Introduction
Osteoarthritis (OA) is the most common degenerative
joint disease among populations that are past middle age (1). In Asian countries, the incidence of
OA in individuals aged >65 years, will increase from 6.8% in
2008 to 16.2% in 2040 (2).
Although the exact mechanism underlying OA pathogenesis remains
unclear, OA is largely defined by cartilage tissue breakdown
resulting from a dysregulated process of tissue homeostasis that is
crucial to this degenerative joint disease. Elevated levels of
catabolic proteases, such as the matrix metalloproteinases (MMPs)
and inflammatory cytokines, such as interleukin-1β (IL-1β) and
tumor necrosis factor α (TNF-α) contribute significantly to the
breakdown of the cartilage extracellular matrix (ECM) (3–7). The
resulting cleavage and release of matrix molecules, such as
glycosaminoglycans (GAGs), type II collagen and hyaluronan
contribute to the overall loss of healthy cartilage tissue
(3,7). Furthermore, cleavage fragments
generated from the catabolism of these ECM molecules exacerbate
disease conditions by promoting further tissue destruction
(7).
While many members of the pool of catabolic
mediators that are involved with OA pathogenesis have been the
subjects of extensive investigation, stromal cell-derived factor-1
(SDF-1) remains relatively less well-known. SDF-1 is an 8-kDa
peptide that regulates cellular activities via interactions with
C-X-C chemokine receptor type 4 (CXCR4) found on chondrocytes
(8). Synovial fluid SDF-1
concentrations are markedly elevated in rheumatoid arthritic and
osteoarthritic joints (9).
Furthermore, pathological concentrations of SDF-1 induce human
chondrocyte death in vitro (10). Binding of SDF-1 to CXCR4 on
chondrocytes results in the release of the OA-associated catabolic
proteases, MMP-3, -9 and -13 (11). However, the mechanism by which
CXCR4 is regulated in chondrocytes remains to be elucidated.
Previous studies have demonstrated that runt-related transcription
factor 2 (Runx2) regulates MMP-13 expression (12). Increased Runx2 has been found in OA
cartilage, which contributes to the increased expression of MMP-13
in human OA chondrocytes (13).
Recently, Zhu et al (14)
demonstrated that pretreatment of a pluripotent mesenchymal C2C12
cell line with SDF-1 siRNA for 2 days led to a marked decrease in
Runx2 protein concentration. The inhibitory effect of SDF-1 siRNA
was largely reversed by the addition of excess recombinant SDF-1,
suggesting that SDF-1 signaling may regulate Runx2 expression.
In attempting to improve understanding of the
pathophysiology of OA with respect to articular cartilage, it is
critical to recognize that cartilage is inherently avascular and,
as such, has significantly lower levels of oxygen (hypoxic) than
many other tissue types (15–16).
Hypoxia often acts as a regulator of certain molecular markers and
thereby alters specific cellular pathways (17). Hypoxia has been demonstrated to
regulate CXCR4 expression in normal and tumor cells (18–19).
It is therefore likely that hypoxia also regulates CXCR4 expression
in chondrocytes. The molecular mechanism underlying the hypoxic
regulation of the SDF-1/CXCR4 signaling pathway in OA chondrocytes
remains to be elucidated. An improved understanding of this process
may result in novel strategies for pharmacological intervention in
OA. In the present study, the effect of hypoxia on CXCR4 expression
in OA chondrocytes was investigated to elucidate a mechanism
through which SDF-1 induces cartilage degradation. In addition, the
efficacy of the commercially available CXCR4 inhibitor, AMD3100
(20) in reducing OA chondrocyte
catabolism under hypoxic conditions was investigated.
Materials and methods
Chondrocyte isolation and primary
culture
Cartilage was obtained from patients undergoing
total knee replacement surgery at the Second Hospital of Shanxi
Medical University (Taiyuan, China) between September 2012 and
December 2013. Cartilage samples were removed from the tibia
plateau and washed in Gibco Dulbecco's modified Eagle's medium
(DMEM; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Chondrocytes were isolated from the cartilage as previously
described (21). Briefly, small
samples of cartilage were minced, digested with 2 mg/ml pronase
(Roche Diagnostics, Basel, Switzerland) in Gibco Hank's balanced
salt solution (HBSS; Thermo Fisher Scientific, Inc.) for 30 min at
37°C, and washed with DMEM. Cartilage samples were digested with 1
mg/ml bacterial collagenase (Type IA; Sigma-Aldrich, St. Louis, MO,
USA) for 6–8 h at 37°C in a shaker. The enzymatic reaction was
terminated with DMEM containing 10% fetal bovine serum (FBS; Gibco;
Thermo Fisher Scientific, Inc.). Residual multicellular aggregates
were removed by filtration and the cells were washed three times in
DMEM. Chondrocytes were plated in DMEM containing 10% FBS,
Invitrogen L-glutamine (2.5 mM; Thermo Fisher Scientific, Inc.),
and antibiotics (100 U/ml penicillin; 0.1 mg/ml streptomycin). Once
cells reached confluence, they were split once (passage 1) and
grown to confluence again. Cells were plated in 8-well chambers
(Nalge Nunc International Corp, Naperville, IL, USA) at
1×105 cells/well or in 100-mm diameter culture dishes
(BD Biosciences, Franklin Lakes, NJ, US) at 1×106
cells/plate. At 70% confluence, certain cells were treated with 5
ng/ml AMD3100 (ApexBio, Boston, MA, USA), 2 h prior to SDF-1
(GenScript, Piscataway, NJ, USA) incubation. Cells were then
cultured under hypoxic condition (2% O2) for two days.
Cells (5×106) were treated with SDF-1 only (40 ng/ml)
and control cells were cultured in the absence of SDF-1. Cells were
incubated in the culture medium for two days and the chondrocyte
culture medium was replaced with fresh medium without
hyaluronidase. Cells were incubated at 37°C in 2 or 5%
O2.
Cartilage explant organ culture
Cartilage explants of uniform size (1.0×1.0×0.2 cm)
were obtained from patients undergoing total knee replacement
surgery. Each explant was divided into three segments. Explants
were cultured at 2% O2 to mimic hypoxic conditions, as
previously described (22,23). One segment was incubated with
AMD3100 (5 ng/ml) prior to SDF-1 (40 ng/ml) treatment, one segment
was incubated with SDF-1 (40 ng/ml) only and the third segment was
cultured in the absence of SDF-1 in DMEM containing 10% FBS at
37°C. The cartilage and conditioned media were collected two days
subsequently.
Immunocytochemistry
Following a two-day incubation, cells cultured in
8-well chambers were fixed at −20°C with 70% ethanol and 50 mM
glycine (pH 2.0) for 20 min. Slides were then washed with
phosphate-buffered saline (PBS) and incubated with a monoclonal
anti-CXCR4 antibody at 1:500 (cat. no. mab173; R&D Systems,
Inc., Minneapolis, MN, USA) for 1 h at 37°C followed by incubation
with a rhodamine-labeled secondary antibody at 1:1,000 (cat. no.
NL557; R&D Systems, Inc.) for 1 h at room temperature. Hoechst
(0.5 mg/ml), a florescent nuclear binding dye, was applied
following washing with PBS. Slides were then washed and mounted in
95% glycerol in PBS. Single or multiple exposure photography was
performed with a Nikon E800 microscope and Nikon Capture NX2 v2.4.7
(Nikon Inc., Melville, NY, USA).
