Introduction
Aortopathy, predominantly manifested as aortic
aneurysm and dissection, represents an important cause of mortality
in industrialized countries. An estimated 20% of patients with
aortopathy have a positive family history, indicating a genetic
predisposition to the disease (1).
Genetics-associated aortopathy can be divided into syndromic and
non-syndromic subgroups, depending on the presence or absence of
manifestations in other organ systems. Well-characterized syndromic
forms with aortic involvement include Marfan syndrome (OMIM
154700), vascular Ehlers-Danlos syndrome (vEDS; OMIM 130050),
Loeys-Dietz syndrome (OMIM 609192) and aneurysms-osteoarthritis
syndrome (OMIM 613795) (2). When
an underlying gene defect is suspected to be responsible for
aortopathy, identifying the causative gene is critical for patient
management and genetic counseling of the family (3). Affected individuals can benefit from
interval screening and timely intervention.
Familial aortopathy is an autosomal dominant
condition with significant genetic heterogeneity, and mutations in
>12 genes have been identified to account for the disorder
(4). Conventional methods to
identify genes underlying aortopathy depend primarily on
genotype-phenotype correlation. For example, patients with ocular,
skeletal and cardiovascular abnormalities are often found to harbor
fibrillin 1 (1) mutations.
However, the clinical manifestations vary substantially, ranging
from mild to severe systemic disease (5). In addition, a significant overlap of
clinical features has been observed in patients with aortopathy
caused by different genes (6). As
a result, Sanger sequencing of all candidate genes is required to
provide a molecular diagnosis, however, the procedure is laborious
and time consuming. The targeted next-generation sequencing method
allows for the simultaneous assessment of multiple genes of
interest and can be an efficient approach to identify the genes
underlying inherited aortopathy in individuals.
Pilot investigations investigating the utilization
of this method in the molecular diagnosis of patients with
aortopathy have shown promising results (7,8). In
the present study, targeted next-generation sequencing was
performed to elucidate the gene defects in a family with
aortopathy, in which the phenotype was atypical.
Materials and methods
Ethics statement and subjects
The present study was performed in accordance with
the review board of Nanjing Drum Tower Hospital (Nanjing, China),
and written informed consent was obtained from all study subjects.
All procedures were performed according to the tenets of the
Declaration of Helsinki.
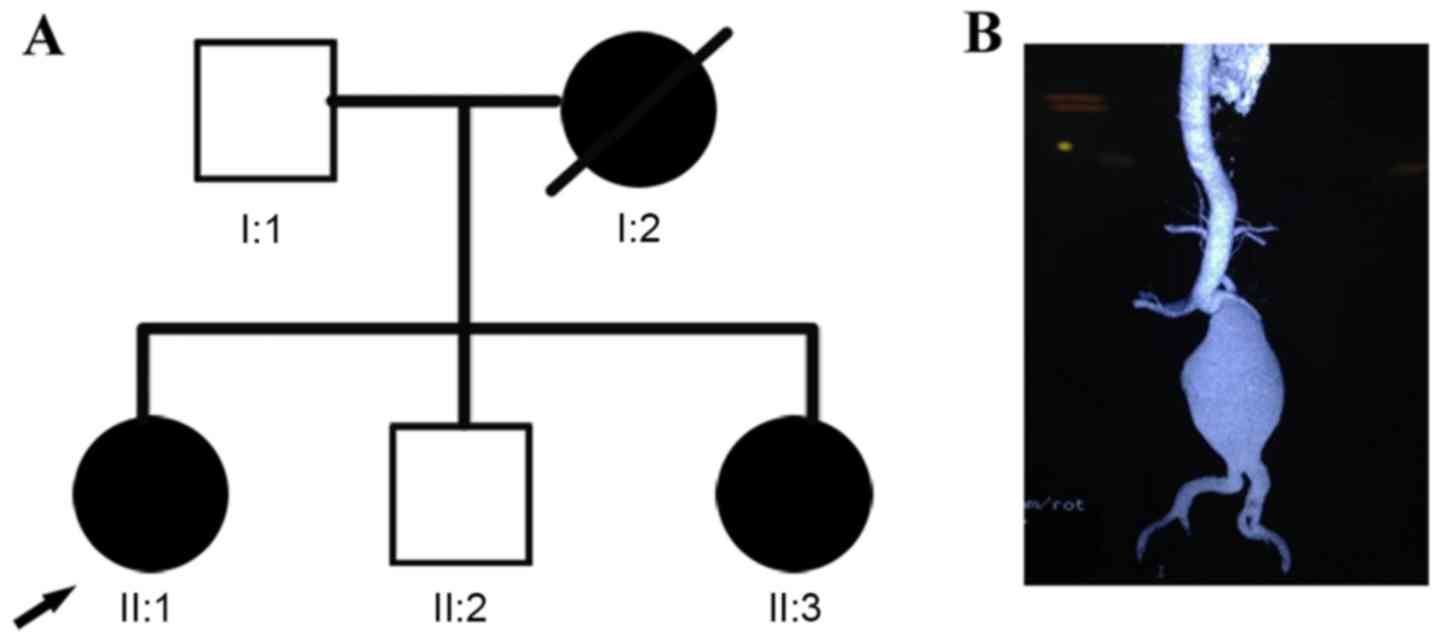
The subjects included in the present study comprised
a non-consanguineous Han Chinese family with suspected inherited
aortopathy from Jiangsu, China (Fig.
