, USA).
Dual luciferase reporter assay
The psiCheck-2 dual-luciferase reporter vector
(Promega Corporation, Madison, WI, USA) harboring an IGF1 wild type
(WT) or mutant (MUT) 3′-untranslated region (UTR; Sangon Biotech,
Co., Ltd., Shanghai, China) was inserted into the BgIII and
KpnI restriction sites at the 3′-end of the Renilla
luciferase gene. For the luciferase assay, 5×105 cells
were cultured to ~80% confluence in 6-well plates. Subsequently,
the cells were co-transfected with the miR-206 mimics or inhibitor,
and either the WT or MUT 3′-UTR of the IGF1 dual luciferase
reporter vector. Following incubation for 6 h with the transfection
reagent/DNA complex, the medium was replaced with fresh complete
medium containing 10% FBS. A total of 48 h post-transfection, a
Dual Luciferase Reporter Gene Assay kit (antibodies-online Inc.,
Atlanta, GA, USA) was used to determine the luciferase activities
in each group on a GloMax® 96 Microplate Luminometer
(Promega Corporation). The activity of Renilla luciferase
was normalized against that of firefly luciferase.
Enzyme-linked immunosorbent assay (ELISA)
determination of IL-1β, TNF-α and Aβ
The culture supernatants were collected from the
treated BV-2 cells, in order to measure the levels of TNF-α and
IL-1β. To measure Aβ peptide 1-42 (Aβ1-42) levels, cell lysates
(the same preparation of lysates as used for western blotting) were
used. Mouse IL-1β ELISA kit, mouse TNF-α ELISA kit and mouse Aβ1-42
ELISA kit were purchased from Nanjing SenBeijia Biotechnology Co.,
Ltd. (Nanjing, China) and were used to measure the levels of IL-1β,
TNF-α and Aβ, according to the manufacturer's protocols. The
optical density was detected at 450 nm using a microplate
absorbance reader (Synergy™ Mx; BioTek, Winooski, VT, USA).
β-secretase activity assay
β-secretase activity in the BV-2 cells was
determined using a β-Secretase Fluorometric Assay kit (Biovision
Inc., Milpitas, CA, USA). Ice-cold extraction buffer (Beyotime
Institute of Biotechnology, Wuhan, China) was used to extract
protein from the treated cells. Following a 20 min incubation on
ice, cell lysates were centrifuged at 10,000 × g for 5 min. The
supernatant was collected and maintained on ice. A 50 µl
sample (total protein, 150 µg) was added to each well in a
96-well plate, followed by 50 µl 2X reaction buffer and 2
µl β-secretase substrate, and the plate was incubated in the
dark at 37°C for 1 h. Fluorescence was detected at excitation and
emission wavelengths of 335 and 495 nm respectively, using a
GloMax® 96 Microplate Luminometer (Promega Corporation).
β-secretase activity is expressed as relative fluorescence units
per µg of protein sample.
Statistical analysis
All experiments were repeated at least three times
with similar results, and the data are expressed as the mean ±
standard deviation. Statistical analyses were performed using
GraphPad Prism 6.0 software (GraphPad Software, Inc., La Jolla, CA,
USA). Data were compared using a one-way analysis of variance with
Dunnett test as a post-hoc comparison, or with the Student's
t-test. Correlation was analyzed using Pearson's correlation.
P<0.05 was considered to indicate a statistically significant
difference.
Results
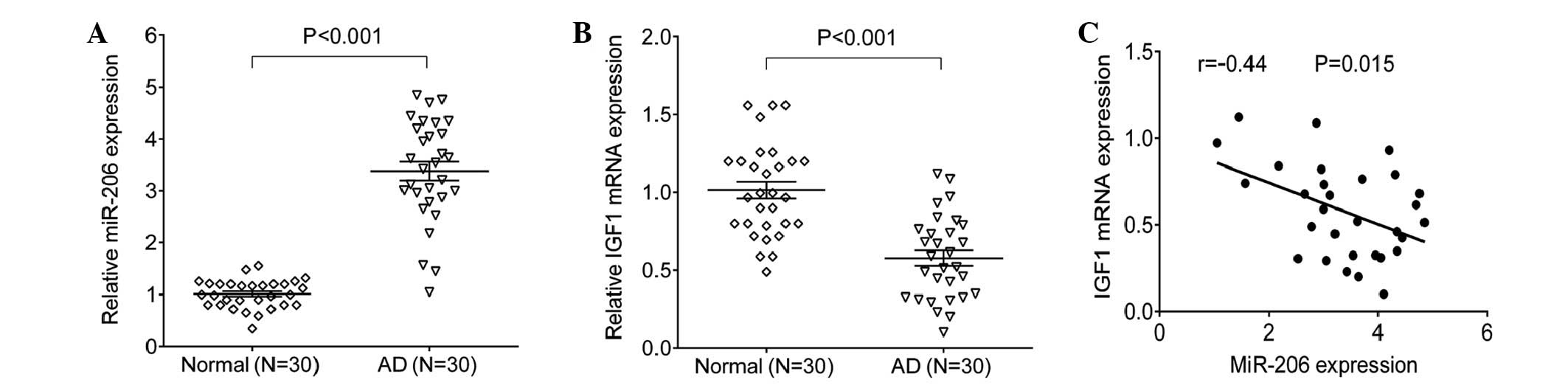
miR-206 expression levels are negatively
correlated with the mRNA expression levels of IGF1 in the blood of
patients with AD
The expression levels of miR-206 and IGF1 were
detected using SYBR green qPCR analysis. Blood samples from 30
patients with AD and 30 normal individuals were tested, the results
indicated that miR-206 was significantly increased in the
peripheral blood samples from patients with AD, as compared with in
the paired normal blood samples (P=0.0004; Fig. 1A). However, as shown in Fig. 1B, the mRNA expression levels of
IGF1 were significantly reduced in the peripheral blood from
patients with AD, as compared with in the normal control
individuals (P=0.0005). In addition, the expression levels of
miR-206 were negatively correlated with the mRNA expression levels
of IGF1 in the peripheral blood samples from patients with AD
(Fig. 1C).
