Introduction
Idiopathic pulmonary fibrosis (IPF) is the most
common type of interstitial lung disease characterized by a
chronic, progressive and irreversible course with median survival
of 3–5 years (1,2). At present there is no efficacious
treatment for lung fibrosis other than lung transplantation. Its
primary pathological characteristics include persistent
inflammatory cell infiltration, diffuse fibrosing alveolitis and
alveolar epithelial cell injury. Fibroblasts become activated, then
transition to the myofibroblastic phenotype. These cells may
contribute to the extracellular matrix and collagen accumulation in
the lungs, gradually replacing normal lung tissue by fibrotic
scarring and honeycombing (3–5). It
has been reported that lung inflammation initiates lung fibrosis,
however, failure to resolve epithelial cell injury is critical to
the pathogenesis of fibrosis. The molecular and cellular mechanisms
underlying the pathogenesis of lung fibrosis remain to be fully
understood. However, evidence in support of a role for
epithelial-mesenchymal transition (EMT) in pulmonary fibrosis has
been presented (6). Enhanced EMT
may lead to excessive production, deposition and contraction of the
extracellular matrix, and pathological fibrogenesis. However, the
molecular mechanisms of EMT involved in the pathogenesis of lung
fibrosis remain to be fully elucidated.
The etiology and pathogenesis of IPF remain unclear,
however the characteristics of this disease can be mimicked using
animal models of pulmonary fibrosis (7). Among these models of IPF, the
bleomycin (BLM)-induced pulmonary fibrosis model is the most
commonly used for studying the disease pathogenesis and
pharmacotherapeutics (8). BLM, a
widely used antineoplastic drug, causes interstitial pulmonary
fibrosis in a dose-dependent manner (9). A single intratracheal administration
of BLM into the lung of an animal has been reported to induce an
inflammatory response, alveolar cell damage, EMT, fibroblast and
myofibroblast dysplasia, in addition to extracellular matrix
remodeling (10). Animal models of
BLM-induced pulmonary fibrosis display similar phenotypes to those
observed in patients with fibrotic lungs. Previous studies have
demonstrated that BLM-induced pulmonary fibrosis is involved in the
secretion of a variety of chemokines, epithelial cell apoptosis,
the transforming growth factor β1 (TGF-β1) pathway and EMT
(11–13).
The phosphoinositide 3-kinase (PI3K)/protein kinase
B (Akt) pathway in cells, a key survival signaling pathway,
regulates numerous biological processes through phosphoryl transfer
(14). This pathway controls
numerous cellular processes including protein synthesis, glucose
metabolism, proliferation and differentiation (15). Its basal activity ensures cell
survival and regulates the cell cycle, whereas inactivation of
PI3K/Akt signaling results in cellular apoptosis (16). Additionally, the PI3K/Akt pathway,
as a form of ‘adaptive strategy’, is involved in the immune
response process of the host cell to counteract viral invasion
(15). A previous study
demonstrated that the PI3K/Akt signaling pathway is upregulated in
human diabetic nephropathy, and serves a crucial pathogenetic role
in glycogen accumulation and tumor development (17). Akt is an effector kinase downstream
from PI3K, which forms focal points for the development of novel
therapeutics. LY294002, as a specific PI3K/Akt inhibitor, has been
reported to significantly ameliorate the PI3K/Akt-mediated cellular
processes by suppressing phosphorylation of Akt (18).
In the present study, the possible involvement of
the PI3K/Akt pathway during BLM-induced lung fibrosis was
investigated in rats. Furthermore, to identify the role of the
PI3K/Akt pathway, PI3K/Akt inhibitor was used to explore the major
role of the PI3K/Akt pathway in BLM-induced pulmonary fibrosis. The
current study predominantly focused on investigating the molecular
mechanisms underlying the pathogenesis of IPF, which will provide a
potentially novel pharmacological target for the treatment of
pulmonary fibrosis.
Materials and methods
Ethics statement
The present study was reviewed and approved by the
Ethics Committee of Animal Care and Experimentation, Hebei Medical
University (Shijiazhuang, China). The protocols were performed in
accordance with the guidelines of National Institutes of Health
(19). Prior to surgery and
treatment, rats were anesthetized with chloral hydrate (300 mg/kg;
intraperitoneal), and necessary efforts were taken to minimize
suffering.
Animals and experimental design
A total of 72 adult male Sprague-Dawley rats, aged
6–8 weeks and weighing 200–220 g, were purchased from Beijing Vital
River Laboratory Animal Technology Co., Ltd. (Beijing, China). The
rats used for experiments were allowed free access to food and
water and housed in a specific pathogen-free room. The animals were
maintained on a 12 h light/dark cycle in a controlled temperature
(20–25°C) and humidity (50±5%) environment. All rats were randomly
divided into three groups: i) Intratracheal saline (control group);
ii) intratracheal BLM (BLM group); iii) intratracheal BLM plus
LY294002 (BLM + LY294002 group). Over the course of the
experiments, the rats were closely monitored for overall health and
activity.
BLM-induced pulmonary fibrosis
model
For the induction of the pulmonary fibrosis model,
BLM (Sanofi-Aventis, Diegem, Belgium) was dissolved in sterile
phosphate-buffered saline (PBS). Subsequent to anesthesia with
intraperitoneal ketamine (80 mg/kg; Zhongshan Biology &
Technology, Beijing, China) and xylazine (15 mg/kg; Zhongshan
Biology & Technology), rats were intratracheally injected with
a single dose of BLM (5.0 mg/kg body weight in 0. 2 ml PBS).
Control animals were given a single intratracheal dose of 0. 2 ml
of PBS solution only. The rats in the LY294002 group were first
treated with BLM according to the protocol described above. On the
third hour following exposure to BLM, rats were treated with
intratracheal administration of LY294002 (0.3 mg/kg; Cell Signaling
Technology, Inc., Danvers, MA, USA). All rats were subsequently
sacrificed using chloral hydrate (300 mg/kg; intraperitoneal:
Baxtor, Deerfield, IL, USA) at 3, 7, 14 and 28 days subsequent to
BLM treatment, and lung tissues and bronchoalveolar lavage (BAL)
fluid (BALF) were collected for histopathology, collagen
quantification, reverse transcription-quantitative polymerase chain
reaction (RT-qPCR) and western blot analysis.