Quantification of GAGs
To determine the concentration of GAG released by
the cultured cartilage explants, the incubation media of the
explants were collected following AMD3100 and/or SDF-1 treatment,
and GAG concentrations were quantified spectrophotometrically by
the 1,9-dimethylmethylene blue (DMMB) assay, using bovine
chondroitin sulfate (Santa Cruz Biotechnology, Inc., Dallas, Texas,
USA) as a standard, according to a previous study (24).
Western blot analysis
Total protein was extracted from cells and
quantified using the BCA Protein Assay Reagent kit (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) according to the manufacturer's
instructions. Subsequently, 40 µg total protein was
separated by electrophoresis (110 V for 2 h) on a 10% SDS-PAGE gel
before being transferred to a nitrocellulose membrane. To detect
hypoxia-inducible factor 1α (HIF-1α) protein, the membrane was
probed with a rabbit anti-human HIF-1α antibody (cat. no. 3716;
Cell Signaling Technology, Inc., Danvers, MA, USA). The antibody
was diluted 1:1,000 in blocking buffer (LI-COR Biosciences,
Lincoln, NE, USA). Goat anti-rabbit IRDye 680 IgG (cat. no.
926-68,071; LI-COR Biosciences) diluted 1:5,000 in blocking buffer
(LI-COR Biosciences) served as the secondary antibody. To detect
Runx2 protein, the membrane was incubated with a rabbit anti-human
Runx2 polyclonal antibody (cat. no. bs-1134R; Bioss, Inc., Woburn,
MA, USA) diluted 1:1,000 in blocking buffer. Goat anti-rabbit IRDye
800 CW IgG at 1:8,000 (cat. no. 926-32,211; LI-COR Biosciences)
served as the secondary antibody. The blots were detected using the
Odyssey Infrared Imaging system (LI-COR Biosciences). To ensure
even loading of samples, the same membrane was probed with a goat
anti-human β-actin polyclonal antibody (cat. no. SC-1616; Santa
Cruz Biotechnology, Inc.) at a dilution of 1:1,000. The relative
intensities of western blot bands were semi-quantified by
densitometry following normalization to b-actin using ImageJ
software (National Institutes of Health; NIH, Bethesda, MA, USA) as
previously described (25).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RT-qPCR was performed as previously reported
(10). Briefly, total RNA was
isolated from the chondrocytes with the Qiagen RNeasy isolation kit
(Qiagen, Inc., Valencia, CA, USA). A total of 1 µg RNA was
transcribed into cDNA using iScripTM (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The resulting cDNA (40 ng/ml) served as the
template to quantify the relative content of mRNA using a
QuantiTect SYBR Green PCR kit (Qiagen, Inc.) with DNA Engine
Opticon 2 Continuous Fluorescence Detection system. (MJ Research,
Waltham, MA, USA). The amplification conditions were as follows:
Pre-incubation, 2 min at 50°C; enzyme activation, 10 min at 95°C;
denaturation, 40 cycles of 95°C for 10 sec; annealing, 55°C for 30
sec; and extension, 72°C for 30 sec. The primers were designed
using Primers Express software v1.0 (BioTools Inc., Edmonton, AB,
Canada) and were as follows: Forward, 5′-AAACTGAGAAGCATGACGGACAA-3′
and reverse, 5′-GCCAACATAGACCACCTTTTCAG-3′ for CXCR4; forward,
5′-TGCTGCATTCTCCTTCAGGA-3′ and reverse, 5′-ATGCATCCAGGGGTCCTGGC-3′
for MMP-13; and forward, 5′-CTTCGCCGTCCATTCACTCC-3′ and reverse,
5′-GTGCATTCGTGGGTTGGAGA-3′ for Runx2. The 18S ribosomal RNA (rRNA;
forward, 5′-CGGCTACCACATCCAAGGAA-3′ and reverse,
5′-GCTGGAATTACCGCGGCT-3′) was amplified at the same time and served
as an internal control. The cycle threshold values for 18S rRNA,
CXCR4, MMP-13 and Runx2 were measured and calculated by IQ5 Optical
System software, version 2.1 (Bio-Rad Laboratories, Inc.,).
Relative transcript levels were calculated as x =
2-ΔΔCq, in which ΔΔCq = ΔE-DC, and ΔE =
Cqexp-Cq18s; ΔC = Cqcontrol-Cq18s
(26).
ELISA
MMP-13 activity in conditioned media was measured
using a double-antibody sandwich ELISA kit (MMP-13 ELISA kit; GE
Healthcare Life Sciences, Chalfont, UK). Assay Diluent RD1-55 (100
µl) was added to each well with 100 µl standard or
sample. The wells were covered with an adhesive strip and incubated
for 2 h at room temperature on a horizontal orbital microplate
shaker set at 500 rpm. Each well was aspirated and washed three
times with 400 µl wash buffer. A total of 200 µl
MMP-13 conjugate was added to each well, covered with a fresh
adhesive strip, and incubated for 2 h at room temperature on the
shaker. The aspiration and washing steps were repeated and 200
µl substrate solution was added to each well and incubated
for 30 min at room temperature in the dark. Stop solution (50
µl) was added to each well and the optical density of each
well was determined within 30 min using a microplate reader
(Packard FluoroCount BF10000) set at 450 nm. IL-1β in conditioned
media collected from cartilage explants was measured using an IL-1β
ELISA kit from Invitrogen (cat. no. KMC0011C; Thermo Fisher
Scientific, Inc.). Culture medium (50 µl) was mixed with 50
µl incubation buffer and 50 µl biotinylated
anti-IL-1β (cat. no. KMC0011C; Thermo Fisher Scientific, Inc.)
solution in the wells. Following incubation at 37°C for 90 min, 100
µl streptavidin-horseradish peroxidase working solution was
added to the wells and incubated at room temperature for 30 min.
The wells were washed with washing solution four times. Following
the addition of 100 µl stabilized chromogen to each well,
the plate was placed in the dark for 30 min at room temperature.
Stop solution (100 µl) was then added and the absorbance was
recorded at 450 nm. All samples were run in duplicate.
Statistical analysis
All quantitative data were expressed as means ±
standard deviation, obtained from at least three independent
experiments and analyzed with SPSS software (version 20.0; IBM
SPSS, Armonk, NY, USA). Independent student's t test was
used for RT-qPCR quantification of CXCR4 expression in
chondrocytes; one-way analysis of variance with Tukey's post-hoc
test was used for group comparisons in all other experiments.
P<0.05 was considered to indicate a statistically significant
difference.
