Introduction
Citrin deficiency (CD) is an autosomal recessive
disorder caused by biallelic mutations in the SLC25A13 gene,
which encodes citrin, the aspartate/glutamate carrier isoform 2
(AGC2) (1–3). In 1999, Kobayashi et al
(4) cloned the SLC25A13
gene, providing a foundation for subsequent investigations of CD.
An increasing number of patients have been diagnosed with CD, not
only in Asia (5–16), but also in Europe (17–20)
and North America (21–24). Currently, CD has been recognized as
a worldwide panethnic disease.
Neonatal intrahepatic cholestasis caused by CD
(NICCD) is a CD phenotype found in neonates and infants, and its
diagnosis relies on the genetic analysis of SLC25A13
(2). However, conventional DNA
analytic approaches, including PCR and Sanger sequencing, cannot
identify all SLC25A13 mutations (25). Obscure mutations are usually large
insertions/deletions (12,24,26),
and the early diagnoses of patients with these types of mutations
require time-consuming and expensive experimental techniques, which
presents a constant challenge. In the present study, a novel large
SLC25A13 deletion was identified in a patient with NICCD
using a diverse range of tools, including cDNA cloning analysis of
the SLC25A13 gene in the peripheral blood lymphocytes (PBLs)
of the infant. The present study reports on the molecular and
clinical findings.
Materials and methods
Subjects and ethical statement
A 6-month-old male infant who was referred to
Department of Pediatrics (First Affiliated Hospital, Jinan
University, Gangzhou, China) on May 20, 2014 and his parents were
enrolled as research subjects. The clinical findings were described
as a case report. The data were predominantly collected from the
Department of Pediatrics and biochemical analysis was performed in
the Department of Laboratory Science. However, partial biochemical
or imaging results from the Children's Hospital Affiliated to the
Capital Institute of Pediatrics (Beijing, China) were provided by
the parents upon request. Written informed consent was obtained
from parents prior to the investigation, which was approved by the
Committee for Medical Ethics of the First Affiliated Hospital,
Jinan University (Guangzhou, China).
Detection of high-frequency mutations
and Sanger sequencing
As described previously (10–12,16),
peripheral blood samples were collected from the subjects, and DNA
was extracted according to the instructions of the genomic DNA
extraction kit (Simgen, Hangzhou, China). The four high-frequency
SLC25A13 mutations c.851_854del4, c.1638_1660dup,
c.615+5G>A and IVS16ins3 kb, were screened using polymerase
chain reaction (PCR)/long and accurate (LA)-PCR and PCR-restriction
fragment length polymorphism procedures. All 18 exons and their
flanking sequences in the SLC25A13 gene were then sequenced
to identify the possible mutations. All PCR amplification in the
present study was conducted by using a Mastercycler nexus PCR
instrument (Eppendorf, Hamburg, Germany) and Sanger sequencing was
performed using a 96-capillary ABI 3730xl DNA Analyzer with BigDye
Terminator version 3.1 Cycle Sequencing kit (Thermo Fisher
Scientific, Waltham, MA, USA), according to the manufacturer's
protocol.
Reverse transcription (RT)-PCR and cDNA cloning
analysis. The cDNA cloning was performed as in our previous study
(12,27). Briefly, PBLs were isolated from the
heparinized blood samples of the infant using lymphocyte separation
medium (ICN Biomedicals, Santa Ana, CA, USA). RNAiso Plus (Takara
Bio, Inc., Otsu, Japan) was then used to extract the total RNA.
