Introduction
Nonalcoholic fatty liver disease (NAFLD) is
currently considered the most common chronic liver disease in
developed countries, and is closely associated with insulin
resistance and other metabolic risk factors, including central
abdominal obesity and dyslipidemia (1). In recent years, the morbidity
associated with NAFLD has increased, whereas the age of onset is
decreasing. However, the exact pathogenesis of NAFLD remains to be
fully elucidated, and at present, there is no specific
pharmacological approach to regulate hepatic steatosis.
Due to insulin resistance, elevated insulin
stimulates blood sugar uptake and storage through binding to the
insulin receptor (IR), which promotes phosphorylation of IR
β-subunit (IRβ) and IR substrate-1 (IRS-1), and activation of
downstream signaling via phosphatidylinositol 3-kinase
(PI3K)-protein kinase B (Akt) (2,3).
C-Jun N-terminal kinase (JNK; including JNK1, JNK2 and JNK3
isoforms) is associated with insulin resistance and is activated by
inflammatory cytokines and free fatty acids (FFAs), which are
associated with type 2 diabetes (2,4,5). A
previous study suggested that JNK activation accelerates lipid
accumulation and causes liver injury (6). Furthermore, activated JNK
phosphorylates its downstream target, IRS-1, on serine 307 to
attenuate insulin sensitivity (7).
Sun et al (8) reported that
activation of the inositol-requiring protein 1 (IRE1)-JNK pathway
is a key factor in impaired hepatic insulin signaling transduction
in the livers of mice fed a high-fructose diet (8). In addition, JNK activation is able to
upregulate the expression levels of Beclin-1 and
microtubule-associated protein 1A/1B light chain 3 (LC3)II in
cancer cells, whereas SP600125 (an inhibitor of JNK) or JNK
knockdown reversed this process (9). Kluwe et al demonstrated that
hepatic fibrosis was modulated by JNK inhibition in hepatic
stellate cells and JNK1-deficient mice (10). These findings suggested that JNK
signaling may serve a crucial role in the initiation and
progression of NAFLD.
Autophagy is a crucial physiological process that
has an important role in cellular homeostasis by regulating
intracellular lipid stores, and eliminating damaged organelles and
misfolded proteins in obesity (11,12).
Autophagy has been reported to be implicated in liver physiology
and pathogenesis (13). Autophagy
dysfunction has been detected in the liver from patients with NAFLD
and in murine models of NAFLD, as well as in lipid-overloaded human
hepatocytes (14–16). A previous study demonstrated that
activation of autophagy by rapamycin ameliorated endoplasmic
reticulum (ER) stress and decreased apoptosis in human
hepatocarcinoma-7 cells (17).
Furthermore, inhibition of autophagy using 3-methyladenine markedly
increased triglyceride levels in hepatocytes treated with oleate
(11). Defective hepatic autophagy
in mouse models of obesity also promoted accumulation of fat in the
liver and aggravated HFD-induced liver injury. In addition, genetic
or molecular suppression of autophagy in various cells promotes ER
stress and results in defective IR signaling. Overexpression of
autophagy related gene 7 (Atg7) by adenovirus-Atg7 injection was
able to alleviate liver condition and insulin resistance in ob/ob
mice and in HFD-fed mice (18).
These findings suggested that autophagy may serve a protective role
in liver injury; however, conflicting results have also been
published regarding the function of autophagy in the progression of
NAFLD (11,19). A previous study in mice with
liver-specific FAK family kinase-interacting protein of 200 kDa
deficiency demonstrated that autophagy inhibition prevents
lipogenesis and reduces hepatic steatosis (14). Similarly, Kim et al
demonstrated that skeletal muscle-specific Atg7 deficiency
ameliorated insulin resistance and reduced diet-induced obesity in
mice fed a HFD (12). The
protective mechanism of autophagy in the pathological process of
NAFLD requires further elucidation to provide guidance for
autophagy-targeting therapeutic strategies for the treatment of
NAFLD.
The JNK signaling pathway is essential for the
autophagic process. Activation of JNK contributes to Beclin-1
expression, mediates dysregulated autophagy modulation and mediates
p53 phosphorylation (20). Wei
et al demonstrated that JNK1-mediated multisite
phosphorylation of B-cell lymphoma 2 (Bcl-2) stimulates
starvation-induced autophagy by disrupting the Bcl-2/Beclin-1
complex (21). A recent study
reported that FFA-stimulated autophagy in INS-1 cells is suppressed
by JNK inhibitor II (SB202190 and SB203580). Furthermore,
conversion of LC3I to LC3II in INS-1 cells treated with
JNK1-targeted small interfering RNA was significantly inhibited
compared with in the control group (22). These findings suggested that
abnormal autophagy interferes with lipid metabolism, and
dysfunctional autophagy may promote the pathogenesis of NAFLD.
However, the precise function of autophagy in lipid metabolism
remains controversial, since lipolytic and lipogenic functions of
autophagy have both been reported.
JNK, autophagy and insulin resistance are all
associated with the pathological process of NAFLD; however, the
interactive relationships among them are not fully understood.
Therefore, understanding the molecular mechanisms by which the JNK
signaling pathway mediates lipid-induced metabolic stress will be
of great significance for the development of novel treatments for
various obesity-associated diseases. In the present study, the
relationships among them were illustrated in vivo. A rat
model of NAFLD was used to investigate the roles of the JNK
signaling pathway in insulin resistance and autophagy. In addition,
the present study examined whether the symptoms of NAFLD in rats
could be alleviated by inhibition of JNK. The findings of the
present study define a core function for JNK in the progression of
NAFLD.
Materials and methods
Animals
Male Sprague Dawley rats (age, 8 weeks; 180–200 g)
were obtained from the Animal Center of Xi'an Jiaotong University
(Xi'an, China). Rats were maintained under standard conditions:
Temperature, 25°C; 12 h light/dark cycle; and ad libitum
standard laboratory feed and water. Rats were randomly divided into
two groups (n=10/group): Normal chow diet (ND) group or HFD group
(D12451; 45 kcal% fat; Research Diets, Inc., New Brunswick, NJ,
USA). The rats received the respective diets for 20 weeks. At the
end of treatment, rats were anesthetized with sodium pentobarbital
(Sigma-Aldrich; Merck Millipore, Darmstadt, Germany; 40 mg/kg body
weight; intraperitoneal injection) were sacrificed by cervical
dislocation for biochemical analysis. Muscle, adipose and liver
samples from the rats were subjected to western blotting.