1A). The index case (II:1; proband) was a 48-year-old woman
with a ruptured abdominal aortic aneurysm (Fig. 1B), who underwent emergency surgery
for repair. During vascular prostheses replacement, blood vessel
friability was noted and the procedure duration was twice that
estimated. The patient developed renal failure due to this extended
duration of surgery. Clinical examination revealed no pronounced
craniofacial, skeletal or skin anomalies. The patient also denied
any tendency to bruise. The proband's sister (II:3) reported a
similar disease experience at the age of 50 years. Her mother (I:2)
succumbed to mortality suddenly at the age of 52 years of an
unknown cardiovascular cause. Peripheral blood (2 ml) was collected
in EDTA-coated tubes from available family members and stored at
−80°C. Genomic DNA was extracted using standard protocols. As a
control group, 100 unrelated, ethnically-matched, healthy
individuals were recruited at the Center of Physical Examination of
Nanjing Drum Tower Hospital.
Illumina library construction
Genomic DNA was purified and quantified using a
NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The generation of a targeted Illumina library
was performed according to the manufacture's protocol (MyGenostics,
Inc., Beijing, China). A final library size of 350–400 bp including
adaptor sequencings was selected.
Targeted capture, sequencing and
bioinformatics analysis
A panel of 428 genes associated with cardiovascular
diseases, including 12 known disease-causing genes [FBN1,
EGF-containing fibulin-like extracellular matrix protein 2,
collagen type III α1 (COL3A1), transforming growth factor β (TGFB)
receptor 1, TGFB receptor 2, TGFB2, TGFB3, small mothers against
decapentaplegic (SMAD) 3, SMAD4, actin α2 smooth muscle aorta,
myosin heavy chain 11 and mysosin light chain kinase], were
selected using a gene capture strategy with the GenCap custom
enrichment kit (MyGenostics, Inc), as previously described
(9). The enriched libraries were
sequenced on an Illumina Solexa HiSeq 2000 sequencer for 100-paired
reads.
Following sequencing, high-quality reads were
retrieved from raw reads by filtering out low-quality reads and
adaptor sequences using the Solexa QA package (10) and Cutadapt program (http://code.google.com/p/cutadapt/),
respectively. Short Oligonucleotide Analysis Package (SOAP) aligner
software (SOAP2.21; soap.genomics.org.cn/soapsnp.html) was then used to
align the clean reads to the reference human genome (hg19)
(11). Polymerase chain reaction
(PCR) duplicates were removed using the Picard program (12). Subsequently, single nucleotide
polymorphisms (SNPs) were determined using the SOAPsnp program
(11), reads were realigned using
Burrows-Wheeler Aligner (13), and
the deletions and insertions (InDels) were detected using Genome
Analysis Toolkit software (14).