Tissue preparation and histological
analysis
The lungs from the rats were dissected and fixed for
48 h in 4% paraformaldehyde. The fixed lungs were sectioned,
embedded in paraffin, cut into 5 µm sections, and stained with
hematoxylin and eosin (H&E) for analysis of lung injury, and
with Elastica-Masson trichrome stain for the assessment of the
collagen deposition, an index of pulmonary fibrosis. The
histological severity of the lung alveolitis and lung fibrosis was
scored as previously described (20).
Hydroxyproline assay
Pulmonary collagen content was determined by the
measurement of hydroxyproline content. Hydroxyproline
quantification was performed as previously described (21). The right lung lobes were incised
and homogenized in 5 ml 0.5 M acetic acid (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) in PBS containing 0.6%
pepsin (Invitrogen; Thermo Fisher Scientific, Inc.). The extracts
were rotated at 4°C overnight and cleared by centrifugation at
15,000 × g for 10 min at 4°C. The hydroxyproline content was
determined according to the protocol of the Hydroxyproline Testing
Kit (Jiancheng, Nanjing, China). The absorbance was measured at 550
nm using a microplate reader.
BAL and enzyme-linked immunosorbent
assay (ELISA) analysis
BAL was performed to collect BALF for analysis of
the cytokine content. The rats were anesthetized with
intraperitoneal ketamine (80 mg/kg) and xylazine (15 mg/kg), the
trachea was then exposed and a plastic cannula was inserted into
the trachea. The lung tissues were lavaged with 5 ml 0.9% saline
solution. The lavage was repeated twice with saline to recover a
total volume of 4–5 ml. It was then centrifuged at 500 × g
for 10 min at 4°C and the supernatant was used for cytokine
measurement. The concentrations of tumor necrosis factor (TNF)-α,
interleukin (IL)-1β, IL-6 and IL-10 in the BALF were determined
using ELISA kits (R&D Systems, Inc., Minneapolis, MN, USA)
according to the manufacturer's instructions.
RNA isolation and RT-qPCR
analysis
Total RNA was isolated from rat lung tissues using
TRIzol reagent (Life Technologies; Thermo Fisher Scientific, Inc.)
and reverse-transcribed using the First Strand cDNA synthesis kit
(Fermentas; Thermo Fisher Scientific, Inc., St. Leon-Rot, Germany)
according to the manufacturer's instructions. RT-qPCR was performed
on a RT-qPCR System (ABI Prism 7300; Applied Biosystems; Thermo
Fisher Scientific, Inc.) instrument with SYBR Premix Ex Taq (Takara
Bio., Inc., Otsu, Japan), starting with 1 ng reverse-transcribed
total RNA. PCR was performed under the following conditions: 95°C
for 10 min, 40 cycles of 95°C for 5 sec and 60°C for 30 sec. The
following primers were used: TNF-α, Forward (F)
5′-TTGACCTCAGCGCTGAGTTG-3′ and reverse (R)
5′-CCTGTAGCCCACGTCGTAGC-3′; IL-1β, F 5′-CAGGATGAGGACATGAGCACC-3′
and R 5′-CTCTGCAGACTCAAACTCCAC-3′; IL-6, F
5′-GTACTCCAGAAGACCAGAGG-3′ and R 5′-TGCTGGTGACAACCACGGCC-3′; IL-10,
F 5′-CATGGCCTTGTAGACACCTTTG-3′ and R 5′-CATCGATTTCTCCCCTGTGAGA-3′.
GAPDH was used as an internal control. Each experiment was
performed in duplicate and repeated three times. The data were
analysed using the 2−ΔΔCq method (22).
Western blot analysis
Lung tissues were lysed in Tissue Protein Lysis
Solution (Life Technologies; Thermo Fisher Scientific, Inc., Gent,
Belgium) supplemented with 5% Proteinase Inhibitor Cocktail
(Sigma-Aldrich; Merck Millipore, Darmstadt, Germany), incubated on
ice for 30 min, and centrifuged at 15,000 × g for 15 min at
4°C. Protein concentrations were determined with the bicinchoninic
acid protein assay reagents (Jiancheng, Nanjing, China). Proteins
from each sample were separated on 10% sodium dodecyl
sulfate-polyacrylimide gel electrophoresis, transferred to
polyvinylidene difluoride membranes for 60 min. Nonspecific binding
sites were blocked with 5% bovine serum albumin (Sigma-Aldrich;
Merck Millipore) for 1 h, then incubated with the following rabbit
anti-rat polyclonal antibodies: Akt (cat. no. sc-32245), p-Akt
(cat. no. sc-20984), epithelial cadherin (E-cad; cat. no.
sc-30956), α-smooth muscle actin (α-SMA; cat. no. sc-28538),
vimentin (Vim; cat. no. sc-31638) and β-actin (cat. no. sc-26351;
all 1:1,000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA)
overnight at 4°C. Next day, the membranes were incubated with
secondary antibodies (cat. no. IBSBIOA-003; 1:5,000; Cell Signaling
Technology, Inc.) at 37°C for 2 h. The immunoreactive bands were
visualized with an enhanced chemiluminescent detection system
(Amersham, Little Chalfont, UK). Blots were scanned by
densitometry, and the integrated density of pixels was quantified
using ImageQuant software, version 5.2 (GE Healthcare Bio-Sciences,
Pittsburgh, PA, USA).