Results
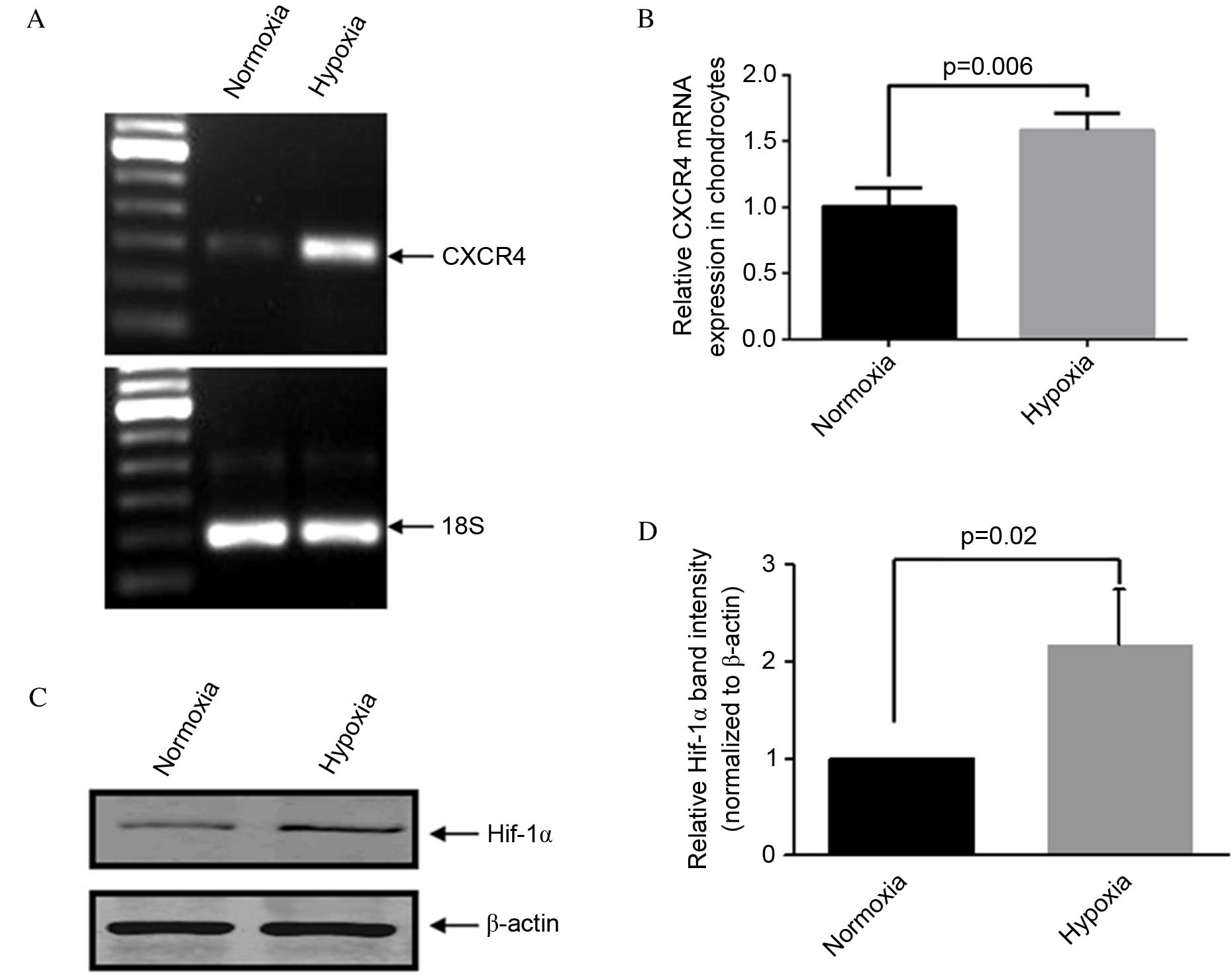
Hypoxia enhances CXCR4 expression in
human OA chondrocytes
Consistent with previously established methods
(22,23), OA articular chondrocytes were
maintained in 5% O2 (normoxia) or exposed to 2%
O2 (hypoxia) for two days. RT-qPCR analysis demonstrated
that CXCR4 expression was significantly higher in OA chondrocytes
cultured under hypoxic compared with normoxic conditions (P=0.006;
Fig. 1A and B). Protein
quantification of HIF-1α via western blotting was performed to
confirm that conditions were hypoxic in the in vitro model
(P=0.02; Fig. 1C and D).
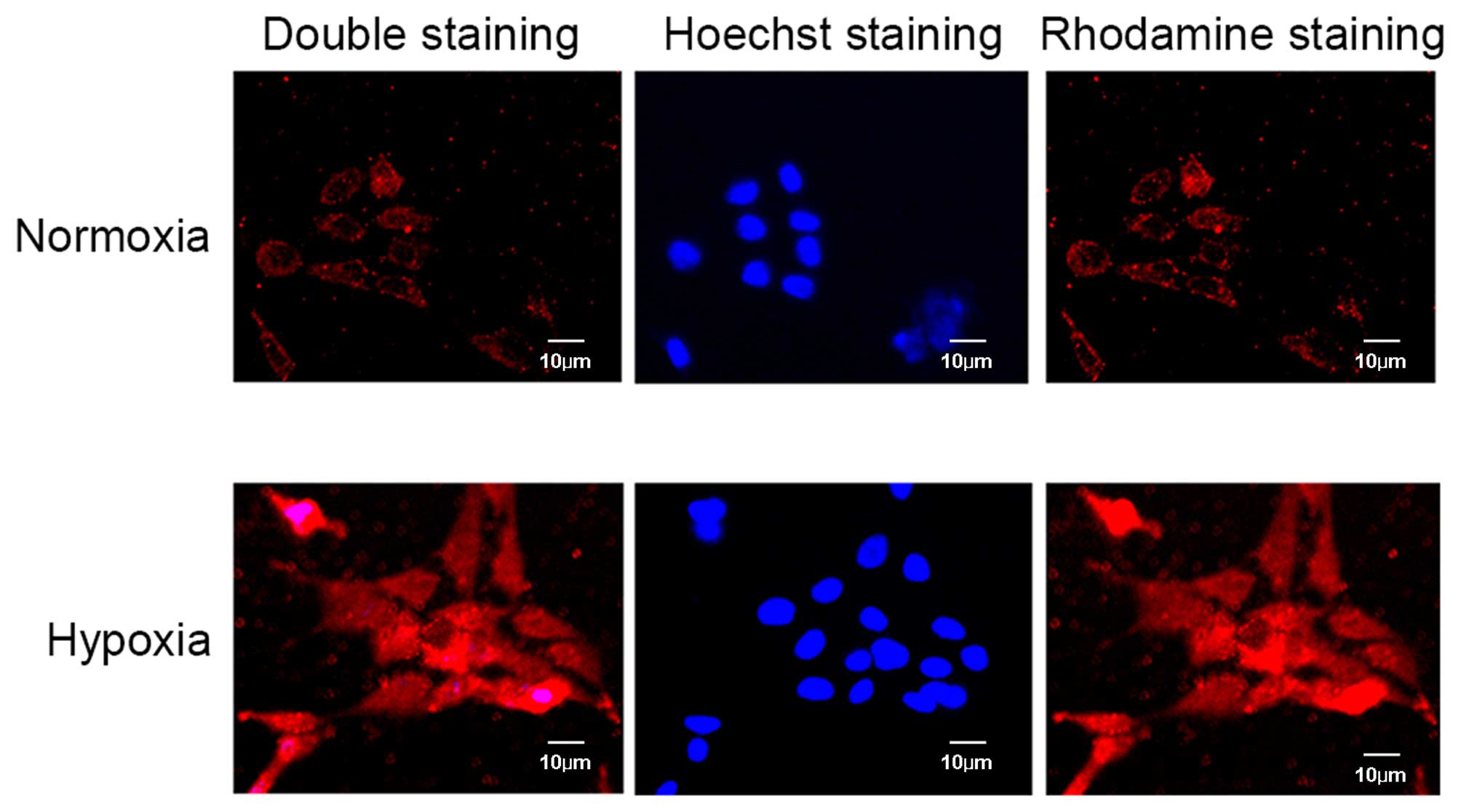
Micrographs of immunocytochemical analysis further identified
strong CXCR4 positive staining in hypoxic conditions compared with
normoxic conditions (Fig. 2).