Rats fed a HFD were intraperitoneally injected with
SP600125 (n=6;30 mg/kg body weight; Calbiochem; EMD Millipore,
Billerica, MA, USA) or phosphate-buffered saline (PBS; n=6) each
day at the end of 20 weeks for an additional 8 weeks. After
treatment, the rats were sacrificed by cervical dislocation for
biochemical analysis. Liver samples from the rats were subjected to
western blotting. The animal experiments were reviewed and approved
by the Animal Care and Use Committee of Shaanxi Provincial People's
Hospital (Xi'an, China), and were conducted in compliance with
Guide for the Care and Use of Laboratory Animals (version 8 in
Chinese, 2012).
Biochemical analyses
Serum alanine aminotransferase (ALT), aspartate
aminotransferase (AST), total cholesterol (T-CHO) and triglyceride
(TG) levels were determined using the cobas automatic analyzer
system (Roche Diagnostics, Basel, Switzerland). Blood samples were
centrifuged at 1,500 × g for 10 min at 4°C. The serum levels
of tumor necrosis factor (TNF)-α, FFA and insulin were measured
using ELISA kits (Assaypro, St. Charles, MO, USA) according to the
manufacturer's protocols.
Glucose tolerance and insulin
resistance tests
Glucose tolerance tests were conducted after the
rats had been fed a ND or HFD for 20 weeks. After 6 h fasting, rats
in the ND and HFD groups were injected intraperitoneally with
glucose (2.0 g/kg body weight). Blood was collected from the tail
vein at various time points (0, 15, 30, 60 and 120 min), and blood
glucose levels were measured using a portable glucose meter
(Glu-test Sensor; Yuwell-Jiangsu Yuyue Medical Equipment &
Supply Co., Ltd., Nanjing, China). To assess whether insulin
resistance occurs in the liver of rats fed a HFD, rats (n=6) were
fasted overnight and treated with an intraperitoneal injection of
insulin (1.5 U/kg). After 30 min, the livers were isolated and
subjected to western blot analysis for detection of p-IRβ, p-IRS-1
and p-Akt.
Western blot analysis
Proteins were isolated from tissues using
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology, Haimen, China). The homogenates were centrifuged at
15,000 × g for 30 min at 4°C and the pellets were discarded.
Protein concentration was measured using a BioSpectrometer kinetic
spectrometer using the Lowry protein assay kit (Pierce; Thermo
Fisher Scientific, Inc., Waltham, MA, USA). A total of 20 µg
protein was separated by 8% SDS-polyacrylamide gel electrophoresis,
with samples being loaded onto each lane of the polyacrylamide
gels. Subsequently, proteins in the gels were blotted onto
polyvinylidene difluoride membranes (EMD Millipore, Bedford, MA,
USA). The membranes were blocked with 5% nonfat milk in TBS
containing 0.1% Tween-20 at room temperature for 1 h, and were then
probed with primary antibodies at 4°C overnight. The following
primary antibodies were used: Anti-Akt (cat. no. 2920; 1:1,000),
anti-phosphorylated (p)-Akt (Ser473) (cat. no. 4060; 1:1,000),
anti-Atg3 (cat. no. 3415; 1:1,000), anti-Atg5 (cat. no. 12994;
1:1,000), anti-LC3 (cat. no. 4108; 1:1,000), anti-Beclin-1 (cat.
no. 3783; all Cell Signaling Technology, Inc., Danvers, MA, USA);
anti-IRS1 (cat. no. 05-784R; 1:2,000), anti-p-IRS1 (Ser307) (cat.
no. 05-1087; 1:2,000) (both EMD Millipore); anti-IRβ (cat. no.
sc-711; 1:500), anti-p-IRβ (Tyr 1150/1151) (cat. no. sc-81500;
1:500), anti-p-JNK1 (Thr183) (cat. no. sc-135642; 1:200), anti-JNK1
(cat. no. sc-571; 1:400) and anti-GAPDH (cat. no. sc-20357;
1:1,000; all Santa Cruz Biotechnology, Inc., Dallas, TX, USA).
Blots were then incubated with corresponding horseradish
peroxidase-conjugated secondary antibodies (Invitrogen; Thermo
Fisher Scientific, Inc.) for 1 hour at room temperature. The blots
were visualized using an enhanced chemiluminescence detection
system (EMD Millipore) and were exposed to film. Quantification was
performed using Image Lab version 2.0 (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA)
Statistical analysis
The results are presented as the mean ± standard
deviation. Data were analyzed by unpaired Student's t-test. One-way
analysis of variance was used to compare the means of ≥3 groups.
All data analyses were performed using SPSS v. 17.0 software (SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
HFD induces impaired insulin
resistance and liver function injury in rats
The body weight of 8-week-old rats fed a ND or HFD
was measured every 4 weeks for 20 weeks. After 8 weeks of feeding,
the body weight of the HFD-fed rats was markedly higher compared
with the ND rats (Fig. 1A). To
investigate the effects of NAFLD on insulin resistance, glucose
concentration was measured in rats intraperitoneally injected with
glucose. After the injection, blood glucose concentration in ND
rats increased slightly, but decreased to fasting levels by 120
min. In HFD-fed rats, the blood glucose concentration increased
gradually and reached its peak at 60 min. Glucose concentration did
not return to normal levels until 120 min (Fig. 1B). Furthermore, the serum levels of
ALT, AST, T-CHO, TG, TNF-α, FFA and insulin were markedly higher in
HFD-fed rats compared with ND-fed rats (Fig. 1C-F).