The identified SNPs and InDels were annotated using the
Exome-assistant program (http://122.228.158.106/exomeassistant). MagicViewer
was used to view the short read alignment, and confirm the
candidate SNPs and InDels (15).
Non-synonymous variants were evaluated using the four algorithms,
PolyPhen (http://genetics.bwh.harvard.edu/pph2/), Sorting
Intolerant From Tolerant [SIFT; (http://sift.jcvi.org/)], Protein Analysis Through
Evolutionary Relationships (PANTHER; www.pantherdb.org) and Pathogenic Mutation Prediction
(Pmut; http://mmb.pcb.ub.es/PMut/) to determine
pathogenicity (16).
Mutation validation and analysis
Genomic DNA from all available family members were
obtained for Sanger sequencing. The PCR samples were visualized on
agarose gels, purified and sequenced on an ABI PRISM 3730 genetic
analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.) using
the terminator cycle sequencing method. Sites of variation were
identified through a comparison of DNA sequences with the
corresponding GenBank (www.ncbi.nlm.nih.gov) reference sequences. Possible
pathogenic effects caused by the rare or novel non-synonymous SNPs
were evaluated using SIFT, PolyPhen2 and MutationTaster (http://www.mutationtaster.org/). Multiple
sequence alignments were performed using ClustalW2 with the default
setting (http://www.ebi.ac.uk/Tools/clustalw2/).
Structure analysis
The 3D structure of the procollagen III C-propeptide
(Protein Data Bank; www.rcsb.org/pdb/; reference, 4AE2) was used to
perform in silico analysis. The crystal structures of the mutant
proteins were predicted using the SWISS-MODEL online server
(17) and displayed using PyMol
software (version 1.5; www.pymol.org).
Results
Targeted next-generation
sequencing
The present study performed targeted capture and
next-generation sequencing of 428 genes implicated in
cardiovascular diseases. The average sequencing depth for the
targeted regions was ~328X. The sample had 93.40% coverage of the
targeted regions. There was 91.20 and 87.50% coverage of the
targeted exons for 10X and 20X, respectively. A total of 782
variants were identified in the sample. Among these, 394 missense,
nonsense, splicing variants and coding InDels were identified,
which more likely to be pathogenic. This was narrowed down to 41 by
excluding variants reported in the dbSNP138 (www.ncbi.nlm.nih.gov/SNP/), 1000 Genomes Project
(www.1000genomes.org), Exome Sequencing
Project (evs.gs.washington.edu/EVS/) and local dataset with
allele frequencies >0.01. This was further narrowed down to 15
by filtering benign variants predicted the by four algorithms,
PolyPhen, SIFT, PANTHER and Pmut. A missense variant, c.3775G>A
(p.A1259T) in exon 48 of COL3A1 (NM_000090), was most likely
disease-causing in this family, as the mutated gene is known to be
involved in inherited aortopathy (Table I). For this site, 51% (193/377)
reads supported for G, whereas 49% (184/377) reads supported for
A.
 | Table I.Number of variants present in the
proband (II:1) at different stages of the filtering process. |
Table I.
Number of variants present in the
proband (II:1) at different stages of the filtering process.