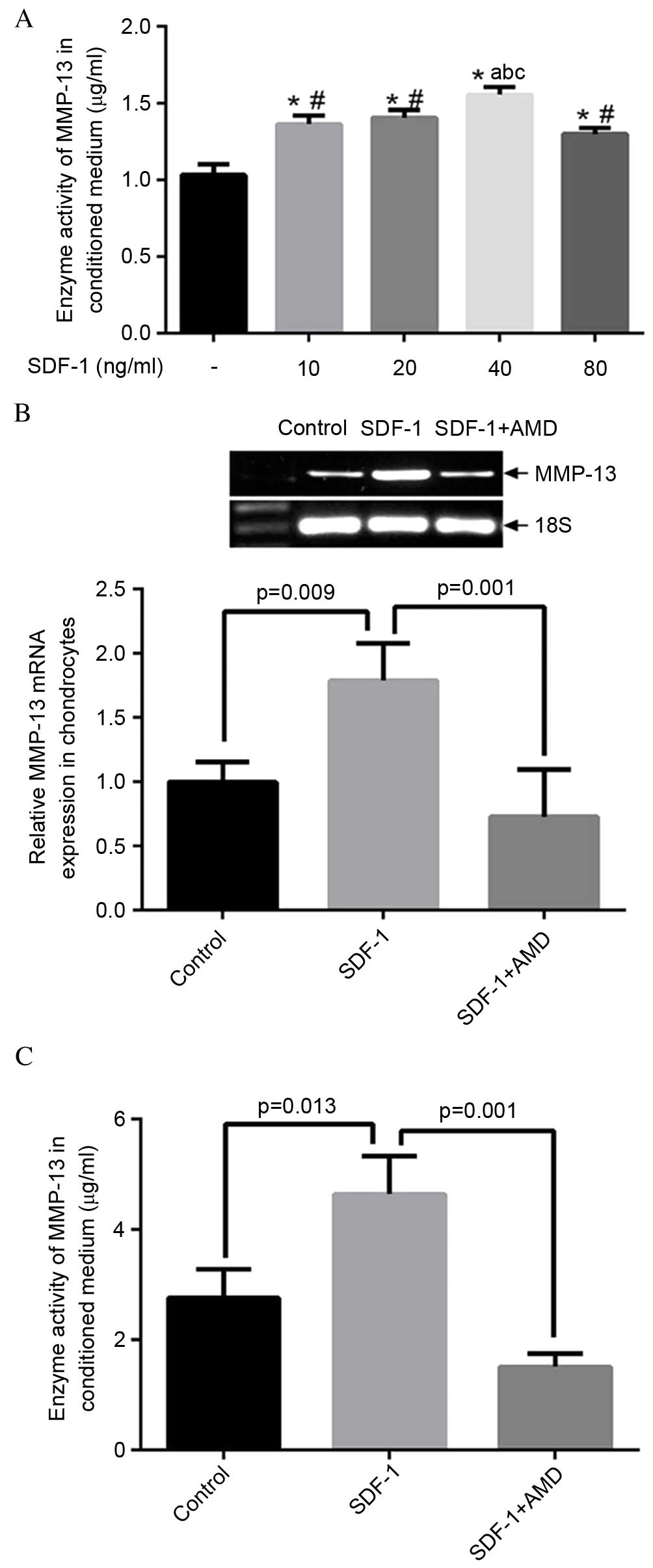
SDF-1 increases MMP-13 expression level
and activity in human OA chondrocyte
The conditioned media were collected for ELISA
analysis 48 h following treatment with SDF-1. As presented in
Fig. 3A, SDF-1 (incubated at
concentrations of 10-80 ng/ml) increased MMP-13 activity in a
dose-dependent manner. Notably, the effect of SDF-1 was greatest at
a dosage of 40 ng/ml. RT-qPCR and ELISA results indicated that
SDF-1 increased the expression level of MMP-13 (P=0.009; Fig. 3B), while suppression of MMP-13
enzyme activity was observed by blocking the SDF-1 pathway with
AMD3100 (P=0.001; Fig. 3C).
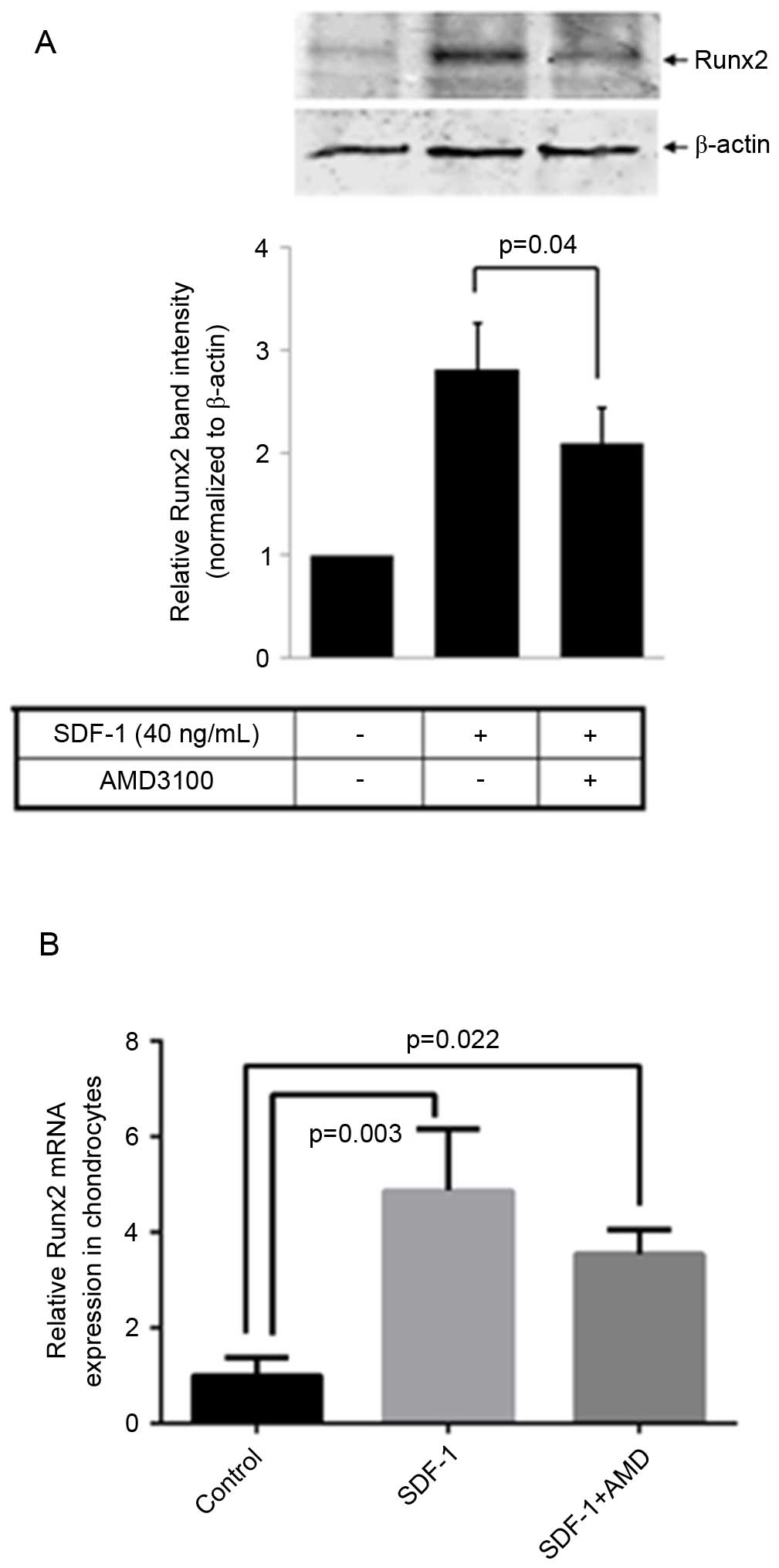
SDF-1 increases Runx2 expression levels
in human cartilage chondrocytes
Total RNA and cell lysates were collected following
48 h treatment with SDF-1 alone or SDF-1 plus AMD3100. Controls
were cultured in the absence of SDF-and AMD3100. Runx2 mRNA and
protein expression levels were determined by RT-qPCR and western
blotting, respectively. The results indicated that SDF-1 increased
Runx2 mRNA (P=0.003) and protein expression levels, while the
upregulation of Runx2 induced by SDF-1 was partially inhibited by
AMD3100 (P=0.04; Fig. 4A and
B).