Following this procedure, RT-nested PCR was performed. The cDNAs
were synthesized from 2 µg of total RNA in the presence of primer
UP-dT (5′-CGGCAGTGGTATCAACGCAGAGTAC(T)18-3′) and 200U
MMLV reverse transcriptase (Promega, Beijing, China). The first PCR
was carried out in a 50 µl volume containing 10µl of 5 × PrimeSTAR
buffer (Mg2+ plus), 0.5 µl of PrimeSTAR HS DNA
polymerase (2.5 U/µl, Takara Bio, Inc., Dalian, Liaoning, China),
1µl of cDNA, 1µl of each primer RAS2
(5′-AACGCACGCTGCCTGGCCGTATC-3′) and RACEA1
(5′-CCACCTTCACAAATTCATGCGCC-3′; 20µM), 4 µl of dNTP mixture (10 mM)
and 32.5 µl PCR-grade water. Following initial denaturation at 94°C
for 3 min, 20 cycles of DNA amplification were performed (98°C for
10 sec, 60°C for 15 sec, 72°C for 4 min), followed by a terminal
extension at 72°C for 7 min. Subsequently, 1 µl of the first PCR
product was subjected to the second PCR for 30 cycles in 50 µl
mixture including 10 µl of 5 × PrimeSTAR buffer (Mg2+
plus), 0.5 µl of PrimeSTAR HS DNA polymerase (2.5 U/µL, Takara Bio,
Inc), 4 µl of dNTP mixture (10 mM), 1 µl of each primer RAS3
(5′-GCCGCCGGGACTAGAAGTGAGC-3′) and Ex18R
(5′-TGCTTCATTCCCAGGAGGGA-3′; 20 µM), and 32.5 µl PCR-grade water.
The PCR thermocycling conditions were: 94°C for 3 min followed by
30 cycles of 98°C for 10 sec, 60°C for 5 sec, 72°C for 2.5 min and
a final extension step at 72°C for 10 min. The products were then
purified by a gel extraction kit (Omega Bio-Tek, Inc., Norcross,
GA, USA) and cloned into the pMD™18-T Vector (Takara Bio, Inc.),
following which they were transformed into DH5α Escherichia
coli competent cells by means of heat shock and cultured for
12–16 h at 37°C. Positive clones were subsequently selected and the
SLC25A13 transcripts were sequenced. The sequencing results
were aligned with the SLC25A13 cDNA sequence, which was
available at http://www.ensembl.org/ by using a
DNAman software version 7.212 (Lynnon Corporation, QC, Canada).
LA-PCR approach
Based upon the findings of the cDNA cloning and
sequencing, the c.851_854del4 mutation was used as a marker for
identifying the obscure mutation in the DNA samples. For LA-PCR,