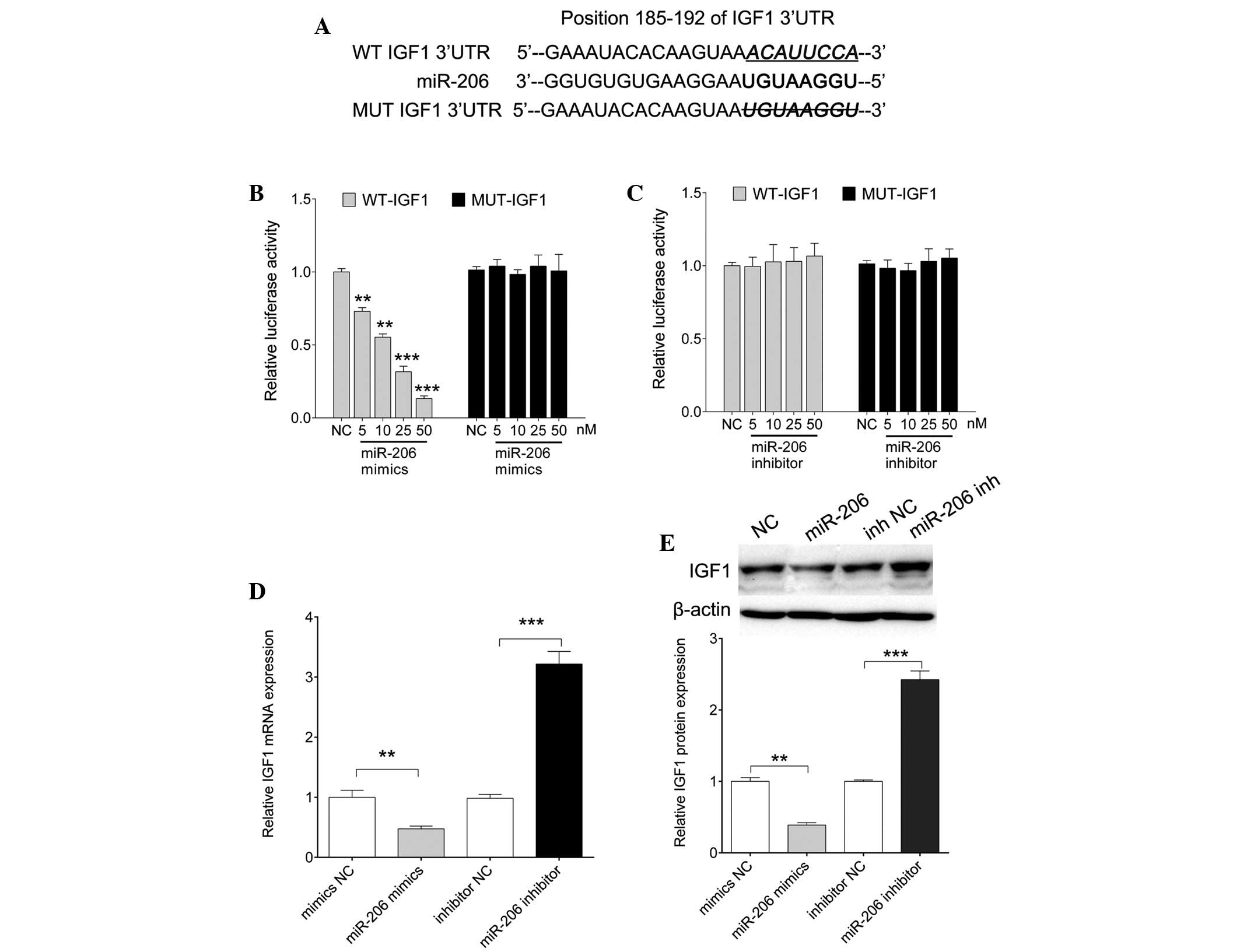
IGF1 is a molecular target of
miR-206
The negative correlation detected between miR-206
and IGF1 in patients with AD promoted us to elucidate whether
miR-206 directly targeted IGF1. Several miRNA public
target-prediction algorithms [Pictar (http://pictar.mdc-berlin.de), TargetScan (http://www.targetscan.org/) and miRanda (http://www.microrna.org/microrna/home.do)] were used
to assess this hypothesis. As shown in Fig. 2A, the binding site between miR-206
and IGF1 was conserved. The present study generated a dual
luciferase reporter containing a putative binding site alongside a
mutant construct (Fig. 2A).