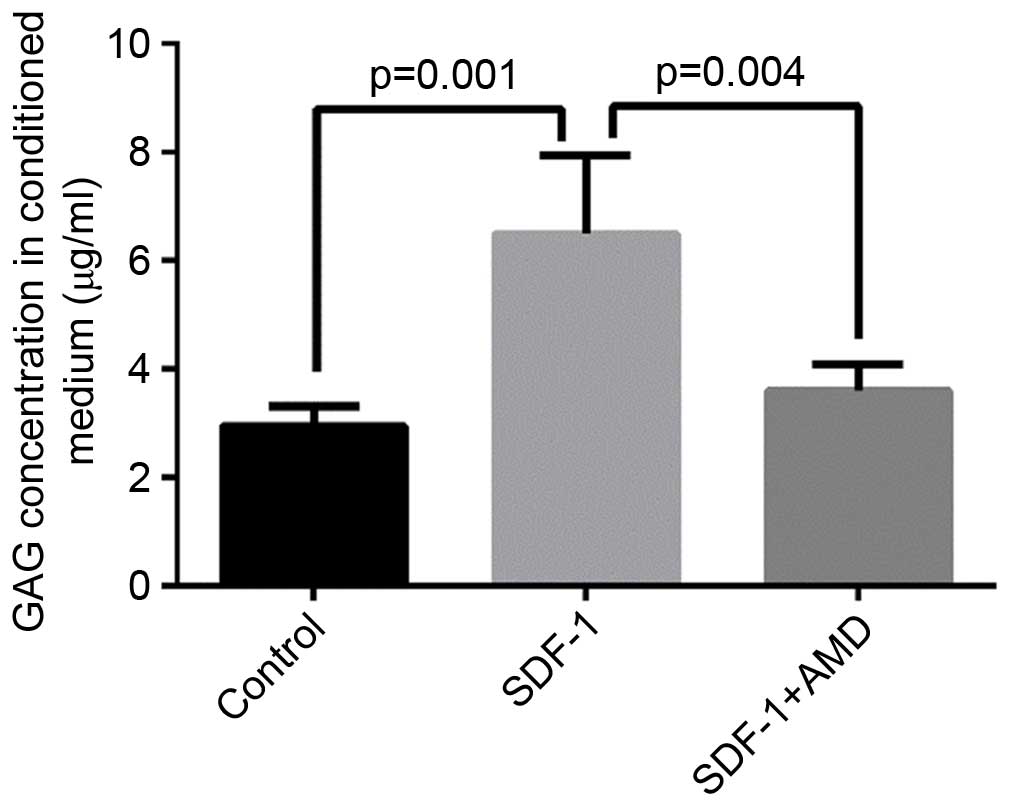
SDF-1 increases GAG release in the
conditioned media
GAG is an important structural component of
proteoglycans and its release is a marker of proteoglycan
catabolism (27). To further
examine whether SDF-1 causes cartilage matrix degradation, the
concentration of GAG in the conditioned media of explants treated
with SDF-1 was determined by spectrophotometry using a DMMB assay,
in which bovine chondroitin sulfate served as a standard. The level
of GAG was significantly higher in media collected from explants
treated with SDF-1 vs. explants cultured in the absence of SDF-1
(P=0.001) or explants pretreated with AMD3100 prior to SDF-1
incubation (P=0.004; Fig. 5).
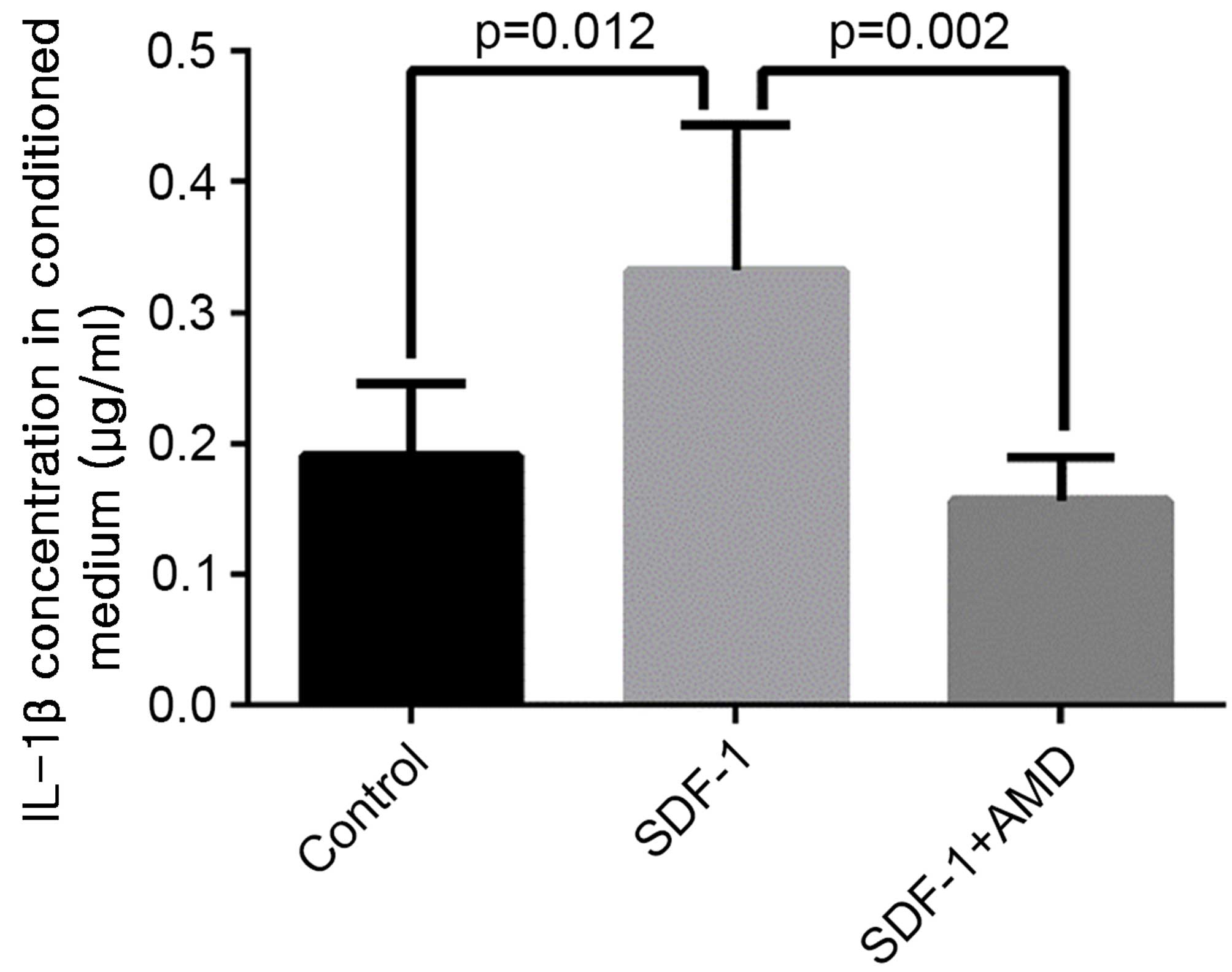
SDF-1 increases IL-1β release in the
conditioned media
The level of IL-1β was significantly higher in media
collected from explants treated with SDF-1 vs. explants cultured in
the absence of SDF-1 (P=0.012) or explants treated with AMD3100
prior to SDF-1 incubation (P=0.002; Fig. 6).