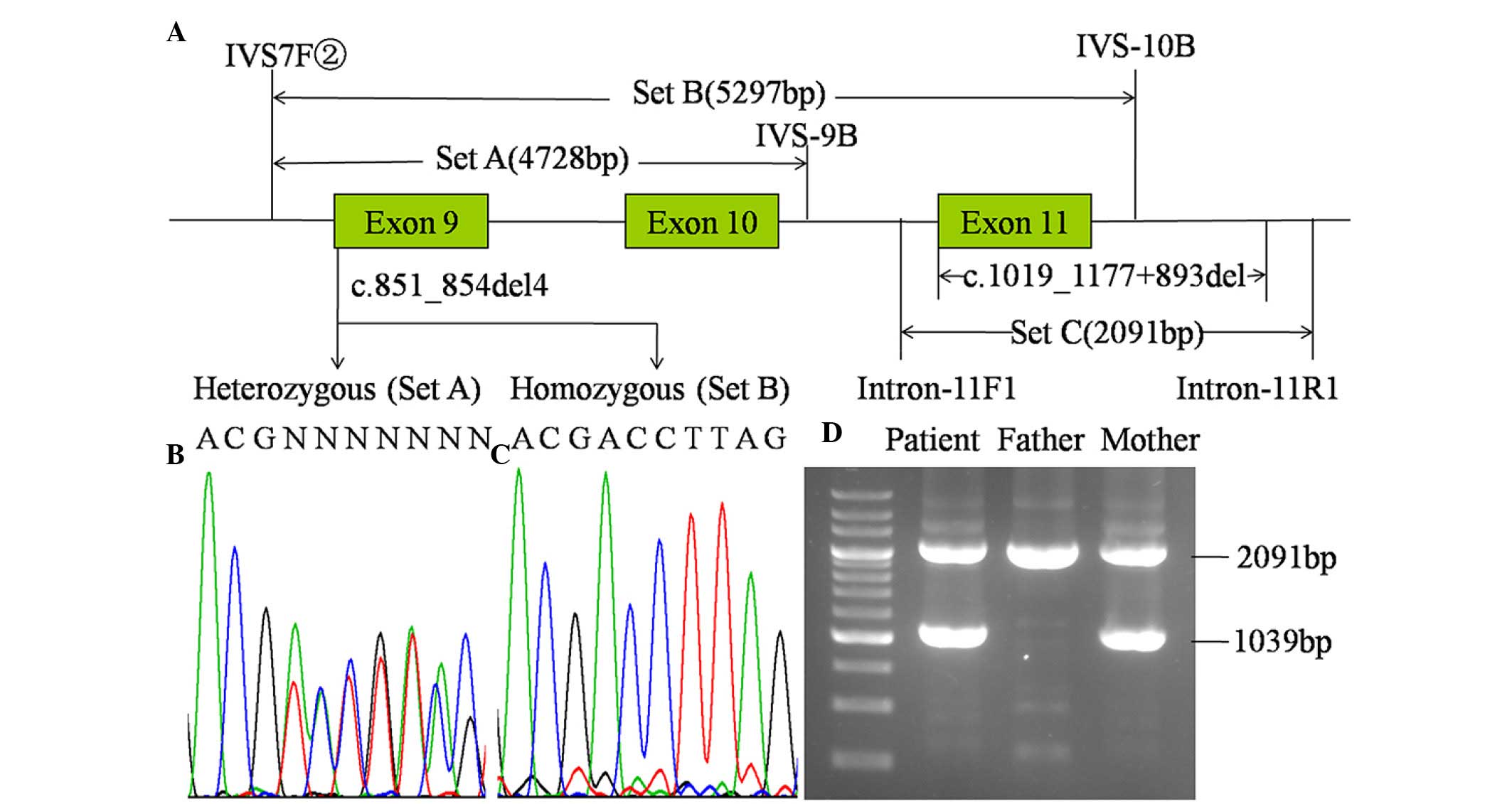
three sets of primers (Fig. 1)
were designed and the PCR kit was purchased from Takara Bio, Inc.
Every 50 µl LA-PCR mixture comprised 0.5 µl LA Taq (5 U/µl), 5 µl
10 × LA Buffer (Mg2+ plus), 1 µl DNA, 6 µl dNTP mixture
(2.5 mM), and 35.5 µl PCR-grade water and the relevant primer pair
(Set A, B and C in Fig. 1,
respectively, with 1 µl of each primer in 20 µM). The temperature
profile was set at 94°C for 4 min, followed by 35 cycles at 98°C
for 11 sec, 62°C for 40 sec, 68°C for 3 min and a final extension
step at 68°C for 10 min. The amplified products were then separated
using electrophoresis, purified and sequenced, respectively. The
electrophoresis was conducted in an 1.5% agarose gel (Bio-Rad
Laboratories, Inc., Hercules, CA, USA), the target products were
purified by a gel extraction kit (Omega Bio-Tek, Inc.).
Results
Case report
A male infant, aged 2.6 months, was admitted to the
Children's Hospital Affiliated to the Capital Institute of
Pediatrics due to prolonged jaundice for >2 months. As the first
child of a non-consanguineous couple, the infant was born at the
gestational age of 38 weeks with a birth weight of 2.35 kg [-2.1
standard deviations (SD)] and a body length of 48 cm (−0.9 SD). The
father and grandmother had hypercholesterolemia. There was no
family history of any other genetic disease.
Physical examination revealed a body weight of 4.7
kg (−2.0 SD), body length of 55 cm (−2.2 SD) and a head
circumference of 37.5 cm (−1.7 SD). Jaundiced skin and sclera were
observed. No positive signs were found in the two lungs or the
heart. The liver was palpable with a soft edge 3 cm below the right
costal margin. Biochemical analysis showed increased levels of
serum γ-glutamyl transpeptidase, total bilirubin, direct bilirubin
and total bile acids, as shown in Table I, indicating the presence of
cholestasis. The levels of cytomegalovirus (CMV) DNA in the urine
and breast milk were 5.57×103 copies/ml and
1.07×104 copies/ml, respectively.
 | Table I.Biochemical alterations over time in
the patient with neonatal intrahepatic cholestasis caused by citrin
deficiency. |
Table I.