Luciferase activity was significantly reduced in cells
co-transfected with miR-206 mimics and WT-IGF1, as the
concentration of miR-206 mimics increased (P=0.0006, 0.008, 0.0002
and 0.0001 for 5, 10, 25 and 50 nM, respectively); however,
decreased luciferase levels were not detected following
co-transfection with the MUT-IGF1 vector. Furthermore, luciferase
activity exhibited no significant alterations in the cells that
were co-transfected with the miR-206 inhibitor and WT-IGF1 or
MUT-IGF1. These data indicate that miR-206 directly targets the
3′-UTR of IGF1. Furthermore, as shown in Fig. 2D and E, ectopic expression of
miR-206 resulted in a significant reduction in IGF1 mRNA (P=0.007)
and protein (P=0.006) expression levels, whereas transfection with
the miR-206 inhibitor restored the expression of IGF1 (P=0.0004 and
0.0002 for mRNA and protein, respectively). These results suggest
that miR-206 may regulate the expression of IGF1 at the
transcriptional level by directly targeting its 3′-UTR.
miR-206 and IGF1 expression are
upregulated and downregulated respectively, in LPS-treated
microglial BV-2 cells
LPS induces the expression of several miRNAs, which
are involved in LPS-mediated inflammation (15). To determine whether miR-206 and
IGF1 are involved in LPS-mediated immune responses in microglial
cells, the expression levels of miR-206 and IGF1 were determined in
LPS-stimulated BV-2 cells (microglial cells of BV-2 mice). The BV-2
cells were stimulated with various concentrations of LPS for a
range of durations. The expression levels of miR-206 were
significantly increased in a time-dependent (P=0.008, 0.0007,
<0.0001 and <0.0001 for 4, 8, 12 and 24 h, respectively;
Fig. 3A) and dose-dependent manner
(P=0.038, 0.008 and <0.0001 for 100, 500 and 1,000 ng/ml,
respectively; Fig. 3B), whereas
the mRNA expression levels of IGF1 were markedly decreased in a
time-dependent (P=0.009, 0.0005, <0.0001 and <0.0001 for 4,