| Filter stage | n |
|---|
| Total variants
identified | 782 |
| Non-synonymous
variants | 394 |
| Variants with allele
frequencies ≤0.01 | 41 |
| Pathogenic variants
predicted by algorithms | 15 |
| Variants in
disease-causing genes of aortopathy |
1 |
Mutation validation
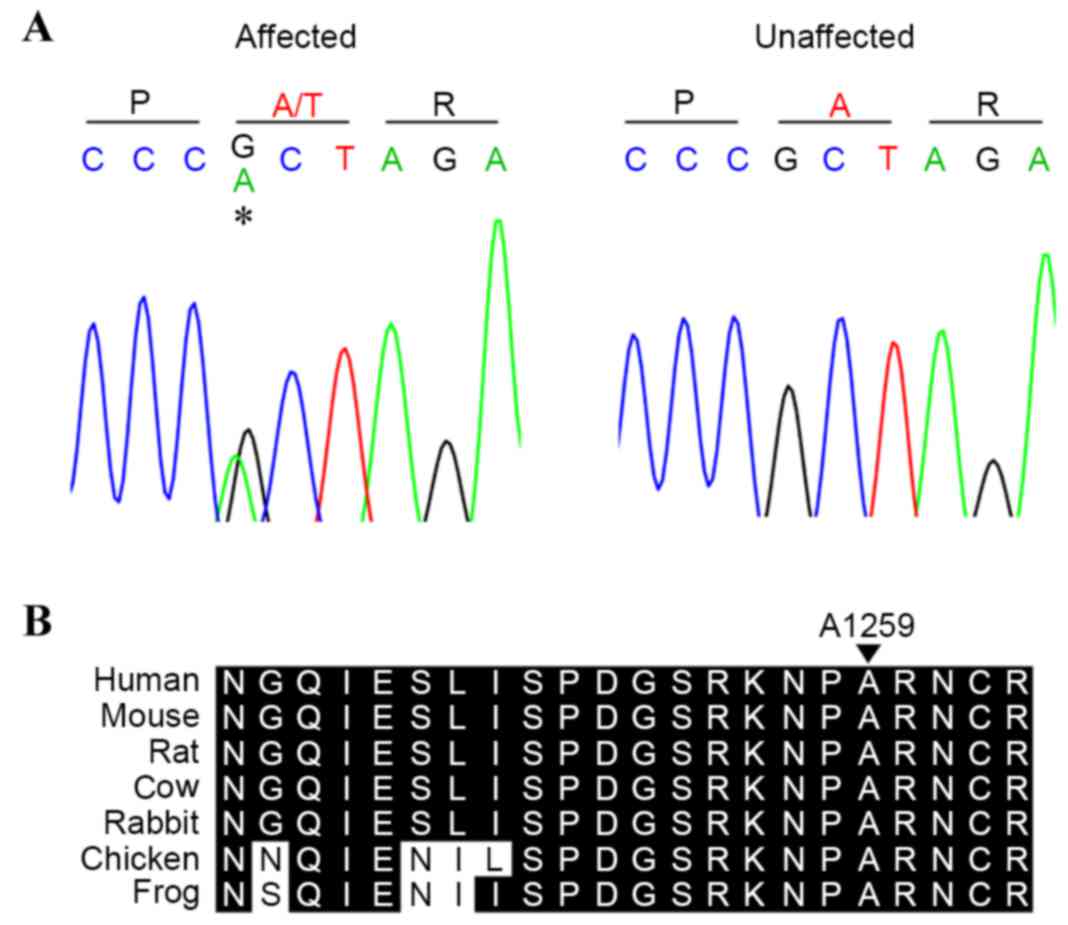
To confirm the above findings, the present study
genotyped four available members of the family (two affected, two
unaffected) to identify the c.3775G>A (p.A1259T) variant by
Sanger sequencing (Fig. 2A). The
affected individuals (II:1 and II:3) were found to harbor the
mutation, suggesting cosegregation of the mutation with development
of the disease. The sequencing results of those unaffected (I:1 and
II:2) excluded the presence of the mutation, suggesting that the
mutation may have been inherited from the proband's affected
mother.
The variant occurred in a conserved amino acid
(Fig. 2B), and was predicted to
have a functional effect by SIFT, PolyPhen2 and MutationTaster. In
addition, the mutation was not found on the 200 unrelated
chromosomes from ethnically matched controls.
Structure analysis
The altered residue was located in the procollagen
C-propeptide region, the stability of which is crucial in
procollagen assembly. The missense mutation resulted in a
substitution of hydrophobic alanine by hydrophilic threonine at a
conserved site. Structure modeling demonstrated the generation of a
novel hydrogen bond between the mutated threonine at residue 1,259
and asparagine at residue 1,261 (Fig.
3). This was likely to alter the spatial conformation of the
region, thus leading to production of abnormal protein.