Biochemical alterations over time in
the patient with neonatal intrahepatic cholestasis caused by citrin
deficiency.
|
|
|
Age
(months) |
|---|
|
|
|
|
|---|
| Biochemical
index | Normal range | 2.6a | 3.1 | 3.3b | 3.5c | 4 | 4.4 | 7.3 | 9.4 | 12 | 18 |
|---|
| ALT (U/l) | 5–40 | 66.0 | 26.00 | 113.0 | 75.7 | 24.0 | 42.0 | 71.0 | 47.00 | 46.0 | 27 |
| AST (U/l) | 5–40 | 132.0 | 52.00 | 578.0 | 134.3 | 47.0 | 58.0 | 63.0 | 48.00 | 36.0 | 47 |
| GGT (U/l) | 8–50 | 171.0 | 152.00 | 215.0 | 165.3 | 139.0 | 58.0 | 22.0 | 25.00 | 21.0 | 21 |
| ALP (U/l) | 20–500 | 810.0 | 396.00 | 395.0 | – | 412.0 | 285.0 | 241.0 | 198.0 | 208.0 | 240 |
| TP (g/l) | 60.0–83.0 | 45.8 | 52.00 | 48.2 | – | 54.1 | 56.3 | 57.8 | 68.20 | 67.9 | 73.4 |
| Alb (g/l) | 35.0–55.0 | 32.2 | 35.30 | 38.2 | – | 39.1 | 40.1 | 45.8 | 50.30 | 50.8 | 50.3 |
| Glb (g/l) | 20.0–30.0 | 13.6 | 16.70 | 10.0 | – | 15.0 | 16.2 | 12.0 | 17.90 | 17.1 | 23.1 |
| Tbil (µmol/l) | 2–19 | 174.7 | 134.90 | 192.8 | 126.2 | 44.9 | 13.3 | 11.4 | 5.90 | 7.0 | 8.0 |
| Dbil (µmol/l) | 0–6 | 99.7 | 60.40 | 83.0 | 66.7 | 18.5 | 4.5 | 2.0 | 1.30 | 1.5 | 2.2 |
| Ibil (µmol/l) | 2.56–20.9 | 75.0 | 74.50 | 109.8 | 59.5 | 25.4 | 8.8 | 9.4 | 4.60 | 5.5 | 5.8 |
| TBA (µmol/l) | 0–10 | 210.2 | 168.83 | 170.9 | 123.2 | 64.5 | 25.6 | 6.9 | 7.36 | 7.3 | 0.5 |
According the above findings, the patient was
suspected to have CMV hepatitis, and breastfeeding was terminated
followed by the commencement of galactose-restricted milk and
ganciclovir treatment. As a result, the urinary CMV DNA levels
became negative; however, the infant remained jaundiced and there
was no marked improvement in biochemistry (Table I). Laparoscopy and cholangiography
were then performed at 3.3 months (in a hospital in Beijing,
China), revealing normal bile ducts, however, liver biopsy showed
microvesicular steatosis, mildly hepatocellular and canalicular
cholestasis, moderate inflammatory cell infiltration in the portal
tracts and perisinusoidal fibrosis. Marginally elevated levels of
citrulline, ornithine and methionine were found on tandem mass
spectrometry (MS-MS) analysis of the serum amino acids; whereas a
high level of 4-hydroxyphenyllactate was identified in the urinary
gas chromatography (GC)-MS assay (both previously performed at a
hospital in Beijing, China).
In view of the above findings, the patient was
suspected to have NICCD, and was switched to a galactose-free,
MCT-enriched formula at the age of 3.5 months. His jaundice
subsided rapidly, and the biochemical abnormalities improved
markedly within half a month. At the 12-month follow-up, the
patient had a body weight of 10.0 kg (−0.1 SD), body length of 74
cm (−1.2 SD) and head circumference of 46 cm (−0.4 SD), and
cholestatic indices were no longer detected on biochemical
assessment. The patient (aged, 6 months) was referred to our
hospital for SLC25A13 genetic analysis.