8, 12 and 24 h, respectively; Fig.
3C) and dose-dependent manner (P=0.029, 0.0007 and <0.0001
for 100, 500 and 1,000 ng/ml, respectively; Fig. 3D), particularly when the cells were
treated with 1 µg/ml LPS for 12 h. These results suggest a
possible involvement for the miR-206/IGF1 pathway in LPS-mediated
immune responses in brain microglia.
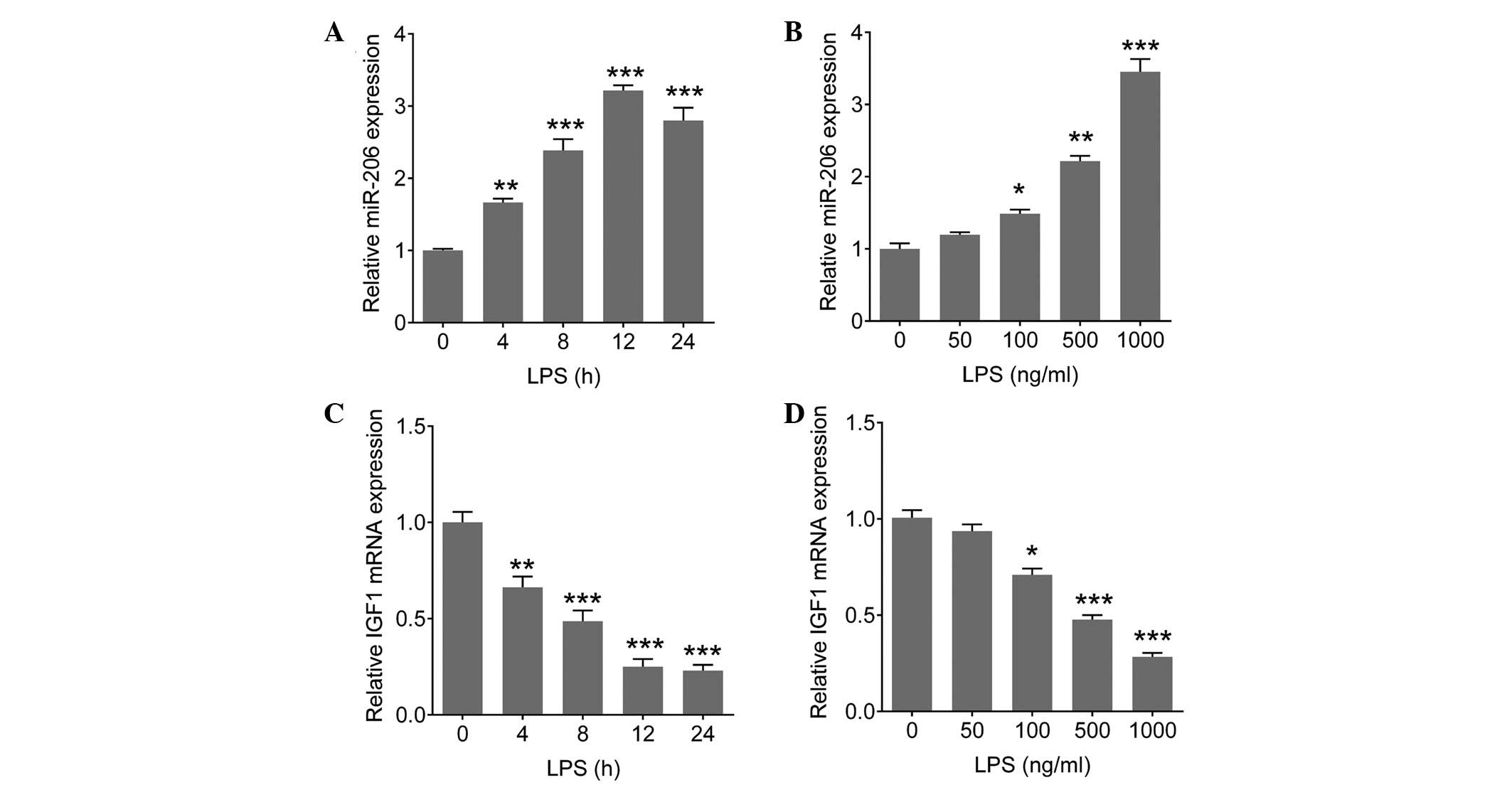 | Figure 3(A and B) Upregulation of miR-206 and
(C and D) downregulation of IGF1 was observed in LPS-treated
microglia. (A and C) BV-2 cells were exposed to 1 µg/ml LPS
for 0, 4, 8, 12 or 24 h. (B and D) BV-2 cells were exposed to LPS
at 0, 50, 100, 500 or 1,000 ng/ml for 12 h. Total RNA was
extracted, and the miR-206 expression levels and IGF1 mRNA
expression levels were detected using quantitative polymerase chain
reaction. Data are presented as the mean ± standard deviation.
*P<0.05, **P<0.01 and
***P<0.001, vs. 0 h or 0 ng/ml group. miR, microRNA;
LPS, lipopolysaccharide; IGF1, insulin-like growth factor 1. |
miR-206 modulates the production of
inflammatory cytokines and amyloidogenesis induced by LPS
To investigate the involvement of miR-206 in
inflammatory cytokine production, miR-206 was upregulated or
downregulated in BV-2 cells by transfection with miR-206 mimics or
an inhibitor for 24 h. Subsequently, the cells were treated with
LPS (1 µg/ml) for an additional 12 h. Upregulation of
miR-206 significantly increased the production of IL-1β and TNF-α,
whereas down-regulation of miR-206 markedly reduced their
expression (P=0.006 for miR-206 and P=0.005 for anti-miR-206, an
P=0.036 for miR-206 and P=0.006 for anti-miR-206; Fig. 4A and B, respectively), as
determined by ELISA.
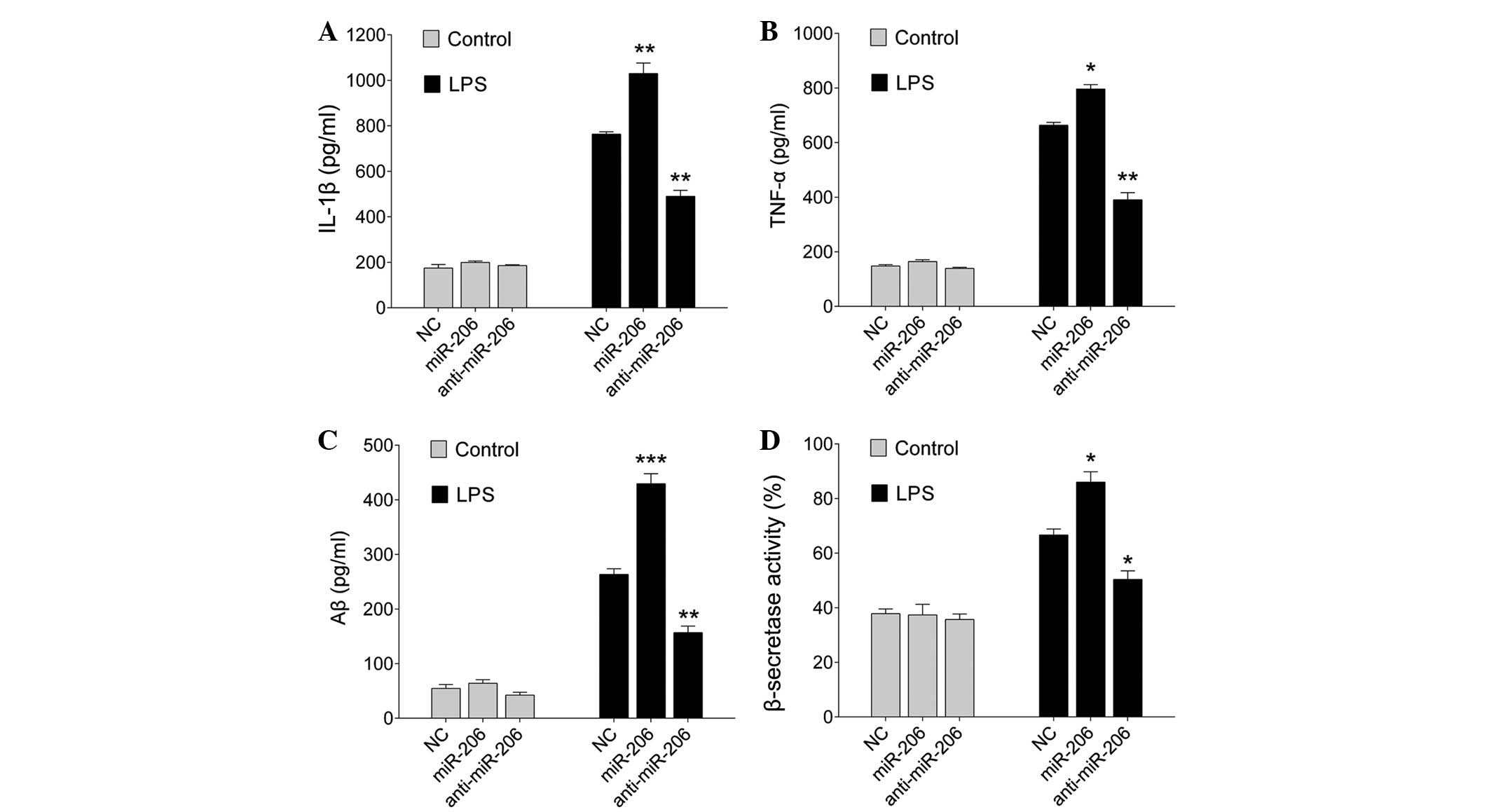 | Figure 4miR-206 enhanced LPS-induced
inflammatory cytokine production and Aβ generation. BV-2 cells were
transfected with miR-206 mimics, a miR-206 inhibitor or the NC for
24 h, and were then exposed to LPS (1 µg/ml) for 12 h.