Discussion
OA is a complex disease and its underlying
pathological mechanisms remain to be elucidated. Genetic,
biochemical and biomechanical factors all contribute to the
pathogenesis of OA (28–30). It is well established that the
inflammatory cytokines, IL-1β and TNF-α are closely associated with
OA-induced cartilage degradation (3,7).
Previous studies have suggested that the SDF-1/CXCR4 signaling
pathway is also an important facet of articular cartilage biology
and may be involved in the pathogenesis of OA (11,31).
SDF-1 belongs to the CXC chemokine family and CXCR4 is a G-protein
coupled receptor that, when activated by SDF-1, triggers a cascade
of molecular events that ultimately regulate many important
cellular functions, including chemotaxis, proliferation, apoptosis
and differentiation (32).
Increased expression of CXCR4 is a defining feature of hypertrophic
chondrocytes (8). Furthermore,
premature terminal differentiation of chondrocytes is indicative of
OA pathogenesis as OA cartilage often consists of large zones of
hypertrophic tissue (33,34).
The aim of the present study was to investigate the
role of CXCR4 activation in the context of OA chondrocyte and
cartilage tissue catabolism. In the current study, cells were
cultured under hypoxic conditions to mimic the physiological state
of hypoxia, a defining characteristic of cartilage tissue, to
examine how these conditions affect CXCR4 expression in OA
chondrocytes. It was demonstrated that hypoxia significantly
increases the CXCR4 expression level in human OA chondrocytes,
which is consistent with the findings of a previous study in which
hypoxia increased CXCR4 expression in a chondrosarcoma cell line
(22). As it has been previously
suggested that synovial fibroblasts exhibit increased SDF-1
production during OA conditions (9), the results of the present study
suggest that the additional insult of hypoxia-induced CXCR4
elevation in chondrocytes may contribute to hyper-activation of the
SDF-1/CXCR4 signaling pathway, thus favoring cartilage catabolism.
To test this hypothesis, the effect of treating OA chondrocytes
cultured in hypoxic conditions with SDF-1 was examined, and
observed to increase MMP-13 mRNA expression and enzyme activity.
This is consistent with a previous study, which demonstrated that
SDF-1 increased MMP-3 and -13 levels in a dose-dependent manner,
although this study was not conducted under hypoxic conditions
(11). Increased MMP-13 production
and activity is a canonical indicator of OA-induced cartilage
catabolism, as it degrades type II collagen, a major building block
of the cartilage ECM (10,35,36).
MMP-13 has been reported to be overexpressed in OA cartilage, and
is a key inducer of events associated with OA (6,20,37).
Consistent with previous findings, increased mRNA
and protein expression levels of Runx2, a well-known regulator of
MMP-13 (13,38–41),
were observed in the present study in response to SDF-1 treatment
of OA chondrocytes (13). Runx2 is
an important transcription factor regulating cartilage maturation
and bone formation (42,43), and it has previously been linked to
chondrocyte hypertrophy, which often occurs in OA. Notably, a
heterozygous Runx2-deficient (Runx2+/−) OA mouse model
demonstrated decreased cartilage destruction, along with a
reduction in MMP-13 expression (44). Furthermore, Runx2 directly
upregulates MMP-13 by binding to a particular C-module-binding
factor/nuclear matrix protein-2/osteoblastic cis-element 2
element in the promoter region of the MMP-13 gene (12,45).
The concomitant upregulation of Runx2 and MMP-13 in the presence of
SDF-1 suggests that SDF-1 signaling is a prerequisite for
Runx2-regulated MMP-13 expression in OA chondrocytes, and that at
least a component of the function of SDF-1/CXCR4 is exerted through
Runx2-mediated regulation of MMP-13 expression. This is further
substantiated by a recent study which demonstrated that knockdown
of SDF-1 signaling in mesenchymal stem/progenitor cells undergoing
chondrogenesis results in significant suppression of Runx2
expression (46).
In addition to increasing Runx2 expression levels
and MMP-13 activity under hypoxic conditions, SDF-1 increased IL-1β
production and GAG release into the conditioned media of cartilage
explants. This suggests that SDF-1 may induce mediators of
OA-associated cartilage catabolism, and in this respect it may be
functionally involved in OA pathogenesis. This is consistent with a
previous study that reported elevated SDF-1 levels in the synovial
fluid of OA patients (9).
Furthermore, while it is reasonable to expect that the state of
hypoxia that is inherent to cartilage tissue elevates CXCR4
expression in chondrocytes, the results of the present study
demonstrate for the first time, to the best of our knowledge, that
AMD3100 effectively prevents the aforementioned catabolic effects
of SDF-1. AMD3100 is a specific inhibitor of CXCR4 that was first
recognized for its potent activity against the HIV infection
(47). A previous study identified
AMD3100 to be effective in treating autoimmune collagen-induced
arthritis in interferon γ receptor-deficient DBA/1 mice (48). The present study reports the novel
discovery that blocking CXCR4 using AMD3100 effectively reduces the
expression of Runx2, inhibits MMP-13 activity and decreases GAG
release under hypoxic conditions. The present study provides
insight into the mechanism of OA pathogenesis, and suggests that
the SDF-1/CXCR4 signaling pathway is hyper-activated in a hypoxic
environment, such as the articular cartilage and induces cartilage
matrix degradation via Runx2-mediated MMP-13 release. Furthermore,
the present study has demonstrated for the first time, to the best
of our knowledge, that SDF-1 stimulates the release of the
inflammatory cytokine, IL-1β from OA cartilage explants; an effect
that is abolished by the CXCR4 inhibitor, AMD3100. Based on the
findings of the present study, blocking the SDF-1/CXCR4 signaling
pathway by AMD3100 may potentially prevent OA progression.