Discussion
vEDS is a rare autosomal dominant disorder of
connective tissue owing to mutations of the COL3A1 gene encoding
type III procollagen, resulting in deficiency of collagen III
protein (18). Type III collagen
is a major component of the skin, blood vessels and hollow organ
walls. This explains why patients affected by vEDS are at increased
risk of vascular, intestinal or uterine rupture (19,20).
Due to these life-threatening complications, the survival rates of
affected patients are significantly reduced. The first major
complication, namely vascular or internal organ rupture, occurs in
25% of patients by the age of 20 years and in >80% of patients
by the age of 40 years, with a median survival duration of 48 years
(19). Therefore, early diagnosis
is necessary for timely precaution measures to avoid such outcomes.
Currently, vEDS is diagnosed on the basis of the presence of at
least two of the following major diagnostic criteria:
Characteristic facial appearance, tendency to bruise easily,
translucent skin with visible veins, history of arterial,
intestinal or uterine rupture. Collagen protein analysis of
cultured fibroblasts or identification of mutations in the COL3A1
gene is required to achieve an ultimate diagnosis. The proband in
the present study presented with minimal signs of vEDS, precluding
from performing direct sequencing of the COL3A1 gene. In accordance
with previously published data, the present study further supported
that vEDS is a heterozygous disease with marked variability of
clinical manifestations (21,22).
To date, >700 COL3A1 mutations have been
identified in vEDS cohorts (23).
Almost two thirds of the mutations are heterozygous missense
substitutions affecting one of the glycine residues of the
(Gly-X-Y) repeat within the triple helical domain. One third of the
mutations are spice-site variants causing exon skipping. These two
types of mutations act in a dominant-negative manner, resulting in
abnormal procollagen peptide production. The remaining COL3A1
mutations are nonsense or frameshift mutations, which cause
premature termination of translation, leading to
haplo-insufficiency of normal protein. Haplo-insufficiency
mutations have been reported to delay the onset of complications
and increase life expectancy, compared with dominant-negative
mutations (24,25). Despite the wide distribution
regarding COL3A1 mutations, variants affecting the C-propeptide
region are rare. Frank et al (26) previously categorized COL3A1
variants into five groups: i) Glycine substitutions, ii)
splice-site and in-frame insertions-deletions, iii) variants
leading to haplo-insufficiency, iv) non-glycine missense variants
within the triple helix and v) non-glycine missense variants or
in-frame InDels in the N- or C-terminal of the protein (26). It was found that patients with
variants leading to haplo-insufficiency, non-glycine missense
variants within the triple helix or non-glycine missense variants
or in-frame InDels in the N- or C-terminal of the protein had a
less typical phenotype and prominent absence of digestive
complication. In line with these observations, the complication of
the proband in the present study occurred at relatively late age
and was limited entirely to vascular events. These findings
provided further evidence that COL3A1 mutations located in the
C-termini had a milder course of the disease and fewer
characteristic features.
Due to the absence of prevalent diagnostic criteria
for patients with vEDS, individuals harboring this specific type of
mutation are unlikely to undergo genetic analysis in clinical
settings. This explains the scarcity of mutations reported in the
C-propeptide region. Previously, targeted region capture combined
with next-generation sequencing was successfully utilized to
uncover pathologic variants of COL3A1 in individuals with
aortopathy with unsuspected vEDS (7,27).
As with the proband in the present study, they were subject to
targeted sequencing primarily based on aortic phenotypes, rather
than other syndromic manifestations. They did not present with the
skin or facial features characteristic of vEDS. Therefore, targeted
next-generation sequencing has been suggested as a useful
diagnostic tool in patients with arotopathy, particularly in those
with an absence of characteristic phenotypes. Of note, one patient
was found to have unusual tissue fragility in the previous study,
which aligned with the findings of the present study and several
previous studies (22,28). Tissue fragility may be a
consequence of collagen abnormality, indicating the presence of
mutations in genes encoding collagen (22,29).