Levels of (A) IL-1β, (B) TNF-α, (C) Aβ and (D) β-secretase were
detected by enzyme-linked immunosorbent assay. Data are presented
as the mean ± standard deviation. *P<0.05,
**P<0.01 and ***P<0.001 vs. NC. miR,
microRNA; LPS, lipopolysaccharide; Aβ, amyloid-β; IL, interleukin;
TNF-α, tumor necrosis factor-α; NC, negative control. |
Microglia are a major source of neuroinflammation,
which induces amyloid generation (16). To assess the involvement of miR-206
in modulating amyloidogenesis, BV-2 cells were transfected with
miR-206 mimics or an inhibitor, and were stimulated with LPS for 12
h. As shown in Fig. 4C and D, when
unstimulated, the cells expressed low levels of Aβ1-42 and
β-secretase, whereas the expression levels of Aβ1-42 and
β-secretase were increased in response to LPS (1 µg/ml)
after 12 h. In addition, miR-206 overexpression increased
LPS-induced Aβ1-42 (P<0.0001 and P=0.008 for miR-206 and
anti-miR-206, respectively; Fig.
4C), and promoted the activation of β-secretases, the
rate-limiting enzymes in Aβ generation (P=0.022 and P=0.037 for
miR-206 and anti- miR-206, respectively; Fig. 4D). Notably, downregulation of
miR-206 decreased LPS-induced Aβ1-42 and the activation of
β-secretases (Fig. 4C and D).
These results indicate that overexpression of miR-206 may have a
pro-inflammatory and pro-amyloidogenesis effect.
IGF1 abrogates the miR-206-modulated
LPS-induced inflammatory response and amyloidogenesis
IGF1 has an important anti-inflammatory role in
activated microglial cells (17).
To further investigate the link between miR-206-mediated regulation
of IGF1 and inflammation, BV-2 cells were transiently transfected
with miR-206 mimics for 24 h, and were then treated with exogenous
IGF1 (5 µg/ml) and exposed to 1 µg/ml LPS for an
additional 12 h. ELISA was used to quantify the levels of IL-1β,
TNF-α, Aβ1-42 and β-secretases. As expected, overexpression of
miR-206, together with IGF1, abrogated the miR-206-modulated
LPS-induced proinflammatory response and amyloidogenesis (P=0.0007,
0.006 and <0.0001; Fig. 5A–D,
respectively). These results indicate that the miR-206/IGF1 pathway
is involved in the process by which LPS induces the production of
proinflammatory cytokines and amyloidogenesis.