Statistical analysis
Data are expressed as the means ± standard
deviation. All tests were performed using SPSS software, version
17.0 (SPSS, Inc., Chicago, IL, USA). Statistical significance was
determined using one-way analysis of variance and the
Student-Newmann-Keuls post hoc test to determine differences among
different groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
Administration of Akt inhibitor
alleviates BLM-induced lung edema and weight
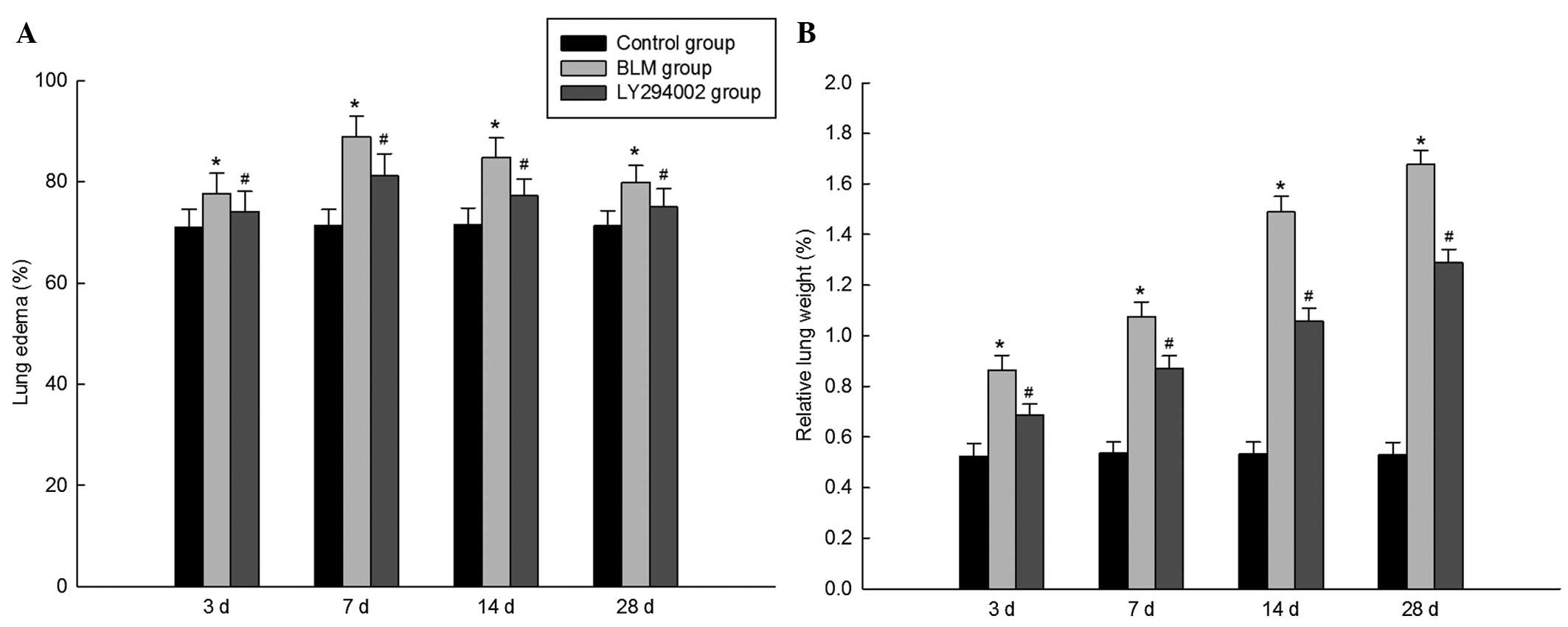
As presented in Fig.
1, clear lung edema was observed in BLM-challenged rats, at the
same time, the relative weights of the lungs were significantly
increased in the BLM group rats. Notably, inhibition of p-Akt,
using tracheal administration of LY294002, significantly alleviated
BLM-induced lung edema and elevation of the relative lung
weight.
Administration of Akt inhibitor
reduces BLM-induced inflammation
To investigate the role of the PI3K/Akt pathway on
BLM-induced pulmonary inflammation and fibrosis, the effects of the
Akt inhibitor on the histopathological alterations observed in the
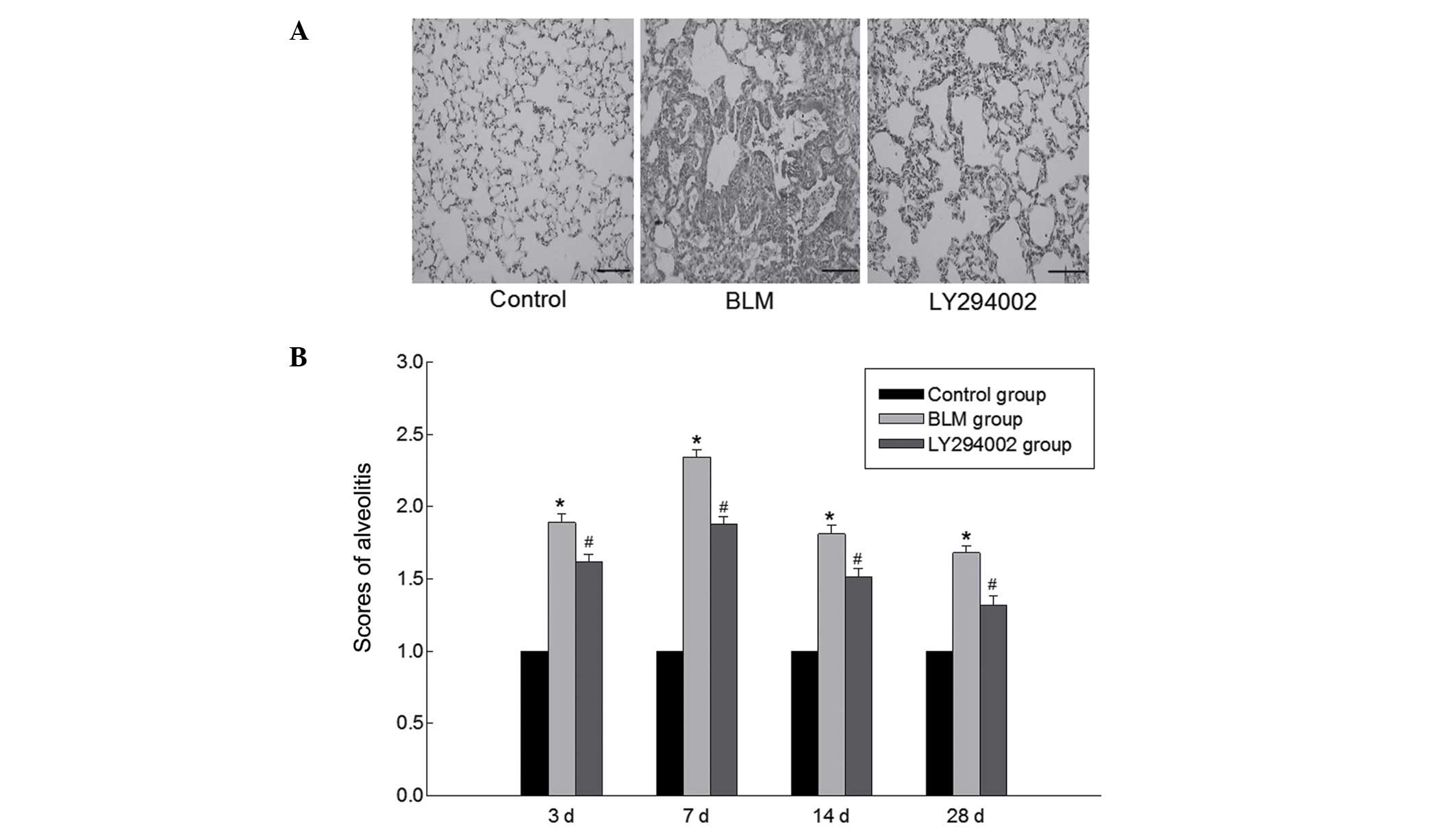
lungs were observed. Fig. 2
presents the H&E-stained lung sections. A histological
evaluation indicated that there were no changes in the control
rats, whereas the section from the BLM group revealed disordered
lung tissue structure, thickened pulmonary interalveolar septa and
infiltrated inflammatory cells. In contrast, the BLM-induced
histological alterations were significantly reduced by treatment
with the Akt inhibitor according to the histological
observations.