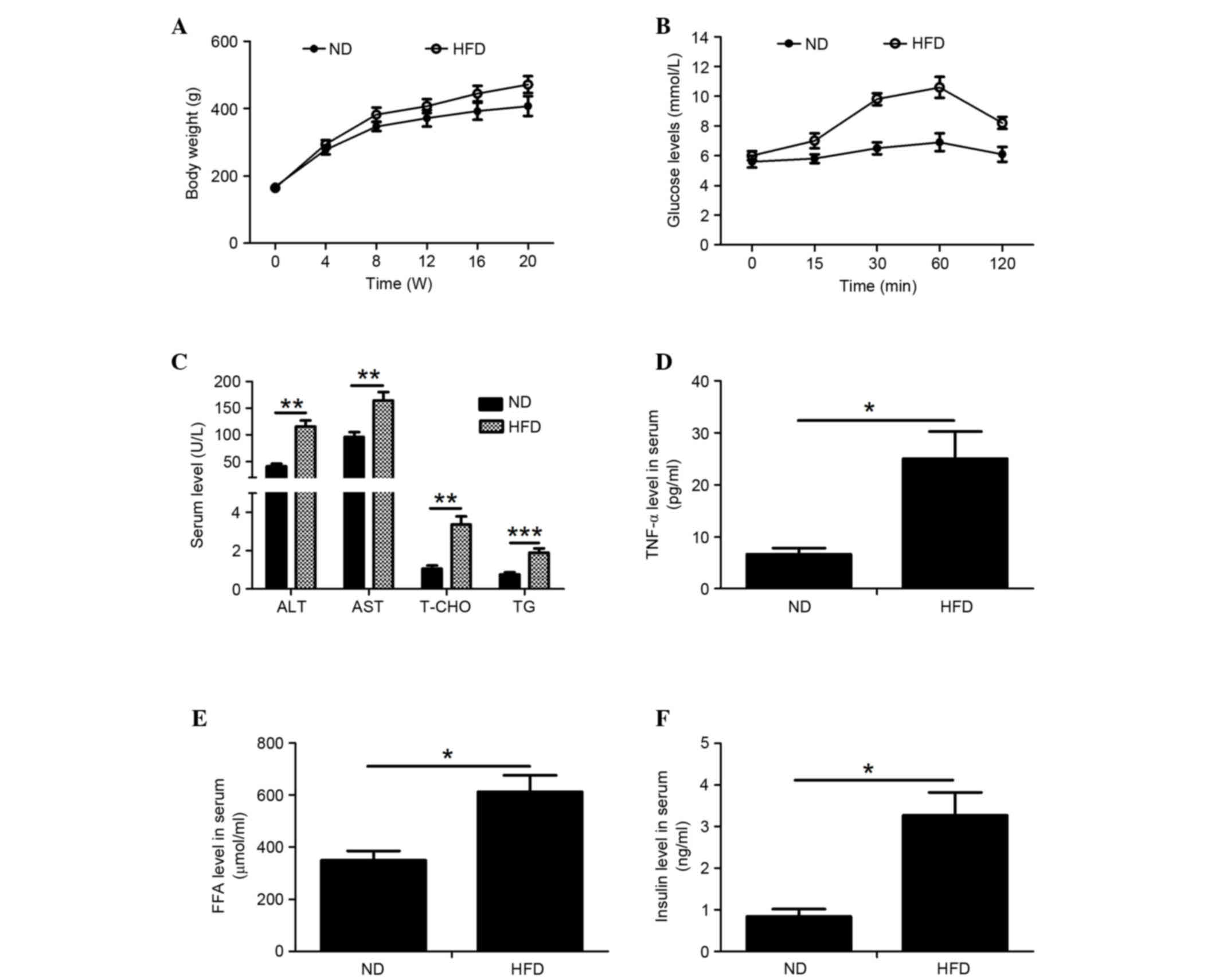 | Figure 1.Effects of a HFD on body weight and
biochemical indicators of nonalcoholic fatty liver disease. (A)
Body weight of rats fed a ND or HFD for 20 weeks. (B) After 20
weeks of feeding, fasting blood glucose levels in the rat models
were determined at 0, 15, 30, 60 and 120 min after glucose
injection (2.0 g/kg body weight). The serum concentrations of (C)
ALT, AST, T-CHO and TG; (D) TNF-α; (E) FFA and (F) insulin in rats
fed a HFD or ND were detected. Data are presented as the mean ±
standard deviation (n=6). *P<0.05, **P<0.01, ***P<0.001.
HFD, high-fat diet; ND, normal diet; ALT, alanine aminotransferase;
AST, aspartate aminotransferase; T-CHO, total cholesterol; TNF-α,
tumor necrosis factor-α; FFA, free fatty acid. |
HFD induces JNK activation, increased
autophagy and insulin resistance in the liver
The JNK pathway is known to be activated by several
factors, including oxidative stress, FFAs and TNF-α. To investigate
the effects of HFD on the JNK pathway, rats were fed a HFD for 20
weeks, and the protein expression levels of p-JNK1 and JNK1 were
detected in muscle, adipose and liver samples from the rats. As
shown in Fig. 2A, the protein
expression levels of p-JNK1 were significantly increased in HFD-fed
rats compared with in the control group. These results indicate
that JNK is activated in muscle, adipose and liver tissues from
HFD-fed rats.