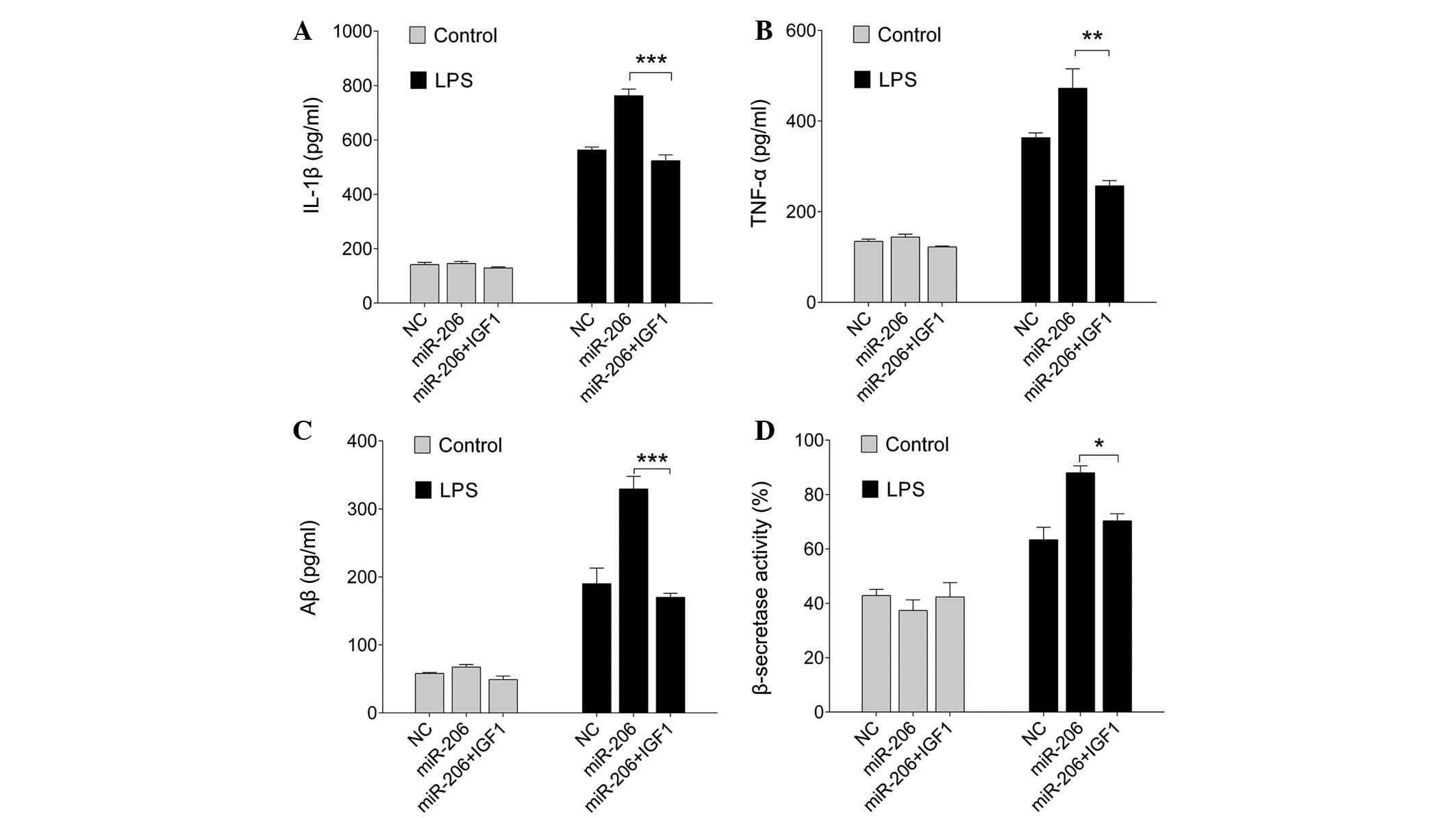 | Figure 5Exogenous IGF1 abrogates the
miR-206-modulated LPS-induced proinflammatory response and Aβ
generation. BV-2 cells were transfected with miR-206 mimics or the
NC, and were then treated with exogenous IGF1 and exposed to 1
µg/ml LPS for a further 12 h. The concentration of (A)
IL-1β, (B) TNF-α and (C) Aβ, and (D) the activity of β-secretases
was measured by enzyme-linked immunosorbent assay. Data are
presented as the mean ± standard deviation. *P<0.05,
**P<0.01 and ***P<0.001. miR, microRNA;
LPS, lipopolysaccharide; Aβ, amyloid-β; IL, interleukin; TNF-α,
tumor necrosis factor-α; NC, negative control. |
Discussion
Activated microglia are associated with the
progression of aging-associated neurodegenerative diseases,
including AD, via the regulation of inflammation through the
generation of IL-1β, TNF-α and other cytokines (18). Aβ deposition activates the
complement system, which, in turn, stimulates microglia to release
neurotoxic materials, including inflammatory factors (19). This positive feedback loop between
Aβ deposition and microglial activation exacerbates the progression
of AD, and the inflammatory response bridges this loop. Previous
studies have indicated that aberrantly expressed miRNAs are
increasingly being implicated in AD by regulation of Aβ,
phosphorylation of tau protein and inflammation, which are the
predominant pathomechanisms of AD (20,21).
In addition, alterations in AD may be associated with the
regulation of various miRNAs in blood and cerebral spinal fluid
(CSF), particularly the brain-specific miRNAs secreted into blood
and CSF, which could be considered as potential AD biomarkers
(21,22). The present study demonstrated that
miR-206 was significantly upregulated in blood samples from
patients with AD compared with in age-matched normal controls. In
animal models of AD, previous studies have reported an increased
expression of miR-206 in brain tissue, CSF and plasma of embryonic
amyloid precursor protein (APP)/presenilin 1 transgenic mice
(23) and Tg2576 mice (24). Furthermore, upregulation of miR-206
has been detected in serum from patients with mild cognitive
impairment (25), and in the
temporal cortex of human AD brains (24). These results indicated that
upregulation of miR-206 in the peripheral circulation truly
reflects the alterations in the AD brain.