Administration of the Akt inhibitor
attenuates BLM-induced fibrosis
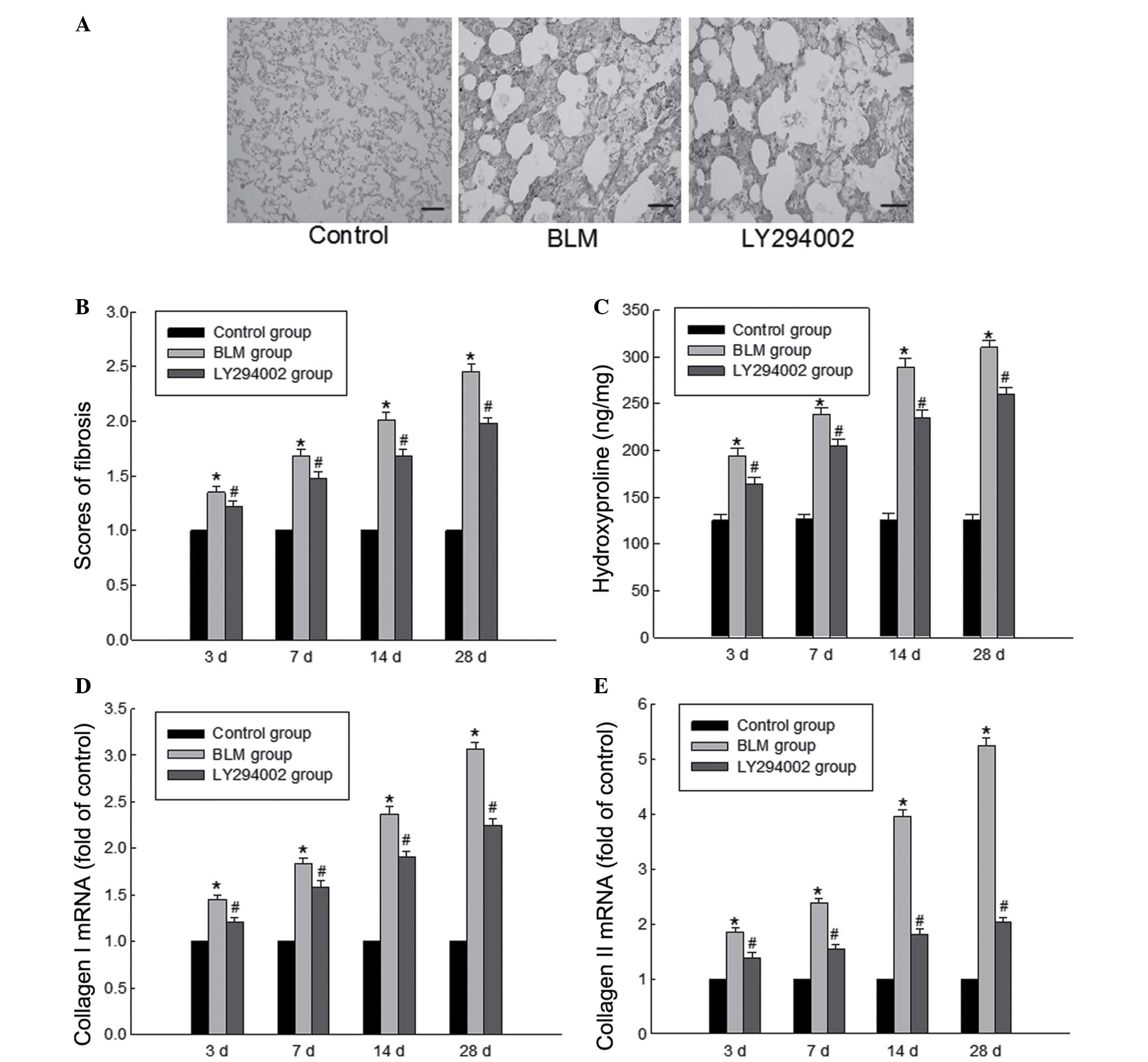
Masson's trichrome staining indicated that there
were large-scale collagen accumulations in the BLM group (Fig. 3A). Excessive deposition of
extracellular matrix was observed in the lung tissues of rats in
the BLM group, which is in accordance with the hallmark
characteristics of pulmonary fibrosis (Fig. 3A). Administration of the Akt
inhibitor significantly suppressed BLM-induced matrix protein
deposition in the lungs of BLM-treated rats. Similarly, a
significant reduction in the fibrosis scoring of these sections was
observed in the rat lungs of the BLM + LY294002 group (Fig. 3B). Furthermore, hydroxyproline is
suggested to be able to act as a primary indicator of the collagen
metabolism in different tissues. In order to more accurately
illustrate the severity of lung fibrosis, the content of
hydroxyproline was assessed to indicate collagen accumulation
within the fibrotic lung tissues. As presented in Fig. 3C, the hydroxyproline content in the
BLM group was significantly greater than that of the BLM + LY294002
group, indicating that the Akt inhibitor significantly reduced
BLM-induced elevation of hydroxyproline content in the lung
tissues. In addition, the gene expression levels of collagen I and
III were measured, and the results indicated a similar trend to
that of the hydroxyproline results (Fig. 3D and E).