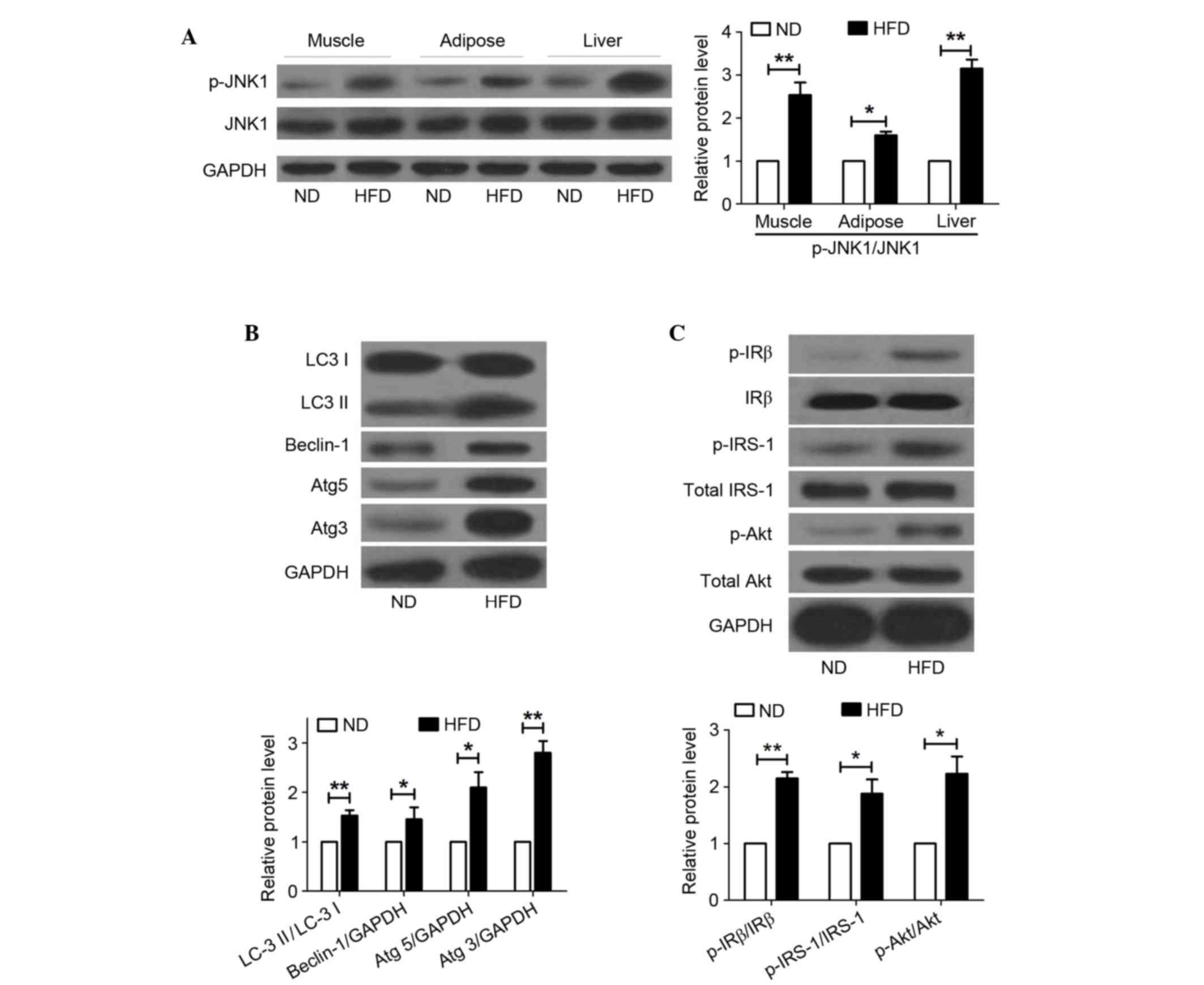 | Figure 2.HFD induces JNK activation, increased
autophagy and insulin resistance in the liver. (A) Western blot
analysis of p-JNK1 and JNK1 in muscle, adipose and liver tissues
from a rat model of nonalcoholic fatty liver disease. (B) Autophagy
indicators were detected in the liver of rats fed a HFD by western
blot analysis. Autophagy levels were assessed by LC3-II/LC3-I
ratio, and Beclin-1, Atg5 and Atg3 protein expression. (C) Insulin
resistance was examined by western blot analysis using p-IRβ, IRβ,
p-IRS-1, total IRS-1, p-Akt and total Akt antibodies. GAPDH was
used as a loading control. Data are presented as the mean ±
standard deviation (n=6). *P<0.05, **P<0.01. HFD, high-fat
diet; ND, normal diet; p-, phosphorylated; JNK1, c-Jun N-terminal
kinase 1; LC3, microtubule-associated protein 1A/1B light chain 3;
Atg, autophagy related gene; IRβ, insulin receptor β-subunit; IRS,
insulin receptor substrate-1; Akt, protein kinase B. |
To investigate the roles of autophagy in NAFLD,
liver samples from rats fed a ND or HFD were subjected to western
blotting. Notably, HFD markedly elevated autophagy in the liver
samples of the NAFLD rat model, as evidenced by LC3 conversion (LC3
I to LC3 II), and upregulation of Beclin-1, Atg5 and Atg3 protein
expression levels (Fig. 2B). These
data indicate that autophagy may be upregulated in NAFLD. It may be
hypothesized that autophagy signals are associated with the
pathogenesis of NAFLD.
To assess whether insulin resistance occurs in the
liver of rats fed a HFD, rats were fasted overnight and treated
with an intraperitoneal injection of insulin (1.5 U/kg). After 30
min, the livers were isolated and subjected to western blot
analysis. The expression levels of p-IRβ, p-IRS-1 and p-Akt were
significantly higher in HFD-fed rats compared with in the control
group, which indicated that HFD increased insulin resistance in the
liver of rats (Fig. 2C).
JNK inhibition suppresses autophagy
and improves the insulin signaling pathway in HFD rats
A previous study reported that autophagy-mediated
insulin receptor downregulation contributes to insulin resistance
in vitro and in vivo (23). In addition, JNK has been shown to
be implicated in impaired insulin signaling transduction via the
IRE1-JNK pathway (8). To
investigate the exact role of JNK in the pathological process of
NAFLD, the present study examined whether SP600125 was able to
reverse pathological events, including abnormal autophagy and
decreased insulin sensitivity, in the livers of HFD-fed rats. JNK1
activity was suppressed in rats by SP600125 treatment (Fig. 3A). Subsequently, protein expression
levels were detected in rats intraperitoneally injected with
SP600125 or PBS, in order to assess changes in autophagy and
insulin signaling. The protein expression levels of LC3 II,
Beclin-1, Atg5, Atg3, p-IRβ, p-IRS-1 and p-Akt were markedly
decreased in the livers of rats treated with SP600125 compared with
in the control group (Fig. 3B and
C). These results suggest that inhibition of JNK1
phosphorylation may suppress autophagy and improve insulin
signaling in a rat model of NAFLD.