Results of DNA sequencing and cDNA
cloning analysis
Screening of high-frequency mutations and Sanger
sequencing of the SLC25A13 gene in the infant revealed a
paternally-inherited mutation, c.851_854del4. Following
SLC25A13 cDNA cloning analysis using PBLs, five alternative
splice variants (ASVs) were identified from the
maternally-inherited SLC25A13 allele, with the common
feature of exon 11 skipping (r.1019_1177del), as shown in Table II. This suggested the existence of
a large insertion or deletion within the DNA fragment spanning the
region between intron 10 and intron 11 in the SLC25A13
gene.
| Table II.SLC25A13 ASVs detected by cDNA
analysis using peripheral blood lymphocytes of the patient with
neonatal intrahepatic cholestasis caused by citrin deficiency. |
Table II.
SLC25A13 ASVs detected by cDNA
analysis using peripheral blood lymphocytes of the patient with
neonatal intrahepatic cholestasis caused by citrin deficiency.
| Allele | Name | ASV | Annotation | Clone (n) | Percentage |
|---|
|
Maternally-inherited | M-01 |
r.213_468del; r.1019_1177del | Exon 4, 5 and 11
skipping | 1 | 16.7 |
|
| M-02 |
r.213_328del; r.1019_1177del | Exon 4 and 11
skipping | 1 | 16.7 |
|
| M-03 |
r.1019_1177del | Exon 11
skipping | 2 | 33.3 |
|
| M-04 |
r.213_328del; r.1019_1177del; |
|
|
|
|
|
|
r.1311_1312ins1311+102_1311+176 | Exon 4 and 11
skipping | 1 | 16.7 |
|
| M-05 |
r.213_468del; r.755_933del; | Exon 4, 5, 8, 9 and
11 | 1 | 16.7 |
|
|
|
r.1019_1177del | skipping |
|
|
|
Paternally-inherited | F-01 |
r.213_328del; r.851_854del4 | Exon 4
skipping | 1 | 100 |
Identification of the large
deletion
The targeted DNA span between intron 10 and intron
11, described above, was then investigated to identify the obscure
mutation of maternal origin. As shown in Fig. 1, the c.851_854del4 mutation was
heterozygous in the PCR product using primer set A, and was
homozygous with primer set B. These results led to the design of
primer set C, with which the LA-PCR amplification procedure yielded
an unexpected product of ~1.0 kb in the infant and the mother in
addition to the expected 2,091 bp band in all family members. As
shown in Fig. 2, direct sequencing
of the unexpected product revealed a large deletion,
c.1019_1177+893del, with a length of 1,052 bp, entirely involving
exon 11 and partially involving intron 11.
Discussion
The intro- and extra-uterine growth retardation,
prolonged jaundice, liver enlargement and biochemical alterations,
which were exhibited by the infant in the present study were all
non-pathognomonic. However, the metabolome and hepatopathologic
findings were consistent with the relevant results in cases of
NICCD reported previously (10,28),
warranting the genetic analysis of SLC25A13 in the present
study. Based on the findings of cDNA cloning analysis using PBLs
and the subsequent LA-PCR analysis and Sanger sequencing
procedures, the maternally-inherited novel deletion,
c.1019_1177+893del, was identified in the patient. To the best of
our knowledge, this mutation has not been reported previously. The
identification of this large deletion, in addition to the
paternally-inherited c.851_854del4 mutation identified, constituted
reliable evidence for a definitive diagnosis of NICCD in the
infant.
The novel large deletion caused the production of
the r.1019_1177del (exon 11 skipping) ASV, which
predictively led to the loss of 53 amino acid residues (codons
340–392) in the citrin protein. According to the functional domains
of citrin (4), the loss of these
residues produces a truncated citrin molecule without the first and
second transmembrane domains. Consequently, the AGC2 function was
impaired in all hepatocytes, resulting in the laboratory and
clinical manifestations observed in the infant with NICCD. Of note,
this pathogenic process, resulting from the novel large deletion,
is consistent with that underlying the IVS11+1G>A mutation
reported previously, which also resulted in exon 11 skipping
(r. 1019_1177del) during the splicing of pre-mRNA (4).