Administration of the Akt inhibitor
upregulates anti-inflammatory cytokines and downregulates
pro-inflammatory cytokines
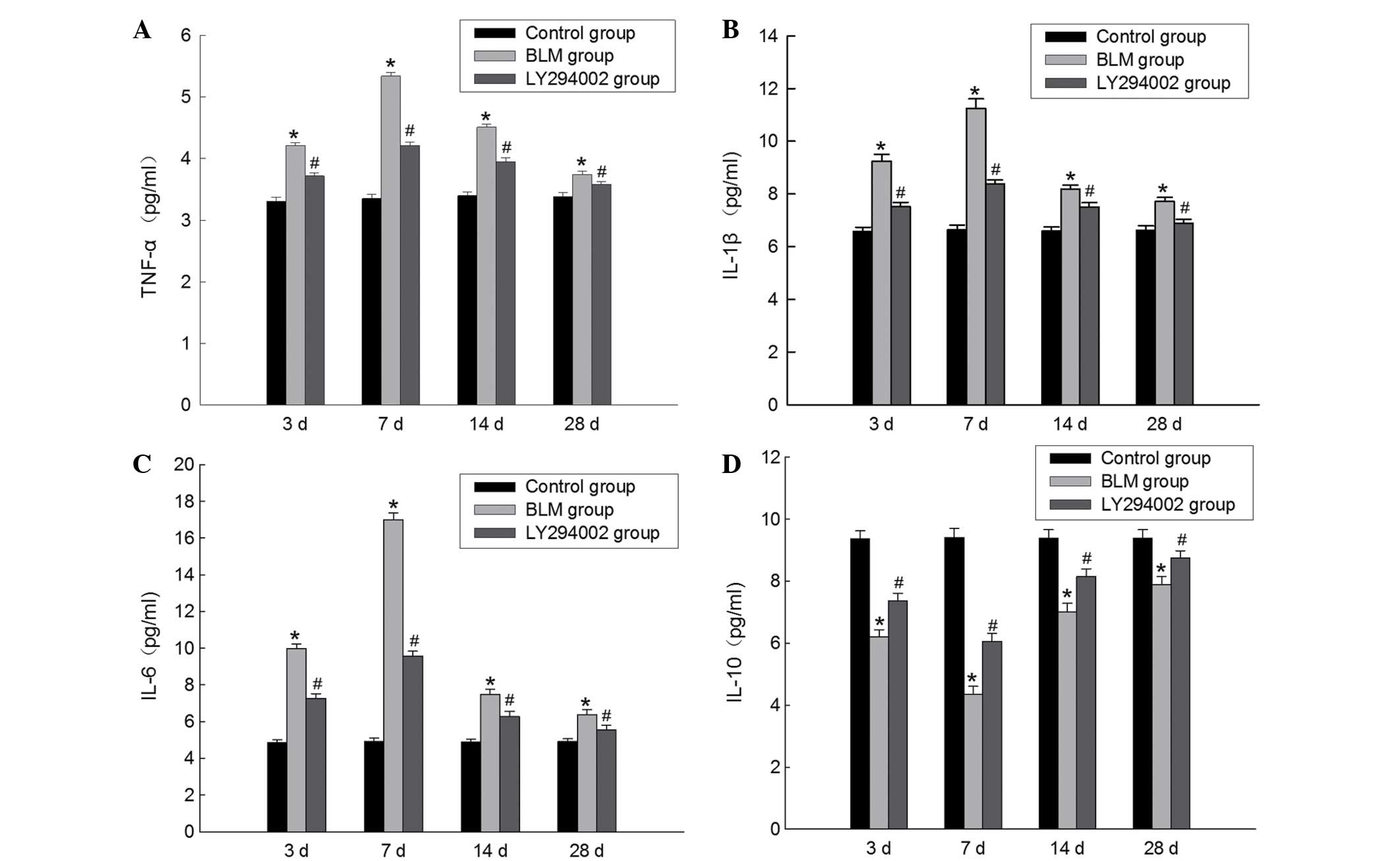
The levels of TNF-α, IL-1β, IL-6 or IL-10 in the
BALF of the rats were detected via ELISA. As presented in Fig. 4, in the BLM group, the
concentrations of TNF-α, IL-1β and IL-6 in the BALF were
significantly increased, while IL-10 levels in the BALF were
significantly reduced compared with the control group. However,
treatment with the Akt inhibitor could reverse the increases in
TNF-α, IL-1β and IL-6 levels and the reductions in the IL-10 levels
in the BALF. The results demonstrated that the Akt inhibitor may
downregulate pro-inflammatory cytokines whereas may upregulate
anti-inflammatory cytokines. Therefore, it is hypothesized that the
PI3K/Akt pathway is involved in the pathogenesis of BLM-induced
pulmonary fibrosis by influencing the expression of inflammatory
factors.
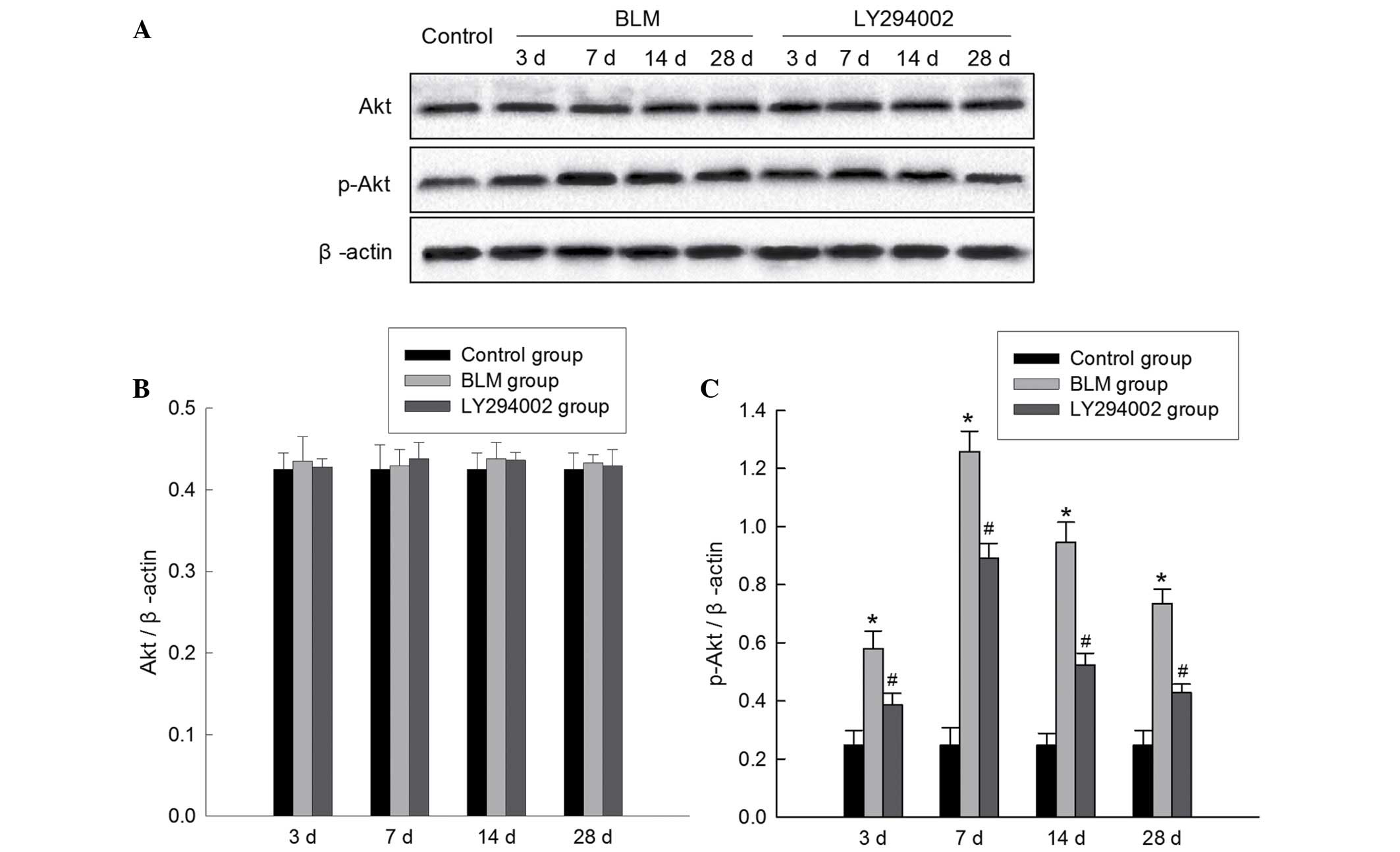
The relative expression of proteins in
the PI3K/Akt signaling pathway
The protein expression levels of Akt and p-Akt were
assessed in lung tisses using western blot analysis. The molecular
weight of the Akt protein is 55 kDa and of the p-Akt protein is 60
kDa. As presented in Fig. 5, BLM
treatment markedly increased the phosphorylation level of Akt