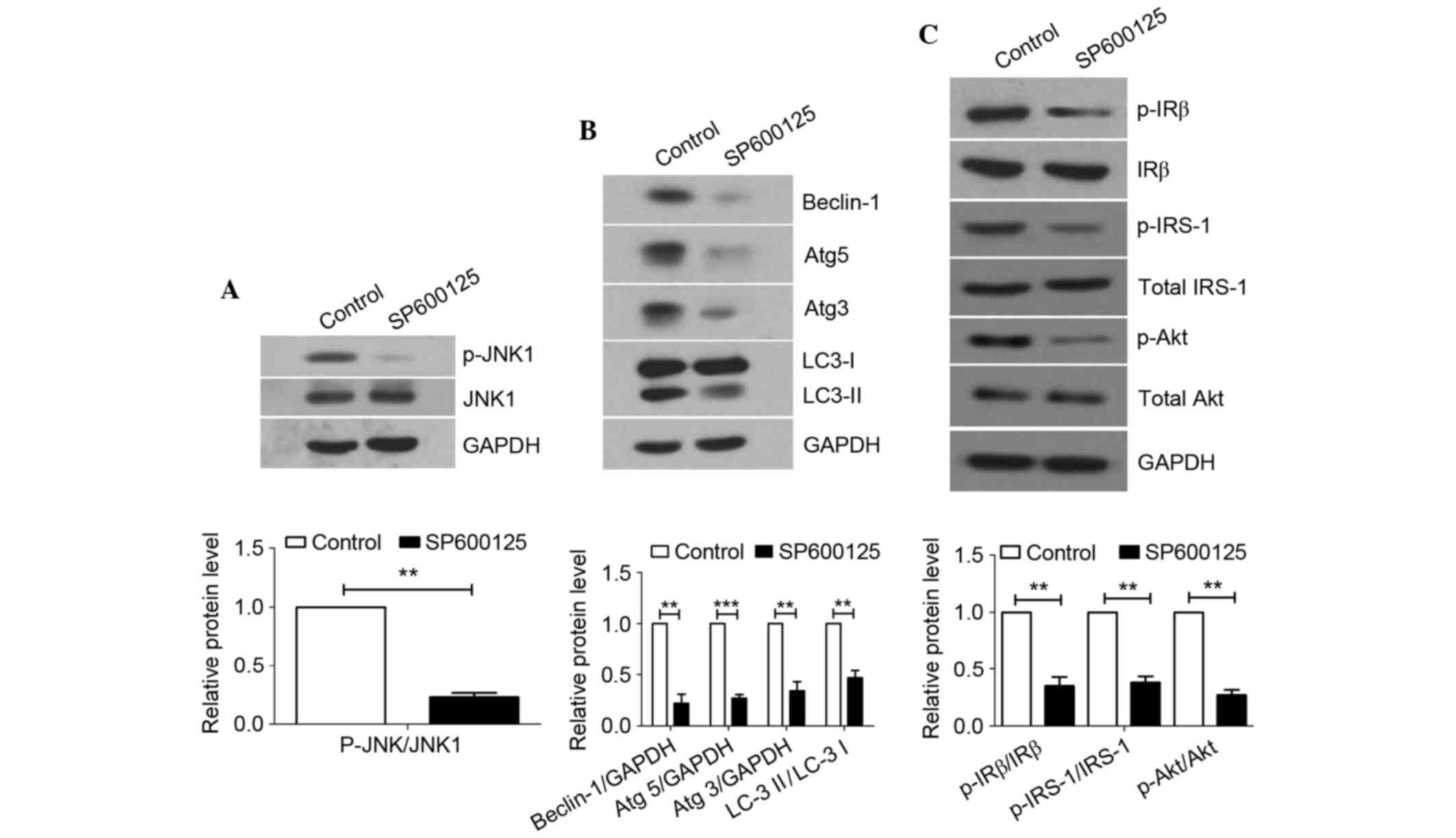 | Figure 3.Effects of SP600125 on autophagy and
the insulin signaling pathway in HFD-fed rats. After 20 weeks of
HFD feeding, the rats were intraperitoneally injected with SP600125
or PBS once a day for 8 weeks, and the rats were then sacrificed.
Total protein samples from the livers of HFD rats treated with or
without SP600125 were subjected to western blotting. (A)
Phosphorylation of JNK was inhibited by SP600125. (B) LC3
conversion, and Beclin-1, Atg5 and Atg3 expression were measured as
markers of autophagy. (C) Expression levels of p-IRβ, p-IRS-1 and
p-Akt in the liver of rat models were measured to observe changes
in insulin resistance. GAPDH was used as a loading control. Data
are presented as the mean ± standard deviation from independent
mice (n=6). **P<0.01, ***P<0.001. HFD, high-fat diet; ND,
normal diet; p-, phosphorylated; JNK1, c-Jun N-terminal kinase 1;
LC3, microtubule-associated protein 1A/1B light chain 3; Atg,
autophagy related gene; IRβ, insulin receptor β-subunit; IRS,
insulin receptor substrate-1; Akt, protein kinase B. |
SP600125 treatment alleviates symptoms
of NAFLD in rats fed a HFD
Serum biochemical indexes in HFD-fed rats were
detected following intraperitoneal injection with SP600125 or PBS
for an additional 8 weeks. The serum levels of ALT, AST, T-CHO, TG,
TNF-α, FFA and insulin were significantly decreased in HFD-fed rats
treated with SP600125 compared with in the control group (Fig. 4A-D). These results indicate that
JNK inhibition may ameliorate NAFLD in HFD-fed rats.