Accumulating clinical evidence has suggested the
therapeutic effectiveness of galactose-free and MCT-enriched
formulas in patients with NICCD (9,16,29,30),
and the clinical findings in the present study further supported
this. A key biochemical alteration of citrin deficiency is the
increased cytosolic NADH/NAD+ ratio in hepatocytes, and
a malate-citrate shuttle may be a form of compensation for the
increased ratio (1). The
pathophysiology of CD may be associated with an energy shortage in
the liver caused by impairment of glycolysis caused by the
increased NADH/NAD+ ratio (29,31).
The metabolism of galactose in hepatocytes further increases this
ratio and reduces the production of mitochondrial acetyl-CoA, a
component of the malate-citrate shuttle. By contrast, MCT
supplementation can produce an excess of acetyl-CoA in the
mitochondria of hepatocytes, providing energy and improving the
cytosolic NADH/NAD+ ratio via the malate-citrate shuttle
(29,31). These mechanisms explain the
clinical and laboratory improvements of the patient examined in the
present study shortly following the introduction of the
galactose-free and MCT-enriched formula.
In conclusion, the present study reported on
clinical and molecular investigations of an infant with NICCD, who
was confirmed to be a compound heterozygote of the c.851_854del4
mutation and a novel large deletion c.1019_1177+893del in the
SLC25A13 gene. These findings confirmed the effectiveness of
the galactose-free and MCT-enriched formula on NICCD therapy,
enriched the SLC25A13 mutational spectrum, and supported the
feasibility of SLC25A13 cDNA cloning analysis using PBLs as
a molecular tool to facilitate the identification of large
SLC25A13 deletion.
Acknowledgements
The present study was financially supported by the
Cultivation Foundation for Scientific Research (grant no. 2014208)
approved by the First Affiliated Hospital, Jinan University, the
Medical Scientific Research Foundation of Guangdong Province (grant
no. A2014385), the Fundamental Research Funds for the Central
Universities (grant no. 21615470), and the National Natural Science
Foundation of China (grant no. 81270957). The authors would like to
thank Mrs Charron Cote (Biomedical Translational Research
Institute, Jinan University, Guangzhou, China) for her grammatical
contributions.
Glossary
Abbreviations
Abbreviations:
|
CD
|
citrin deficiency
|
|
NICCD
|
neonatal intrahepatic cholestasis
caused by citrin deficiency
|
|
AGC2
|
aspartate/glutamate carrier isoform
2
|
|
PBLs
|
peripheral blood lymphocytes
|
|
PCR
|
polymerase chain reaction
|
|
LA-PCR
|
long and accurate-PCR
|
|
RT-PCR
|
reverse transcription-PCR
|
|
ALT
|
alanine transaminase
|
|
AST
|
aspartate transaminase
|
|
GGT
|
γ-glutamyl transpeptidase
|
|
TBA
|
total bile acid
|
|
Tbil
|
total bilirubin
|
|
Dbil
|
direct bilirubin
|
|
Ibil
|
indirect bilirubin
|
|
MCT
|
medium-chain triglyceride
|
|
GC-MS
|
gas chromatography-mass
spectrometry
|
|
MS-MS
|
tandem mass spectrometry
|
|
ASVs
|
alternative splice variants
|
|
CMV
|
cytomegalovirus
|
|
NADH
|
reduced form of nicotinamide-adenine
dinucleotide
|
References
|
1
|
Saheki T and Kobayashi K: Mitochondrial
aspartate glutamate carrier (citrin) deficiency as the cause of
adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal
hepatitis (NICCD). J Hum Genet. 47:333–341. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kobayashi K, Saheki T and Song YZ: Citrin
DeficiencyPagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong
CT, Smith RJH and Stephens K: GeneReviews® [Internet].