Tian et al (23) and Lee et al (24) demonstrated that brain-derived
neurotrophic factor (BDNF), a neuroprotec-tive factor, was a target
of miR-206. Similar to BDNF, IGFs, including IGF1 and IGF2, are the
key regulators of memory, cognition and inflammation in the central
nervous system (26,27). IGF2 was previously shown to reduce
the number of hippocampal Aβ40- and Aβ42-positive amyloid plaques
in APP mice (28,29). Furthermore, IGF2 may increase the
protein levels of hippocampal BDNF and IGF1 (28). Delivery of IGF1 into the
hippocampus of the APP mouse model Tg2576 was able to rescue
behavioral deficits, promote dendritic spine formation and restore
normal hippocampal excitatory synaptic transmission (29). In addition, microglia-specific
deletion of the gene encoding the prostaglandin E2 receptor, a
proinflammatory factor implicated in preclinical AD development,
restored microglial chemotaxis and Aβ clearance, increased
cytoprotective IGF1 expression, and was able to prevent memory
deficits (30). The present study
demonstrated that IGF1 was markedly reduced in blood samples from
patients with AD. Notably, IGF1 was negatively correlated with
miR-206 in human AD blood samples. As determined by dual luciferase
reporter gene assay, miR-206 directly targeted the 3′-UTR of IGF1.
In the present study, microglia were exposed to various
concentrations of LPS for a range of durations; the results
demonstrated that LPS induced miR-206 upregulation and IGF1
downregulation in a time- and dose- dependent manner. Lee et
al (24) reported that miR-206
inhibition prevented the detrimental effects of Aβ42 in Tg2576
neurons in vitro, and third ventricle or intranasal
administration of a miR-206 inhibitor into the cerebral ventricles
of AD mice improved their memory function, and enhanced hippocampal
synaptic density and neurogenesis. These results indicated that
miR-206/IGF1 signaling may have a key role in microglia-mediated
inflammation in AD.
In the present study, miR-206 expression was
upregulated by transfecting microglial BV-2 cells with miR-206
mimics. The results revealed that following LPS treatment,
increased miR-206 expression enhanced the release of
proinflammatory cytokines, including IL-1β and TNF-α, and also
increased the activity of β-secretase and the secretion of Aβ from
microglia. Conversely, downregulation of miR-206 reduced the
release of IL-1β and TNF-α, attenuated the activity of β-secretase
and decreased the secretion of Aβ. Notably, exogenous IGF1
treatment abolished the effects of miR-206 on inflammation and Aβ
generation in LPS-exposed microglia. Besides microglia, IGF1
treatment has been reported to reverse the Aβ-induced neurotoxic
effects on survival of septal neurons (26). In addition, overexpression of
miR-206 in astrocytes led to increased expression of inflammatory
cytokines, including IL-6, IL-1β and chemokine (C-C motif) ligand 5
upon exposure to LPS, whereas knockdown of miR-206 had the opposite
effects (9). A previous study
suggested that loss of serum IGFI input to the brain may be an
early biomarker of disease onset in AD mice (12).
In conclusion, the present study suggested that
miR-206/IGF1 signaling may regulate microglial inflammation and
amyloidogenesis, which are critical processes for the development
of AD. In future experiments, we aim to determine the possible
anti-AD effects of miR-206/IGF1 signaling in an animal model of AD.
Taken together, these data indicated that targeting the small
molecule miR-206, and treatment with exogenous IGF1 may be
considered a novel therapeutic strategy for the treatment of
inflammatory neurodegenerative diseases such as AD.
References
|
1
|
Wu Z and Nakanishi H: Connection between
periodontitis and Alzheimer's disease: Possible roles of microglia
and leptomeningeal cells. J Pharmacol Sci. 126:8–13. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Latta CH, Sudduth TL, Weekman EM, Brothers
HM, Abner EL, Popa GJ, Mendenhall MD, Gonzalez-Oregon F, Braun K
and Wilcock DM: Determining the role of IL-4 induced
neuroinflammation in microglial activity and amyloid-β using BV2
microglial cells and APP/PS1 transgenic mice. J Neuroinflammation.
12:412015. View Article : Google Scholar
|
|
3
|
Zhang F and Jiang L: Neuroinflammation in
Alzheimer's disease. Neuropsychiatr Dis Treat. 11:243–256. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Doens D and Fernandez PL: Microglia
receptors and their implications in the response to amyloid β for
Alzheimer's disease pathogenesis. J Neuroinflammation. 11:482014.
View Article : Google Scholar
|
|
5
|
Guedes J, Cardoso AL and Pedroso de Lima
MC: Involvement of microRNA in microglia-mediated immune response.
Clin Dev Immunol. 2013:1868722013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Su W, Hopkins S, Nesser NK, Sopher B,
Silvestroni A, Ammanuel S, Jayadev S, Möller T, Weinstein J and
Garden GA: The p53 transcription factor modulates microglia
behavior through microRNA-dependent regulation of c-Maf. J Immunol.