protein, while no significant alterations were observed in the
levels of total Akt protein. The expression levels of p-Akt were
the greatest 7 days subsequent to BLM injection. The increased
phosphorylation of Akt in lung tissue following BLM challenge was
significantly reduced in the BLM + LY294002 group. These results
indicated that the PI3K/Akt signaling pathway was involved in the
pathogenesis of BLM-induced pulmonary fibrosis.
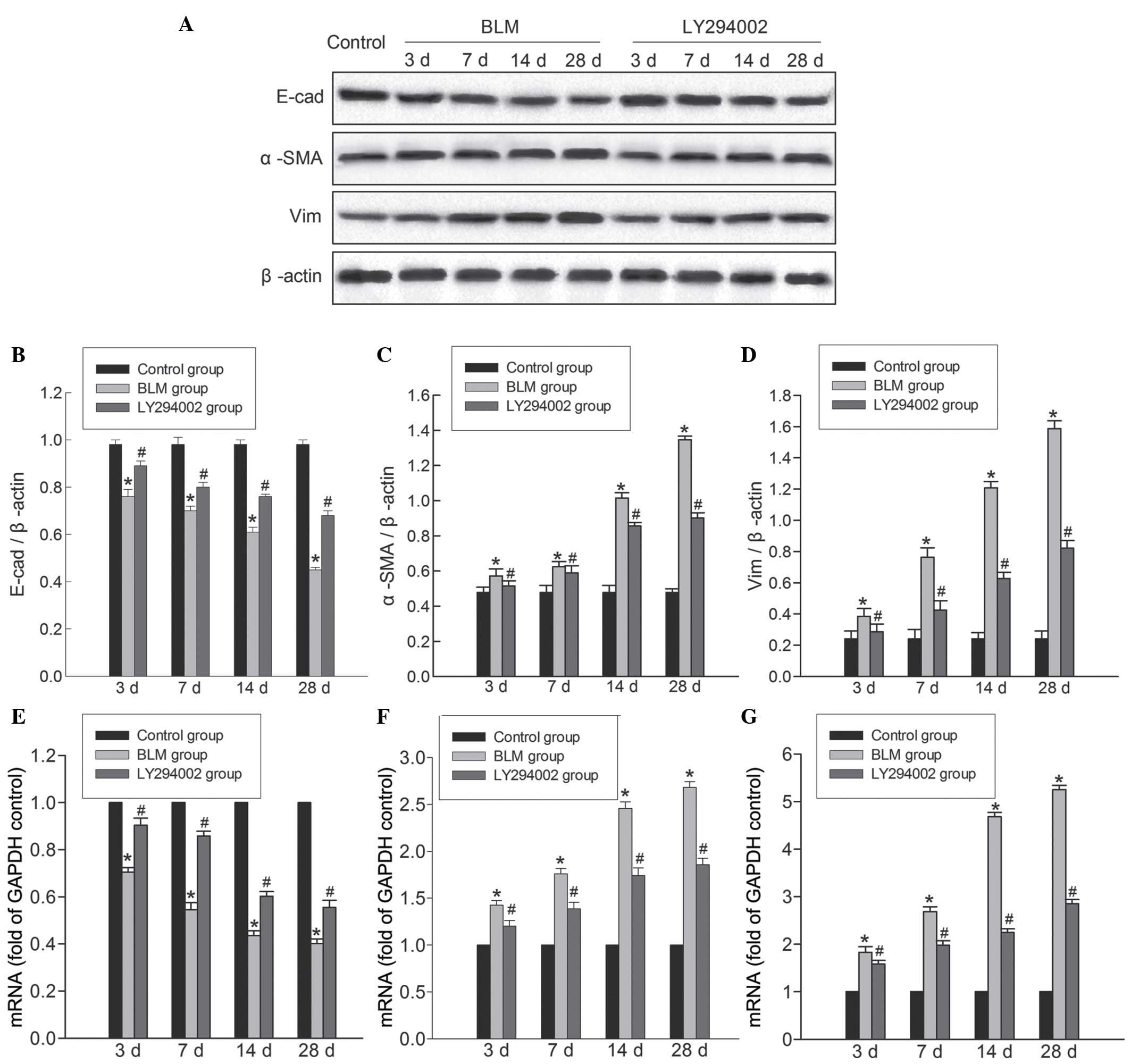
Administration of Akt inhibitor
reverses BLM-induced EMT
In order to investigate the involvement of the
PI3K/Akt pathway in mediating BLM-induced EMT, the epithelial
marker E-cad and the mesenchymal markers α-SMA and Vim were
analyzed by RT-qPCR and western blotting. The molecular weight of
the E-cad protein is 120 kDa, for the α-SMA protein is 42 kDa and
for Vim protein is 57 kDa. As presented in Fig. 6, E-cad expression was downregulated
in the lungs following BLM stimulation, while the expression of
α-SMA and Vim were upregulated. Notably, blocking of the PI3K/Akt
pathway significantly increased the expression of epithelial marker
genes and proteins, whereas the expression of mesenchymal marker
genes and proteins were reduced. Taken together, these results
indicate that the Akt inhibitor attenuated fibrosis subsequent to
BLM treatment, potentially via inhibition of BLM-induced EMT.