Seattle (WA): University of Washington, Seattle. 1993–2014, 2005
Sep 16 [updated 2014 Jul 31].
|
|
3
|
Palmieri F: Mitochondrial transporters of
the SLC25 family and associated diseases: A review. J Inherit Metab
Dis. 37:565–575. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kobayashi K, Sinasac DS, Iijima M, Boright
AP, Begum L, Lee JR, Yasuda T, Ikeda S, Hirano R, Terazono H, et
al: The gene mutated in adult-onset type II citrullinaemia encodes
a putative mitochondrial carrier protein. Nat Genet. 22:159–163.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bijarnia-Mahay S, Häberle J, Rüfenacht V,
Shigematsu Y, Saxena R and Verma IC: Citrin deficiency: A treatable
cause of acute psychosis in adults. Neurol India. 63:220–222. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ko JM, Kim GH, Kim JH, Kim JY, Choi JH,
Ushikai M, Saheki T, Kobayashi K and Yoo HW: Six cases of citrin
deficiency in Korea. Int J Mol Med. 20:809–815. 2007.PubMed/NCBI
|
|
7
|
Lee BH, Jin HY, Kim GH, Choi JH and Yoo
HW: Nonalcoholic fatty liver disease in 2 siblings with adult-onset
type ii citrullinemia. J Pediatr Gastroenterol Nutr. 50:682–685.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ngu HL, Zabedah MY and Kobayashi K:
Neonatal intrahepatic cholestasis caused by citrin deficiency
(NICCD) in three Malay children. Malays J Pathol. 32:53–57.
2010.PubMed/NCBI
|
|
9
|
Ohura T, Kobayashi K, Tazawa Y, Abukawa D,
Sakamoto O, Tsuchiya S and Saheki T: Clinical pictures of 75
patients with neonatal intrahepatic cholestasis caused by citrin
deficiency (NICCD). J Inherit Metab Dis. 30:139–144. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Song YZ, Li BX, Chen FP, Liu SR, Sheng JS,
Ushikai M, Zhang CH, Zhang T, Wang ZN, Kobayashi K, et al: Neonatal
intrahepatic cholestasis caused by citrin deficiency: Clinical and
laboratory investigation of 13 subjects in mainland of China. Dig
Liver Dis. 41:683–689. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song YZ, Deng M, Chen FP, Wen F, Guo L,
Cao SL, Gong J, Xu H, Jiang GY, Zhong L, et al: Genotypic and
phenotypic features of citrin deficiency: Five-year experience in a
Chinese pediatric center. Int J Mol Med. 28:33–40. 2011.PubMed/NCBI
|
|
12
|
Song YZ, Zhang ZH, Lin WX, Zhao XJ, Deng
M, Ma YL, Guo L, Chen FP, Long XL, He XL, et al: SLC25A13 gene
analysis in citrin deficiency: Sixteen novel mutations in East
Asian patients, and the mutation distribution in a large pediatric
cohort in China. PLoS One. 8:e745442013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thong MK, Boey CC, Sheng JS, Ushikai M and
Kobayashi K: Neonatal intrahepatic cholestasis caused by citrin
deficiency in two Malaysian siblings: Outcome at one year of life.
Singapore Med J. 51:e12–e14. 2010.PubMed/NCBI
|
|
14
|
Treepongkaruna S, Jitraruch S, Kodcharin
P, Charoenpipop D, Suwannarat P, Pienvichit P, Kobayashi K and
Wattanasirichaigoon D: Neonatal intrahepatic cholestasis caused by
citrin deficiency: Prevalence and SLC25A13 mutations among Thai
infants. BMC Gastroenterol. 12:1412012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yeh JN, Jeng YM, Chen HL, Ni YH, Hwu WL
and Chang MH: Hepatic steatosis and neonatal intrahepatic
cholestasis caused by citrin deficiency (NICCD) in Taiwanese
infants. J Pediatr. 148:642–646. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zeng HS, Zhao ST, Deng M, Zhang ZH, Cai
XR, Chen FP and Song YZ: Inspissated bile syndrome in an infant
with citrin deficiency and congenital anomalies of the biliary
tract and esophagus: Identification and pathogenicity analysis of a
novel SLC25A13 mutation with incomplete penetrance. Int J Mol Med.
34:1241–1248. 2014.PubMed/NCBI
|
|
17
|
Avdseva-Tzavella DM, Ivanova MB, Todorov
TP, Todorova AP, Panteleeva EI, Tincheva SS, Lazarova EA, Kathom
HM, Yaneva PG and Tincheva RS: First Bulgarian case of citrin
deficiency caused by one novel and one recurrent mutation in the
SLC25A13 gene. Genet Couns. 25:271–276. 2014.PubMed/NCBI
|
|
18
|
Fiermonte G, Parisi G, Martinelli D, De
Leonardis F, Torre G, Pierri CL, Saccari A, Lasorsa FM, Vozza A,
Palmieri F and Dionisi-Vici C: A new Caucasian case of neonatal
intrahepatic cholestasis caused by citrin deficiency (NICCD): A
clinical, molecular, and functional study. Mol Genet Metab.