Previous studies have demonstrated that animals
tolerated AMD3100 well at a dose of 160 µg/per day, a dose
that has been reported safe in animals and humans (49–51).
The severity of OA cartilage degeneration is attenuated by blocking
SDF-1/CXCR4 signaling by continuous infusion of the CXCR4 blocker,
AMD3100 via osmotic minipump, i.p (31). These data present a novel
therapeutic target for the prevention and treatment of OA. However,
the potential risk of intra-articular injection of AMD3100 has not
been evaluated. Further research is required to evaluate the
potential risk of intra-articular injection of AMD3100 and to
establish a method of ensuring AMD3100 is released slowly.
In conclusion, the findings of the present study
indicate that hypoxia upregulates CXCR4 expression in OA
chondrocytes, that SDF-1/CXCR4 may be crucial in OA pathogenesis
and that AMD3100 may abolish SDF-1-induced catabolic effects. Thus,
blocking SDF-1 interaction with its receptor, CXCR4 may be
applicable to developing chondro-protective therapy to treat
inflammatory chemokine-induced arthritis. Further studies
addressing these points are necessary to evaluate the therapeutic
value of AMD3100 in large animal models of OA prior to clinical
development.
Acknowledgments
The present study was supported by grants from the
following: NIH/National Institute of Arthritis and Musculoskeletal
and Skin Diseases (grant no. R01AR059142), NIH/National Center for
Research Resources (grant no. P20RR024484), National Natural
Science Foundation of China (grant nos. 81171676, 31271033 and
81572098), Shanxi Province Science and Technology Research Projects
(grant no. 20150313012-6), The Second Hospital of Shanxi Medical
University Doctor Funds (grant no. 20140406) and Research Project
Supported by Shanxi Scholarship Council of China (grant no.
2016-122). The content is solely the responsibility of the authors
and does not necessarily represent the official view of the NIH.
The authors gratefully acknowledge Dr Ericka M. Bueno, for help
with the paper preparation and editorial services.
References
|
1
|
Arden N and Nevitt MC: Osteoarthritis:
Epidemiology. Best Pract Res Clin Rheumatol. 20:3–25. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Miller ME, Rejeski WJ, Messier SP and
Loeser RF: Modifiers of change in physical functioning in older
adults with knee pain: The Observational Arthritis Study in Seniors
(OASIS). Arthritis Rheum. 45:331–339. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Martel-Pelletier J, Boileau C, Pelletier
JP and Roughley PJ: Cartilage in normal and osteoarthritis
conditions. Best Pract Res Clin Rheumatol. 22:351–384. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aida Y, Maeno M, Suzuki N, Shiratsuchi H,
Motohashi M and Matsumura H: The effect of IL-1beta on the
expression of matrix metalloproteinases and tissue inhibitors of
matrix metalloproteinases in human chondrocytes. Life Sci.
77:3210–3221. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Daheshia M and Yao JQ: The interleukin
1beta pathway in the pathogenesis of osteoarthritis. J Rheumatol.
35:2306–2312. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mitchell PG, Magna HA, Reeves LM,
Lopresti-Morrow LL, Yocum SA, Rosner PJ, Geoghegan KF and Hambor
JE: Cloning, expression, and type II collagenolytic activity of
matrix metal-loproteinase-13 from human osteoarthritic cartilage. J
Clin Invest. 97:761–768. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jayasuriya CT, Goldring MB, Terek R and
Chen Q: Matrilin-3 induction of IL-1 receptor antagonist is
required for up-regulating collagen II and aggrecan and
down-regulating ADAMTS-5 gene expression. Arthritis Res Ther.
14:R1972012. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wei L, Kanbe K, Lee M, Wei X, Pei M, Sun
X, Terek R and Chen Q: Stimulation of chondrocyte hypertrophy by
chemokine stromal cell-derived factor 1 in the chondro-osseous
junction during endochondral bone formation. Dev Biol. 341:236–245.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kanbe K, Takagishi K and Chen Q:
Stimulation of matrix metalloprotease 3 release from human
chondrocytes by the interaction of stromal cell-derived factor 1
and CXC chemokine receptor 4. Arthritis Rheum. 46:130–137. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wei L, Sun X, Kanbe K, Wang Z, Sun C,
Terek R and Chen Q: Chondrocyte death induced by pathological
concentration of chemokine stromal cell-derived factor-1. J
Rheumatol. 33:1818–1826. 2006.PubMed/NCBI
|
|
11
|
Kanbe K, Takemura T, Takeuchi K, Chen Q,
Takagishi K and Inoue K: Synovectomy reduces stromal-cell-derived
factor-1 (SDF-1) which is involved in the destruction of cartilage
in osteoarthritis and rheumatoid arthritis. J Bone Joint Surg Br.
86:296–300. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jiménez MJ, Balbín M, López JM, Alvarez J,
Komori T and López-Otín C: Collagenase 3 is a target of Cbfa1, a
transcription factor of the runt gene family involved in bone
formation. Mol Cell Biol. 19:4431–4442. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang X, Manner PA, Horner A, Shum L, Tuan
RS and Nuckolls GH: Regulation of MMP-13 expression by RUNX2 and
FGF2 in osteoarthritic cartilage. Osteoarthritis Cartilage.
12:963–973. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhu W, Boachie-Adjei O, Rawlins BA,
Frenkel B, Boskey AL, Ivashkiv LB and Blobel CP: A novel regulatory
role for stromal-derived factor-1 signaling in bone morphogenic
protein-2 osteogenic differentiation of mesenchymal C2C12 cells. J
Biol Chem. 282:18676–18685. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lund-Olesen K: Oxygen tension in synovial
fluids. Arthritis Rheum. 13:769–776. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou S, Cui Z and Urban JP: Factors
influencing the oxygen concentration gradient from the synovial
surface of articular cartilage to the cartilage-bone interface: A
modeling study. Arthritis Rheum. 50:3915–3924. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Koay EJ and Athanasiou KA: Hypoxic
chondrogenic differentiation of human embryonic stem cells enhances
cartilage protein synthesis and biomechanical functionality.
Osteoarthritis Cartilage. 16:1450–1456. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schioppa T, Uranchimeg B, Saccani A,
Biswas SK, Doni A, Rapisarda A, Bernasconi S, Saccani S, Nebuloni
M, Vago L, et al: Regulation of the chemokine receptor CXCR4 by
hypoxia. J Exp Med. 198:1391–1402. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Staller P, Sulitkova J, Lisztwan J, Moch
H, Oakeley EJ and Krek W: Chemokine receptor CXCR4 downregulated by
von Hippel-Lindau tumour suppressor pVHL. Nature. 425:307–311.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
De Clercq E: The bicyclam AMD3100 story.
Nat Rev Drug Discov. 2:581–587. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pulsatelli L, Dolzani P, Piacentini A,
Silvestri T, Ruggeri R, Gualtieri G, Meliconi R and Facchini A:
Chemokine production by human chondrocytes. J Rheumatol.
26:1992–2001. 1999.PubMed/NCBI
|
|
22
|
Sun X, Wei L, Chen Q and Terek RM:
CXCR4/SDF1 mediate hypoxia induced chondrosarcoma cell invasion
through ERK signaling and increased MMP1 expression. Mol Cancer.