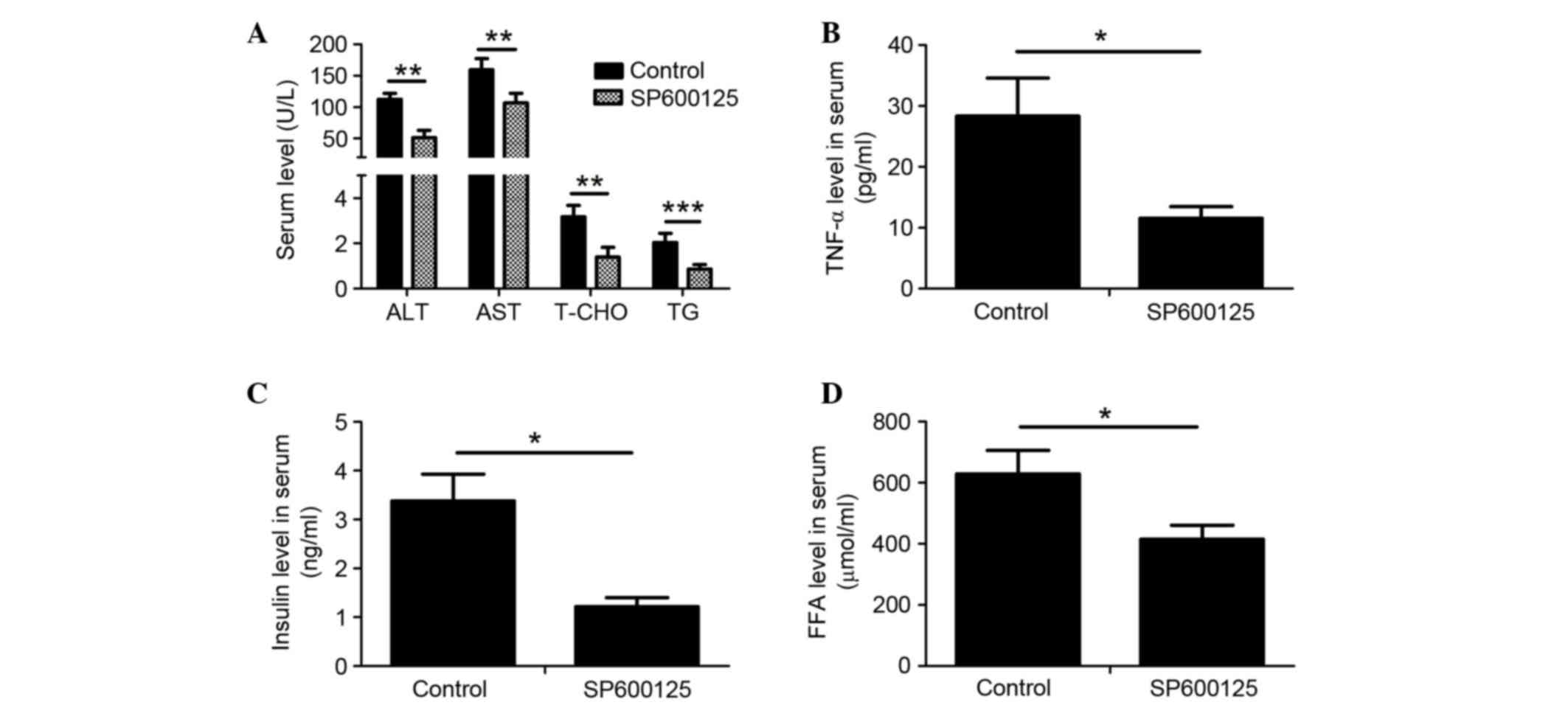 | Figure 4.Treatment with SP600125 ameliorated
symptoms of nonalcoholic fatty liver disease in HFD-fed rats. The
serum levels of (A) ALT, AST, T-CHO and TG; (B) TNF-α; (C) insulin
and (D) FFA were determined in HFD-fed rats injected with SP600125
or PBS. Data are presented as the mean ± SD (n=6). *P<0.05,
**P<0.01, ***P<0.001. HFD, high-fat diet; ND, normal diet;
ALT, alanine aminotransferase; AST, aspartate aminotransferase;
T-CHO, total cholesterol; TNF-α, tumor necrosis factor-α; FFA, free
fatty acid. |
Discussion
Although previous studies have demonstrated that
JNK, autophagy and insulin resistance are correlated, the role of
JNK in aberrant autophagy and insulin resistance in the
pathogenesis of NAFLD has not yet been addressed. The present study
hypothesized that the JNK signaling pathway may be a key modulator
of autophagy function in HFD-induced insulin resistance. The
results demonstrated that JNK inhibition attenuated insulin
resistance and autophagic activity in vivo.
It is widely known that obesity and insulin
resistance increase the risk of NAFLD. After years of research, the
association between lipids and insulin resistance is widely
accepted. Insulin resistance is closely associated with the
pathological progression of NAFLD, and JNK is a crucial molecule in
the insulin signaling pathway. JNK is activated by almost all forms
of metabolic stress, which have been implicated in insulin
resistance. Previous studies have suggested that TNF-α, FFAs and
reactive oxygen species activate JNK, thus promoting initiation and
progression of insulin resistance (24,25).
In the present study, in HFD-fed obese rats, JNK activity was
significantly increased in insulin-sensitive tissues, such as liver
and adipose. Activated JNK phosphorylates the serine residues of
IRS1, thus contributing to obesity-induced insulin resistance.
Furthermore, JNK is critically involved in the promotion of
diet-induced fatty liver and metabolic inflammation. JNK1, but not
JNK2, has been reported to serve as a crucial molecular link
between obesity, metabolic inflammation and disorders of glucose
homeostasis (26). Notably,
knockout of JNK1 markedly reduced insulin resistance in HFD-fed
JNK1-null mice; conversely, mice with JNK2 deficiency fed a HFD
were obese and insulin-resistant (27). Özcan et al reported that
obesity-induced ER stress may lead to suppression of insulin
receptor signaling via hyperactivation of JNK (28). Growing evidence has suggested that
JNK may be considered a potential therapeutic target for the
treatment of insulin resistance and diabetes. Therefore,
elucidating the exact mechanisms by which JNK triggers insulin
resistance and results in NAFLD is extremely urgent. To explore
this complex pathogenesis, the present study measured JNK activity
in the muscle, adipose and liver tissues of rats fed a ND or HFD.