Discussion
The PI3K/Akt signaling pathway in the host cell
serves a critical regulatory role in numerous cellular processes
including translation, metabolism, RNA processing, apoptosis and
autophagy (15). A previous study
demonstrated that the activation of the PI3K/Akt pathway is a
crucial molecular event in a variety of diseases, including in
cancer development and maintenance via the regulation of cell
survival, cellular growth and cell cycle progression (23). In the present study, male
Sprague-Dawley rats were subjected to intratracheal injection of
BLM (5 mg/kg; 0.2 ml) to induce the pulmonary fibrosis model. The
results of western blotting demonstrated that BLM administration
markedly upregulated the phosphorylation level of Akt protein,
while no significant alterations in the total Akt protein level
were observed, indicating that the PI3K/Akt pathway is invovled in
lung fibrosis. In the current study, LY294002, a specific Akt
inhibitor, was used to further identify the roles of the PI3K/Akt
pathway in the pathogenesis of pulmonary fibrosis. The data
indicated that inhibition of the PI3K/Akt pathway may markedly
block BLM-induced increases in p-Akt expression, while no effects
on Akt protein levels in the lung tissues were observed.
In addition, the effects of the Akt inhibitor on
BLM-induced inflammation and fibrosis were investigated. The
inflammatory alterations were evaluated with H&E staining of
lung tissues and via ELISA in BALF. The histopathological
observations indicated that treatment with the Akt inhibitor
ameliorated inflammatory cell infiltration and promoted lung injury
repair. In addition, inhibition of the PI3K/Akt pathway could
suppress the overproduction of proinflammatory cytokines including
TNF-α, IL-1β and IL-6 in the BALF, and enhanced the release of the
anti-inflammatory cytokine IL-10. These observations indicated that
blocking the PI3K/Akt pathway exhibited positive anti-inflammatory
activity in the development of BLM-induced pulmonary fibrosis.
Additionally, the fibrotic alterations to lung tissue following
LY294002 treatment together with BLM were observed. The results
indicated that the Akt inhibitor suppressed myofibroblast expansion
and fibronectin matrix formation in lung tissues, reduced the
collagen content, reduced the increase of collagen I and collagen
III levels, and preserved pulmonary compliance. The suggested that
the Akt inhibitor exerts an anti-fibrotic effect in pulmonary
fibrosis. Therefore, it was concluded that the PI3K/Akt pathway
served a crucial role in BLM-induced pulmonary fibrosis, via the
regulation of inflammation and fibrosis.
Whether the PI3K/Akt pathway influences lung
fibrosis-associated EMT remains unclear and requires further
investigation. A previous study demonstrated that EMT is associated
with liver fibrosis and regeneration (24). An additional study observed that
EMT, a phenotypic change in which epithelial cells acquire
mesenchymal characteristics, served a specific role in fibrogenesis
and in the progression of cancer by biophysical signaling
mechanisms (25). It has been
previously demonstrated in in vitro studies and animal
models that TGF-β leads to the induction of EMT in the genetically
engineered type II alveolar epithelial cell line RLE/Abca3 and in
BLM-induced pulmonary fibrosis in mice (26,27).
The present study was conducted to examine the possibility that the
PI3K/Akt pathway modulated BLM-induced EMT. EMT is activated in
lung tissues in BLM-challenged rats, characterized by the loss of
epithelial characteristics (E-cad) and the acquisition of a
mesenchymal phenotype (α-SMA and Vim). Previous studies have
identified that the Akt inhibitor reversed the reductions of E-cad,
and prevented the increased expression of α-SMA and Vim that induce
fibrosis-associated EMT (28–30).
Therefore, it is suggested that EMT of alveolar epithelial cells
occurs in the pathogenesis of lung fibrosis and is partly reversed
by the Akt inhibitor, indicating that the PI3K/Akt pathway may
promote EMT progress, contributing to the pathogenesis of
fibrosis.
In conclusion, the results of the current study
suggest an involvement of the PI3K/Akt signaling pathway in the
pathogenesis of pulmonary fibrosis, contributing to fibrogenesis.
Blocking the PI3K/Akt pathway could attenuate BLM-induced
inflammation and fibrosis and reverse the process of lung
fibrosis-associated EMT. Thus, pharmacological blockade of the
PI3K/Akt pathway may be considered as a potential therapeutic
strategy for the treatment of pulmonary fibrosis.
Glossary
Abbreviations
Abbreviations:
|
IPF
|
idiopathic pulmonary fibrosis
|
|
PI3K
|
phosphatidylinositol 3-kinase
|
|
EMT
|
epithelial-mesenchymal transition
|
|
BLM
|
bleomycin
|
|
BALF
|
bronchoalveolar lavage fluid
|
|
E-cad
|
epithelial cadherin
|
|
α-SMA
|
α smooth muscle actin
|
|
vim
|
vimentin
|
References
|
1
|
Guiot J, Corhay JL and Louis R: Idiopathic
pulmonary fibrosis. Rev Med Liege. 69:605–610. 2014.PubMed/NCBI
|
|
2
|
Kim HJ, Perlman D and Tomic R: Natural
history of idiopathic pulmonary fibrosis. Respir Med. 109:661–670.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gharaee-Kermani M, Gyetko MR, Hu B and
Phan SH: New insights into the pathogenesis and treatment of
idiopathic pulmonary fibrosis: A potential role for stem cells in
the lung parenchyma and implications for therapy. Pharm Res.
24:819–841. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fernandez IE and Eickelberg O: New
cellular and molecular mechanisms of lung injury and fibrosis in
idiopathic pulmonary fibrosis. Lancet. 380:680–688. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
King TE Jr, Pardo A and Selman M:
Idiopathic pulmonary fibrosis. Lancet. 378:1949–1961. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yan W, Xiaoli L, Guoliang A, Zhonghui Z,
Di L, Ximeng L, Piye N, Li C and Lin T: SB203580 inhibits
epithelial-mesenchymal transition and pulmonary fibrosis in a rat
silicosis model. Toxicol Lett. 259:28–34. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Harrison JH Jr and Lazo JS: High dose
continuous infusion of bleomycin in mice: A new model for
drug-induced pulmonary fibrosis. J Pharmacol Exp Ther.