104:501–506. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hutchin T, Preece MA, Hendriksz C,
Chakrapani A, McClelland V, Okumura F, Song YZ, Iijima M, Kobayashi
K, Saheki T, et al: Neonatal intrahepatic cholestasis caused by
citrin deficiency (NICCD) as a cause of liver disease in infants in
the UK. J Inherit Metab Dis. 32:(Suppl 1). S151–S155. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vitoria I, Dalmau J, Ribes C, Rausell D,
García AM, López-Montiel J and Rubio V: Citrin deficiency in a
Romanian child living in Spain highlights the worldwide
distribution of this defect and illustrates the value of
nutritional therapy. Mol Genet Metab. 110:181–183. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dimmock D, Kobayashi K, Iijima M, Tabata
A, Wong LJ, Saheki T, Lee B and Scaglia F: Citrin deficiency: A
novel cause of failure to thrive that responds to a high-protein,
low-carbohydrate diet. Pediatrics. 119:e773–e777. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dimmock D, Maranda B, Dionisi-Vici C, Wang
J, Kleppe S, Fiermonte G, Bai R, Hainline B, Hamosh A, O'Brien WE,
et al: Citrin deficiency, a perplexing global disorder. Mol Genet
Metab. 96:44–49. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ricciuto A and Buhas D: A novel citrin
deficiency mutation in a cholestatic infant. J Pediatr
Gastroenterol Nutr. 59:e522014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wong LJ, Dimmock D, Geraghty MT, Quan R,
Lichter-Konecki U, Wang J, Brundage EK, Scaglia F and Chinault AC:
Utility of oligonucleotide array-based comparative genomic
hybridization for detection of target gene deletions. Clin Chem.
54:1141–1148. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tokuhara D, Iijima M, Tamamori A, Ohura T,
Takaya J, Maisawa S, Kobayashi K, Saheki T and Yamano T: Novel
diagnostic approach to citrin deficiency: Analysis of citrin
protein in lymphocytes. Mol Genet Metab. 90:30–36. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tabata A, Sheng JS, Ushikai M, Song YZ,
Gao HZ, Lu YB, Okumura F, Iijima M, Mutoh K, Kishida S, et al:
Identification of 13 novel mutations including a retrotransposal
insertion in SLC25A13 gene and frequency of 30 mutations found in
patients with citrin deficiency. J Hum Genet. 53:534–545. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang ZH, Lin WX, Deng M, Zhao XJ and Song
YZ: Molecular analysis of SLC25A13 gene in human peripheral blood
lymphocytes: Marked transcript diversity, and the feasibility of
cDNA cloning as a diagnostic tool for citrin deficiency. Gene.
511:227–234. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kimura A, Kage M, Nagata I, Mushiake S,
Ohura T, Tazawa Y, Maisawa S, Tomomasa T, Abukawa D, Okano Y, et
al: Histological findings in the livers of patients with neonatal
intrahepatic cholestasis caused by citrin deficiency. Hepatol Res.
40:295–303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hayasaka K, Numakura C, Toyota K and
Kimura T: Treatment with lactose (galactose)-restricted and
medium-chain triglyceride-supplemented formula for neonatal
intrahepatic cholestasis caused by citrin deficiency. JIMD Rep.
2:37–44. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang ZH, Lin WX, Deng M, Zhao ST, Zeng
HS, Chen FP and Song YZ: Clinical, molecular and functional
investigation on an infant with neonatal intrahepatic cholestasis
caused by citrin deficiency (NICCD). PLoS One. 9:e892672014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hayasaka K, Numakura C and Watanabe H:
Treatment and pathomechanism of citrin deficiency. Brain Nerve.
67:739–747. 2015.(In Japanese). PubMed/NCBI
|