In conclusion, the present study identified a
missense mutation in COL3A1 in a family with aortopathy by
performing targeted next-generation sequencing. Despite the lack of
characteristic vEDS manifestations, co-segregation analysis, the
absence in public variation databases, conservation scores,
bioinformatics prediction and alteration in crystal structure all
provided support for the pathogenic role of the variant. The
results of the present study suggested that targeted gene capture
combined with next-generation sequencing serves as a useful
technique in the genetic diagnosis of aortopathy, particularly in
the content of atypical phenotype.
Acknowledgements
This study was supported by the Nanjing Municipal
Science and Technology Commission (grant no. BL2012035), the
Medical Science and Technology Development Foundation, Nanjing
Department of Health (grant no. YKK13085), and the Natural Science
Foundation of Jiangsu Province, China (grant no. BK20140103).
References
|
1
|
Elefteriades JA and Pomianowski P:
Practical genetics of thoracic aortic aneurysm. Prog Cardiovasc
Dis. 56:57–67. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jondeau G and Boileau C: Familial thoracic
aortic aneurysms. Curr Opin Cardiol. 29:492–498. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Erbel R, Aboyans V, Boileau C, Bossone E,
Bartolomeo RD, Eggebrecht H, Evangelista A, Falk V, Frank H,
Gaemperli O, et al: 2014 ESC guidelines on the diagnosis and
treatment of aortic diseases: Document covering acute and chronic
aortic diseases of the thoracic and abdominal aorta of the adult.
The task force for the diagnosis and treatment of aortic diseases
of the European society of cardiology (ESC). Eur Heart J.
35:2873–2926. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pyeritz RE: Heritable thoracic aortic
disorders. Curr Opin Cardiol. 29:97–102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Milewicz DM, Chen H, Park ES, Petty EM,
Zaghi H, Shashidhar G, Willing M and Patel V: Reduced penetrance
and variable expressivity of familial thoracic aortic
aneurysms/dissections. Am J Cardiol. 82:474–479. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Isselbacher EM, Cardenas CL Lino and
Lindsay ME: Hereditary influence in thoracic aortic aneurysm and
dissection. Circulation. 133:2516–2528. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Proost D, Vandeweyer G, Meester JA,
Salemink S, Kempers M, Ingram C, Peeters N, Saenen J, Vrints C,
Lacro RV, et al: Performant mutation identification using targeted
next generation sequencing of 14 thoracic aortic aneurysm genes.
Hum Mutat. 36:808–814. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ziganshin BA, Bailey AE, Coons C, Dykas D,
Charilaou P, Tanriverdi LH, Liu L, Tranquilli M, Bale AE and
Elefteriades JA: Routine genetic testing for thoracic aortic
aneurysm and dissection in a clinical setting. Ann Thorac Surg.
100:1604–1611. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu J, Matthaei H, Maitra A, Dal Molin M,
Wood LD, Eshleman JR, Goggins M, Canto MI, Schulick RD, Edil BH, et
al: Recurrent GNAS mutations define an unexpected pathway for
pancreatic cyst development. Sci Transl Med. 3:92ra662011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cox MP, Peterson DA and Biggs PJ:
SolexaQA: At-a-glance quality assessment of illumina
second-generation sequencing data. BMC Bioinformatics. 11:4852010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li R, Yu C, Li Y, Lam TW, Yiu SM,
Kristiansen K and Wang J: Soap2: An improved ultrafast tool for
short read alignment. Bioinformatics. 25:1966–1967. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R: 1000 Genome
Project Data Processing Subgroup: The sequence alignment/map format
and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li H and Durbin R: Fast and accurate short
read alignment with burrows-wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hou H, Zhao F, Zhou L, Zhu E, Teng H, Li
X, Bao Q, Wu J and Sun Z: Magicviewer: Integrated solution for
next-generation sequencing data visualization and genetic variation
detection and annotation. Nucleic Acids Res. 38:(Web Server issue).