9:172010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sun X, Charbonneau C, Wei L, Yang W, Chen
Q and Terek RM: CXCR4-targeted therapy inhibits VEGF expression and
chondrosarcoma angiogenesis and metastasis. Mol Cancer Ther.
12:1163–1170. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Farndale RW, Buttle DJ and Barrett AJ:
Improved quantitation and discrimination of sulphated
glycosaminoglycans by use of dimethylmethylene blue. Biochim
Biophys Acta. 883:173–177. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guan YJ, Yang X, Wei L and Chen Q:
MiR-365: A mechanosensitive microRNA stimulates chondrocyte
differentiation through targeting histone deacetylase 4. FASEB J.
25:4457–4466. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
27
|
Lohmander S: Proteoglycans of joint
cartilage. Structure, function, turnover and role as markers of
joint disease. Baillieres Clin Rheumatol. 2:37–62. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Krasnokutsky S, Attur M, Palmer G, Samuels
J and Abramson SB: Current concepts in the pathogenesis of
osteoarthritis. Osteoarthritis Cartilage. (16 Suppl 3): S1–S3.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Goldring MB: Osteoarthritis and cartilage:
The role of cytokines. Curr Rheumatol Rep. 2:459–465. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guilak F: Biomechanical factors in
osteoarthritis. Best Pract Res Clin Rheumatol. 25:815–823. 2011.
View Article : Google Scholar
|
|
31
|
Wei F, Moore DC, Li Y, Zhang G, Wei X, Lee
JK and Wei L: Attenuation of osteoarthritis via blockade of the
SDF-1/CXCR4 signaling pathway. Arthritis Res Ther. 14:R1772012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ganju RK, Brubaker SA, Meyer J, Dutt P,
Yang Y, Qin S, Newman W and Groopman JE: The alpha-chemokine,
stromal cell-derived factor-1alpha, binds to the transmembrane
G-protein-coupled CXCR-4 receptor and activates multiple signal
transduction pathways. J Biol Chem. 273:23169–23175. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dreier R: Hypertrophic differentiation of
chondrocytes in osteoarthritis: The developmental aspect of
degenerative joint disorders. Arthritis Res Ther. 12:2162010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
van der Kraan PM and van den Berg WB:
Chondrocyte hypertrophy and osteoarthritis: Role in initiation and
progression of cartilage degeneration? Osteoarthritis Cartilage.
20:223–232. 2012. View Article : Google Scholar
|
|
35
|
Massova I, Kotra LP, Fridman R and
Mobashery S: Matrix metalloproteinases: Structures, evolution, and
diversification. FASEB J. 12:1075–1095. 1998.PubMed/NCBI
|
|
36
|
Knäuper V, Will H, López-Otin C, Smith B,
Atkinson SJ, Stanton H, Hembry RM and Murphy G: Cellular mechanisms
for human procollagenase-3 (MMP-13) activation. Evidence that
MT1-MMP (MMP-14) and gelatinase a (MMP-2) are able to generate
active enzyme. J Biol Chem. 271:17124–17131. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Billinghurst RC, Dahlberg L, Ionescu M,
Reiner A, Bourne R, Rorabeck C, Mitchell P, Hambor J, Diekmann O,
Tschesche H, et al: Enhanced cleavage of type II collagen by
collagenases in osteoarthritic articular cartilage. J Clin Invest.
99:1534–1545. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Enomoto H, Enomoto-Iwamoto M, Iwamoto M,
Nomura S, Himeno M, Kitamura Y, Kishimoto T and Komori T: Cbfa1 is
a positive regulatory factor in chondrocyte maturation. J Biol
Chem. 275:8695–8702. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ueta C, Iwamoto M, Kanatani N, Yoshida C,
Liu Y, Enomoto-Iwamoto M, Ohmori T, Enomoto H, Nakata K, Takada K,
et al: Skeletal malformations caused by overexpression of Cbfa1 or
its dominant negative form in chondrocytes. J Cell Biol.
153:87–100. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kim IS, Otto F, Zabel B and Mundlos S:
Regulation of chondrocyte differentiation by Cbfa1. Mech Dev.
80:159–170. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zheng Q, Zhou G, Morello R, Chen Y,
Garcia-Rojas X and Lee B: Type X collagen gene regulation by Runx2
contributes directly to its hypertrophic chondrocyte-specific
expression in vivo. J Cell Biol. 162:833–842. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ducy P, Zhang R, Geoffroy V, Ridall AL and
Karsenty G: Osf2/Cbfa1: A transcriptional activator of osteoblast
differentiation. Cell. 89:747–754. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Karsenty G and Wagner EF: Reaching a
genetic and molecular understanding of skeletal development. Dev
Cell. 2:389–406. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kamekura S, Kawasaki Y, Hoshi K, Shimoaka
T, Chikuda H, Maruyama Z, Komori T, Sato S, Takeda S, Karsenty G,
et al: Contribution of runt-related transcription factor 2 to the
pathogenesis of osteoarthritis in mice after induction of knee
joint instability. Arthritis Rheum. 54:2462–2470. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pendás AM, Balbín M, Llano E, Jiménez MG
and López-Otín C: Structural analysis and promoter characterization
of the human collagenase-3 gene (MMP13). Genomics. 40:222–233.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Guang LG, Boskey AL and Zhu W: Regulatory
role of stromal cell-derived factor-1 in bone morphogenetic
protein-2-induced chondrogenic differentiation in vitro. Int J
Biochem Cell Biol. 44:1825–1833. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
De Clercq E, Yamamoto N, Pauwels R, Baba
M, Schols D, Nakashima H, Balzarini J, Debyser Z, Murrer BA,
Schwartz D, et al: Potent and selective inhibition of human
immunodeficiency virus (HIV)-1 and HIV-2 replication by a class of
bicyclams interacting with a viral uncoating event. Proc Natl Acad
Sci USA. 89:5286–5290. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ma Q, Jones D, Borghesani PR, Segal RA,
Nagasawa T, Kishimoto T, Bronson RT and Springer TA: Impaired
B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron
migration in CXCR4- and SDF-1-deficient mice. Proc Natl Acad Sci
USA. 95:9448–9453. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Matthys P, Hatse S, Vermeire K, Wuyts A,
Bridger G, Henson GW, De Clercq E, Billiau A and Schols D: AMD3100,
a potent and specific antagonist of the stromal cell-derived
factor-1 chemokine receptor CXCR4, inhibits autoimmune joint
inflammation in IFN-gamma receptor-deficient mice. J Immunol.
167:4686–4692. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hendrix CW, Flexner C, MacFarland RT,
Giandomenico C, Fuchs EJ, Redpath E, Bridger G and Henson GW:
Pharmacokinetics and safety of AMD-3100, a novel antagonist of the
CXCR-4 chemokine receptor, in human volunteers. Antimicrob Agents
Chemother. 44:1667–1673. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hendrix CW, Collier AC, Lederman MM,
Schols D, Pollard RB, Brown S, Jackson JB, Coombs RW, Glesby MJ,
Flexner CW, et al: Safety, pharmacokinetics, and antiviral activity
of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1
infection. J Acquir Immune Defic Syndr. 37:1253–1262. 2004.
View Article : Google Scholar : PubMed/NCBI
|