Consistent with the results of previous studies, JNK was activated
in HFD rats. Furthermore, western blot analysis indicated that
autophagy, and IRS-1 and Akt phosphorylation were induced in the
liver of rats fed a HFD. The present study focused on the effects
of JNK inhibition on autophagy and insulin resistance occurring in
NAFLD.
Autophagy is essential for regulating the
degradation of lipid droplets when a nutritional deficiency occurs
in cells. Autophagic dysfunction may lead to excessive lipid
accumulation in the liver, thus contributing to the emergence and
progression of NAFLD (29).
Autophagy-related gene expression, autophagosomes and the protein
levels of autophagy markers have been shown to be significantly
increased in obese adipose tissue, along with insulin resistance
(30). Conversely, hepatic
triglyceride content was increased in rats with an adipose-specific
deletion of Atg7 (31). The
mammalian target of rapamycin (mTOR) pathway is associated with
insulin signaling in several cell lines. A previous study
demonstrated that increased activation of mTOR and S6 kinase beta-1
(S6K1) may promote serine phosphorylation of IRS-1, thus resulting
in the pathogenesis of hepatic insulin resistance in obese rats
(32). Mitochondrial uncoupling
protein 2 protects against palmitic acid-induced liver injury by
enhancing the function of autophagy (33). The present study demonstrated that
serine 307 of IRS-1 was hyperphosphorylated, accompanied by
increased p-Akt (Ser473), in HFD-fed rats. Furthermore, activated
autophagic flux was observed in the livers of rats fed a HFD. It
may be hypothesized that the JNK-mediated autophagy pathway was of
great importance in insulin resistance. These results may provide
insight into potential NAFLD therapies.
JNK has been reported to regulate autophagy in
Drosophila and mammalian cells, in response to various
stressors (34). Using a JNK
inhibitor and a dominant-negative mutant of JNK, Shimizu et
al demonstrated that inactivated JNK can suppress autophagic
cell death (20). A previous study
demonstrated that autophagy is inhibited by increased insulin via
the mTOR signaling pathway (17).
Further associations between dysfunctional autophagy and insulin
resistance must be identified. In the present study, the effects of
JNK inhibition on autophagy and insulin resistance were detected in
HFD-fed rats. The results suggested that JNK inhibition by SP600125
attenuated autophagy and prevented insulin resistance in the liver
of rats fed a HFD. The molecular mechanism by which SP600125
inhibits autophagy and improves insulin resistance in the liver of
HFD-fed rats remains to be elucidated.
In conclusion, the present study is the first, to
the best of our knowledge, to demonstrate that JNK-mediated
autophagy was involved in insulin resistance. Further experiments
are required to explore the molecular mechanisms by which p-JNK
interferes with autophagy-related gene expression, and subsequently
contributes to insulin resistance. The present study also provided
evidence suggesting that obesity is closely associated with
abnormally elevated JNK1 activity. The results of the present study
suggested that selective downregulation of JNK activity presents an
attractive opportunity for the treatment of human obesity, insulin
resistance, type 2 diabetes and NAFLD. JNK may be considered a
potential drug target for the prevention and treatment of
NAFLD.
Glossary
Abbreviations
Abbreviations:
|
NAFLD
|
nonalcoholic fatty liver disease
|
|
JNK
|
c-Jun N-terminal kinase
|
|
IR
|
insulin receptor
|
|
HFD
|
high-fat diet
|
|
IRβ
|
insulin receptor β-subunit
|
|
IRS-1
|
insulin receptor substrate-1
|
|
PI3K
|
phosphatidylinositol 3-kinase
|
|
Akt
|
protein kinase B
|
|
Atg7
|
autophagy related gene 7
|
|
ALT
|
alanine aminotransferase
|
|
AST
|
aspartate aminotransferase
|
|
T-CHO
|
total cholesterol
|
|
TG
|
triglyceride
|
|
FFAs
|
free fatty acids
|
|
TNF-α
|
tumor necrosis factor-α
|
|
mTOR
|
mammalian target of rapamycin
|
References
|
1
|
Chang E, Park CY and Park SW: Role of
thiazolidinediones, insulin sensitizers, in non-alcoholic fatty
liver disease. J Diabetes Investig. 4:517–524. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hirosumi J, Tuncman G, Chang L, Görgün CZ,
Uysal KT, Maeda K, Karin M and Hotamisligil GS: A central role for
JNK in obesity and insulin resistance. Nature. 420:333–336. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brännmark C, Nyman E, Fagerholm S,
Bergenholm L, Ekstrand EM, Cedersund G and Strålfors P: Insulin
signaling in type 2 diabetes: Experimental and modeling analyses
reveal mechanisms of insulin resistance in human adipocytes. J Biol
Chem. 288:9867–9880. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sethi JK and Hotamisligil GS: The role of
TNF alpha in adipocyte metabolism. Semin Cell Dev Biol. 10:19–29.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Boden G: Role of fatty acids in the
pathogenesis of insulin resistance and NIDDM. Diabetes. 46:3–10.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schattenberg JM, Singh R, Wang Y,
Lefkowitch JH, Rigoli RM, Scherer PE and Czaja MJ: JNK1 but not
JNK2 promotes the development of steatohepatitis in mice.
Hepatology. 43:163–172. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hiratani K, Haruta T, Tani A, Kawahara J,
Usui I and Kobayashi M: Roles of mTOR and JNK in serine
phosphorylation, translocation and degradation of IRS-1. Biochem
Biophys Res Commun. 335:836–842. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun RQ, Wang H, Zeng XY, Chan SM, Li SP,
Jo E, Leung SL, Molero JC and Ye JM: IRE1 impairs insulin signaling
transduction of fructose-fed mice via JNK independent of excess
lipid. Biochim Biophys Acta. 1852:156–165. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li DD, Wang LL, Deng R, Tang J, Shen Y,
Guo JF, Wang Y, Xia LP, Feng GK, Liu QQ, et al: The pivotal role of
c-Jun NH2-terminal kinase-mediated Beclin 1 expression during
anticancer agents-induced autophagy in cancer cells. Oncogene.