243:1185–1194. 1987.PubMed/NCBI
|
|
8
|
Moeller A, Ask K, Warburton D, Gauldie J
and Kolb M: The bleomycin animal model: A useful tool to
investigate treatment options for idiopathic pulmonary fibrosis?
Int J Biochem Cell Biol. 40:362–382. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Adamson IY and Bowden DH: The pathogenesis
of bleomycin-induced pulmonary fibrosis in mice. Am J Pathol.
77:185–197. 1974.PubMed/NCBI
|
|
10
|
Moore BB and Hogaboam CM: Murine models of
pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol.
294:L152–L160. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim HS, Go H, Akira S and Chung DH:
TLR2-mediated production of IL-27 and chemokines by respiratory
epithelial cells promotes bleomycin-induced pulmonary fibrosis in
mice. J Immunol. 187:4007–4017. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Degryse AL, Tanjore H, Xu XC, Polosukhin
VV, Jones BR, Boomershine CS, Ortiz C, Sherrill TP, McMahon FB,
Gleaves LA, et al: TGFβ signaling in lung epithelium regulates
bleomycin-induced alveolar injury and fibroblast recruitment. Am J
Physiol Lung Cell Mol Physiol. 300:L887–L897. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen YL, Zhang X, Bai J, Gai L, Ye XL,
Zhang L, Xu Q, Zhang YX, Xu L, Li HP and Ding X: Sorafenib
ameliorates bleomycin-induced pulmonary fibrosis: Potential roles
in the inhibition of epithelial-mesenchymal transition and
fibroblast activation. Cell Death Dis. 4:e6652013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yin MM, Cui YR, Wang L, Wang JY, Gao Y and
Xi JY: Progress on PI3K/Akt signaling pathway regulating
self-renewal and pluripotency of embryonic stem cells. Sheng Li Xue
Bao. 66:223–230. 2014.(In Chinese). PubMed/NCBI
|
|
15
|
Diehl N and Schaal H: Make yourself at
home: Viral hijacking of the PI3K/Akt signaling pathway. Viruses.
5:3192–3212. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Diab S, Fidanzi C, Léger DY, Ghezali L,
Millot M, Martin F, Azar R, Esseily F, Saab A, Sol V, et al:
Berberis libanotica extract targets NF-κB/COX-2, PI3K/Akt and
mitochondrial/caspase signalling to induce human erythroleukemia
cell apoptosis. Int J Oncol. 47:220–230. 2015.PubMed/NCBI
|
|
17
|
Ribback S, Cigliano A, Kroeger N, Pilo MG,
Terracciano L, Burchardt M, Bannasch P, Calvisi DF and Dombrowski
F: PI3K/AKT/mTOR pathway plays a major pathogenetic role in
glycogen accumulation and tumor development in renal distal tubules
of rats and men. Oncotarget. 30:13036–13048. 2015. View Article : Google Scholar
|
|
18
|
Xu X, Li H, Hou X, Li D, He S, Wan C, Yin
P, Liu M, Liu F and Xu J: Punicalagin induces Nrf2/HO-1 expression
via upregulation of PI3K/AKT pathway and inhibits LPS-induced
oxidative stress in RAW264.7 macrophages. Mediators Inflamm.
2015:3802182015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen H, Mei YQ and Solomon MA: Experiences
of establishing an abdominal heart transplantation model in rats.
Zhonghua Yi Xue Za Zhi. 92:1715–1718. 2012.(In Chinese). PubMed/NCBI
|
|
20
|
Szapiel SV, Elson NA, Fulmer JD,
Hunninghake GW and Crystal RG: Bleomycin-induced interstitial
pulmonary disease in the nude, athymic mouse. Am Rev Respir Dis.
120:893–899. 1979.PubMed/NCBI
|
|
21
|
Huang LS, Berdyshev E, Mathew B, Fu P,
Gorshkova IA, He D, Ma W, Noth I, Ma SF, Pendyala S, et al:
Targeting sphingosine kinase 1 attenuates bleomycin-induced
pulmonary fibrosis. FASEB J. 27:1749–1760. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mabuchi S, Kuroda H, Takahashi R and
Sasano T: The PI3K/AKT/mTOR pathway as a therapeutic target in
ovarian cancer. Gynecol Oncol. 137:173–179. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee SJ, Kim KH and Park KK: Mechanisms of
fibrogenesis in liver cirrhosis: The molecular aspects of
epithelial-mesenchymal transition. World J Hepatol. 6:207–216.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
O'Connor JW and Gomez EW: Biomechanics of
TGFβ-induced epithelial-mesenchymal transition: Implications for
fibrosis and cancer. Clin Transl Med. 3:232014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Takano M, Yamamoto C, Yamaguchi K, Kawami
M and Yumoto R: Analysis of TGF-β1- and drug-induced
epithelial-mesenchymal transition in cultured alveolar epithelial
cell line RLE/Abca3. Drug Metab Pharmacokinet. 30:111–118. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
PLOS ONE Staff, . Correction: Melatonin
inhibits endoplasmic reticulum stress and epithelial-mesenchymal
transition during bleomycin-induced pulmonary fibrosis in mice.
PloS One. 10:e01193812015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou N, Lu F, Liu C, Xu K, Huang J, Yu D
and Bi L: IL-8 induces the epithelial-mesenchymal transition of
renal cell carcinoma cells through the activation of AKT signaling.
Oncol Lett. 12:1915–1920. 2016.PubMed/NCBI
|
|
29
|
Shan B, Man H, Liu J, Wang L, Zhu T, Ma M,
Xv Z, Chen X, Yang X and Li P: TIM-3 promotes the metastasis of
esophageal squamous cell carcinoma by targeting
epithelial-mesenchymal transition via the Akt/GSK-3β/Snail
signaling pathway. Oncol Rep. 36:1551–1561. 2016.PubMed/NCBI
|
|
30
|
Yi GZ, Liu YW, Xiang W, Wang H, Chen ZY,
Xie SD and Qi ST: Akt and β-catenin contribute to TMZ resistance
and EMT of MGMT negative malignant glioma cell line. J Neurol Sci.
367:101–106. 2016. View Article : Google Scholar : PubMed/NCBI
|