192:358–366. 2014. View Article : Google Scholar
|
|
7
|
Fenn AM, Smith KM, Lovett-Racke AE,
Guerau-de-Arellano M, Whitacre CC and Godbout JP: Increased
micro-RNA 29b in the aged brain correlates with the reduction of
insulin-like growth factor-1 and fractalkine ligand. Neurobiol
Aging. 34:2748–2758. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Louafi F, Martinez-Nunez RT and
Sanchez-Elsner T: MicroRNA-155 targets SMAD2 and modulates the
response of macrophages to transforming growth factor-{beta}. J
Biol Chem. 285:41328–41336. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Duan X, Zohaib A, Li Y, Zhu B, Ye J, Wan
S, Xu Q, Song Y, Chen H and Cao S: MiR-206 modulates
lipopolysaccharide-mediated inflammatory cytokine production in
human astrocytes. Cell Signal. 27:61–68. 2015. View Article : Google Scholar
|
|
10
|
Fernandez AM and Torres-Alemán I: The many
faces of insulin-like peptide signalling in the brain. Nat Rev
Neurosci. 13:225–239. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Carro E, Trejo JL, Gomez-Isla T, LeRoith D
and Torres-Aleman I: Serum insulin-like growth factor I regulates
brain amyloid-beta levels. Nat Med. 8:1390–1397. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Trueba-Sáiz A, Cavada C, Fernandez AM,
Leon T, Gonzalez DA, Fortea OJ, Fortea Ormaechea J, Lleó A, Del Ser
T, Nuñez A and Torres-Aleman I: Loss of serum IGF-I input to the
brain as an early biomarker of disease onset in Alzheimer mice.
Transl Psychiatry. 3:e3302013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang W, Yu JT, Tan L, Liu QY, Wang HF and
Ma XY: Insulin-like growth factor 1 (IGF1) polymorphism is
associated with Alzheimer's disease in Han Chinese. Neurosci Lett.
531:20–23. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
15
|
Hsieh CH, Rau CS, Jeng JC, Chen YC, Lu TH,
Wu CJ, Wu YC, Tzeng SL and Yang JC: Whole blood-derived microRNA
signatures in mice exposed to lipopolysaccharides. J Biomed Sci.
19:692012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Theriault P, ElAli A and Rivest S: The
dynamics of monocytes and microglia in Alzheimer's disease.
Alzheimers Res Ther. 7:412015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Suh HS, Zhao ML, Derico L, Choi N and Lee
SC: Insulin-like growth factor 1 and 2 (IGF1, IGF2) expression in
human microglia: Differential regulation by inflammatory mediators.
J Neuroinflammation. 10:372013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McGeer PL and McGeer EG: Targeting
microglia for the treatment of Alzheimer's disease. Expert Opin
Ther Targets. 19:497–506. 2015. View Article : Google Scholar
|
|
19
|
Cai Z, Hussain MD and Yan LJ: Microglia,
neuroinflammation and beta-amyloid protein in Alzheimer's disease.
Int J Neurosci. 124:307–321. 2014. View Article : Google Scholar
|
|
20
|
Tan L, Yu JT, Hu N and Tan L: Non-coding
RNAs in Alzheimer's disease. Mol Neurobiol. 47:382–393. 2013.
View Article : Google Scholar
|
|
21
|
Schonrock N and Gotz J: Decoding the
non-coding RNAs in Alzheimer's disease. Cell Mol Life Sci.
69:3543–3559. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Muller M, Kuiperij HB, Claassen JA,
Kusters B and Verbeek MM: MicroRNAs in Alzheimer's disease:
Differential expression in hippocampus and cell-free cerebrospinal
fluid. Neurobiol Aging. 35:152–158. 2014. View Article : Google Scholar
|
|
23
|
Tian N, Cao Z and Zhang Y: MiR-206
decreases brain-derived neurotrophic factor levels in a transgenic
mouse model of Alzheimer's disease. Neurosci Bull. 30:191–197.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee ST, Chu K, Jung KH, Kim JH, Huh JY,
Yoon H, Park DK, Lim JY, Kim JM, Jeon D, et al: MiR-206 regulates
brain-derived neurotrophic factor in Alzheimer disease model. Ann
Neurol. 72:269–277. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xie B, Zhou H, Zhang R, Song M, Yu L, Wang
L, Liu Z, Zhang Q, Cui D, Wang X and Xu S: Serum miR-206 and
miR-132 as potential circulating biomarkers for mild cognitive
impairment. J Alzheimers Dis. 45:721–731. 2015.PubMed/NCBI
|
|
26
|
Jarvis K, Assis-Nascimento P, Mudd LM and
Montague JR: Beta-amyloid toxicity and reversal in embryonic rat
septal neurons. Neurosci Lett. 423:184–188. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Luo YW, Xu Y, Cao WY, Zhong XL, Duan J,
Wang XQ, Hu ZL, Li F, Zhang JY, Zhou M, et al: Insulin-like growth
factor 2 mitigates depressive behavior in a rat model of chronic
stress. Neuropharmacology. 89:318–324. 2015. View Article : Google Scholar
|
|
28
|
Mellott TJ, Pender SM, Burke RM, Langley
EA and Blusztajn JK: IGF2 ameliorates amyloidosis, increases
cholinergic marker expression and raises BMP9 and neurotrophin
levels in the hippocampus of the APPswePS1dE9 Alzheimer's disease
model mice. PLoS One. 9:e942872014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pascual-Lucas M, Viana da Silva S, Di
Scala M, Garcia-Barroso C, González-Aseguinolaza G, Mulle C,
Cuadrado-Tejedor M and Garcia-Osta A: Insulin-like growth factor 2
reverses memory and synaptic deficits in APP transgenic mice. EMBO
Mol Med. 6:1246–1262. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Johansson JU, Woodling NS, Wang Q, Panchal
M, Liang X, Trueba-Saiz A, Brown HD, Mhatre SD, Loui T and
Andreasson KI: Prostaglandin signaling suppresses beneficial
microglial function in Alzheimer's disease models. J Clin Invest.
125:350–364. 2015. View
Article : Google Scholar
|