W732–W736. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jin ZB, Mandai M, Yokota T, Higuchi K,
Ohmori K, Ohtsuki F, Takakura S, Itabashi T, Wada Y, Akimoto M, et
al: Identifying pathogenic genetic background of simplex or
multiplex retinitis pigmentosa patients: A large scale mutation
screening study. J Med Genet. 45:465–472. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Arnold K, Bordoli L, Kopp J and Schwede T:
The SWISS-MODEL workspace: A web-based environment for protein
structure homology modelling. Bioinformatics. 22:195–201. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Beighton P, De Paepe A, Steinmann B,
Tsipouras P and Wenstrup RJ: Ehlers-danlos syndromes: Revised
nosology, villefranche, 1997. Ehlers-Danlos National Foundation
(USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet.
77:31–37. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pepin M, Schwarze U, Superti-Furga A and
Byers PH: Clinical and genetic features of Ehlers-Danlos syndrome
type iv, the vascular type. N Engl J Med. 342:673–680. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Oderich GS, Panneton JM, Bower TC, Lindor
NM, Cherry KJ, Noel AA, Kalra M, Sullivan T and Gloviczki P: The
spectrum, management and clinical outcome of Ehlers-Danlos syndrome
type iv: A 30-year experience. J Vasc Surg. 42:98–106. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jorgensen A, Fagerheim T, Rand-Hendriksen
S, Lunde PI, Vorren TO, Pepin MG, Leistritz DF and Byers PH:
Vascular Ehlers-Danlos Syndrome in siblings with biallelic COL3A1
sequence variants and marked clinical variability in the extended
family. Eur J Hum Genet. 23:796–802. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wendorff H, Pelisek J, Zimmermann A, Mayer
K, Seidel H, Weirich G, Hausser I, Siegel C, Zernecke A and
Eckstein HH: Early venous manifestation of Ehlers-Danlos syndrome
type iv through a novel mutation in COL3A1. Cardiovasc Pathol.
22:488–492. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shalhub S, JH III Black, Cecchi AC, Xu Z,
Griswold BF, Safi HJ, Milewicz DM and McDonnell NB: Molecular
diagnosis in vascular Ehlers-Danlos syndrome predicts pattern of
arterial involvement and outcomes. J Vasc Surg. 60:160–169. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Leistritz DF, Pepin MG, Schwarze U and
Byers PH: COL3A1 haploinsufficiency results in a variety of
Ehlers-Danlos syndrome type IV with delayed onset of complications
and longer life expectancy. Genet Med. 13:717–722. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pepin MG, Schwarze U, Rice KM, Liu M,
Leistritz D and Byers PH: Survival is affected by mutation type and
molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type
IV). Genet Med. 16:881–888. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Frank M, Albuisson J, Ranque B, Golmard L,
Mazzella JM, Bal-Theoleyre L, Fauret AL, Mirault T, Denarié N,
Mousseaux E, et al: The type of variants at the COL3A1 gene
associates with the phenotype and severity of vascular
Ehlers-Danlos syndrome. Eur J Hum Genet. 23:16575–1664. 2015.
View Article : Google Scholar
|
|
27
|
Campens L, Callewaert B, Mosquera L Muino,
Renard M, Symoens S, De Paepe A, Coucke P and De Backer J: Gene
panel sequencing in heritable thoracic aortic disorders and related
entities: Results of comprehensive testing in a cohort of 264
patients. Orphan J Rare Dis. 10:92015. View Article : Google Scholar
|
|
28
|
Abayazeed A, Hayman E, Moghadamfalahi M
and Cain D: Vascular type Ehlers-Danlos syndrome with fatal
spontaneous rupture of a right common iliac artery dissection: Case
report and review of literature. J Radiol Case Rep. 8:63–69.
2014.PubMed/NCBI
|
|
29
|
Monroe GR, Harakalova M, van der Crabben
SN, Majoor-Krakauer D, Bertoli-Avella AM, Moll FL, Oranen BI,
Dooijes D, Vink A, Knoers NV, et al: Familial Ehlers-Danlos
syndrome with lethal arterial events caused by a mutation in
COL5A1. Am J Med Genet A. 167:1196–1203. 2015. View Article : Google Scholar : PubMed/NCBI
|