28:886–898. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kluwe J, Pradere JP, Gwak GY, Mencin A, De
Minicis S, Osterreicher CH, Colmenero J, Bataller R and Schwabe RF:
Modulation of hepatic fibrosis by c-Jun-N-terminal kinase
inhibition. Gastroenterology. 138:347–359. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Singh R, Kaushik S, Wang Y, Xiang Y, Novak
I, Komatsu M, Tanaka K, Cuervo AM and Czaja MJ: Autophagy regulates
lipid metabolism. Nature. 458:1131–1135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim KH, Jeong YT, Oh H, Kim SH, Cho JM,
Kim YN, Kim SS, Kim do H, Hur KY, Kim HK, et al: Autophagy
deficiency leads to protection from obesity and insulin resistance
by inducing Fgf21 as a mitokine. Nat Med. 19:83–92. 2013.PubMed/NCBI
|
|
13
|
Rautou PE, Mansouri A, Lebrec D, Durand F,
Valla D and Moreau R: Autophagy in liver diseases. J Hepatol.
53:1123–1134. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma D, Molusky MM, Song J, Hu CR, Fang F,
Rui C, Mathew AV, Pennathur S, Liu F, Cheng JX, et al: Autophagy
deficiency by hepatic FIP200 deletion uncouples steatosis from
liver injury in NAFLD. Mol Endocrinol. 27:1643–1654. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li L, Hai J, Li Z, Zhang Y, Peng H, Li K
and Weng X: Resveratrol modulates autophagy and NF-κB activity in a
murine model for treating non-alcoholic fatty liver disease. Food
Chem Toxicol. 63:166–173. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Christian P, Sacco J and Adeli K:
Autophagy: Emerging roles in lipid homeostasis and metabolic
control. Biochim Biophys Acta. 1831:819–824. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gonzalez-Rodriguez A, Mayoral R, Agra N,
Valdecantos MP, Pardo V, Miquilena-Colina ME, Vargas-Castrillón J,
Lo Iacono O, Corazzari M, Fimia GM, et al: Impaired autophagic flux
is associated with increased endoplasmic reticulum stress during
the development of NAFLD. Cell Death Dis. 5:e11792014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang L, Li P, Fu S, Calay ES and
Hotamisligil GS: Defective hepatic autophagy in obesity promotes ER
stress and causes insulin resistance. Cell Metab. 11:467–478. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shibata M, Yoshimura K, Furuya N, Koike M,
Ueno T, Komatsu M, Arai H, Tanaka K, Kominami E and Uchiyama Y: The
MAP1-LC3 conjugation system is involved in lipid droplet formation.
Biochem Biophys Res Commun. 382:419–423. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shimizu S, Konishi A, Nishida Y, Mizuta T,
Nishina H, Yamamoto A and Tsujimoto Y: Involvement of JNK in the
regulation of autophagic cell death. Oncogene. 29:2070–2082. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wei Y, Pattingre S, Sinha S, Bassik M and
Levine B: JNK1-mediated phosphorylation of Bcl-2 regulates
starvation-induced autophagy. Mol Cell. 30:678–688. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Komiya K, Uchida T, Ueno T, Koike M, Abe
H, Hirose T, Kawamori R, Uchiyama Y, Kominami E, Fujitani Y and
Watada H: Free fatty acids stimulate autophagy in pancreatic
β-cells via JNK pathway. Biochem Biophys Res Commun. 401:561–567.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou L, Zhang J, Fang Q, Liu M, Liu X, Jia
W, Dong LQ and Liu F: Autophagy-mediated insulin receptor
down-regulation contributes to endoplasmic reticulum stress-induced
insulin resistance. Mol Pharmacol. 76:596–603. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
D'Adamo E, Cali AM, Weiss R, Santoro N,
Pierpont B, Northrup V and Caprio S: Central role of fatty liver in
the pathogenesis of insulin resistance in obese adolescents.
Diabetes Care. 33:1817–1822. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kodama Y and Brenner DA: c-Jun N-terminal
kinase signaling in the pathogenesis of nonalcoholic fatty liver
disease: Multiple roles in multiple steps. Hepatology. 49:6–8.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Solinas G and Karin M: JNK1 and IKK beta:
Molecular links between obesity and metabolic dysfunction. FASEB J.
24:2596–2611. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Singh R, Wang Y, Xiang Y, Tanaka KE,
Gaarde WA and Czaja MJ: Differential effects of JNK1 and JNK2
inhibition on murine steatohepatitis and insulin resistance.
Hepatology. 49:87–96. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Özcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi
NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH and Hotamisligil
GS: Endoplasmic reticulum stress links obesity, insulin action and
type 2 diabetes. Science. 306:457–461. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Amir M and Czaƒja MJ: Autophagy in
nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol.
5:159–166. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kovsan J, Blüher M, Tarnovscki T, Klöting
N, Kirshtein B, Madar L, Shai I, Golan R, Harman-Boehm I, Schön MR,
et al: Altered autophagy in human adipose tissues in obesity. J
Clin Endocrinol Metab. 96:E268–E277. 2010.PubMed/NCBI
|
|
31
|
Zhang Y, Goldman S, Baerga R, Zhao Y,
Komatsu M and Jin S: Adipose-specific deletion of autophagy-related
gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl
Acad Sci USA. 106:19860–19865. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Khamzina L, Veilleux A, Bergeron S and
Marette A: Increased activation of the mammalian target of
rapamycin pathway in liver and skeletal muscle of obese rats:
Possible involvement in obesity-linked insulin resistance.
Endocrinology. 146:1473–1481. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lou J, Wang Y, Wang X and Jiang Y:
Uncoupling protein 2 regulates palmitic acid-induced hepatoma cell
autophagy. Biomed Res Int. 2014:8104012014.PubMed/NCBI
|
|
34
|
Sekine Y, Takeda K and Ichijo H: The
ASK1-MAP kinase signaling in ER stress and neurodegenerative
diseases. Curr Mol Med. 6:87–97. 2006. View Article : Google Scholar : PubMed/NCBI
|














