Introduction
Focal cortical dysplasia (FCD) is the most frequent
malformation of cortical development, which may result in
drug-refractory epilepsy (1). The
frequency of FCD in patients submitted to surgery for refractory
epilepsy varies between 12 and 40% (2,3). A
definitive diagnosis of FCD is generally made following surgical
treatment for refractory epilepsy, based on neuropathological
findings of the resected cortical tissue (4). FCD is characterized by numerous
alterations, which may be divided into two major groups. The first
group is characterized by abnormalities of the cortical
architecture through columnar disorganization and laminar
interruption, which is observed by alterations in the composition
of the sixth tangential layer. The second group is defined by
cytological abnormalities, with hypertrophic neuronal cells
observed outside the normal anatomic location at layer V and/or the
presence of balloon cells. Balloon cells possess a poorly defined
membrane with single or multiple nuclei and an eosinophilic
cytoplasm, which are characteristics of neuronal and glial cells;
this condition is primarily diagnosed as Taylor's FCD or FCD Type
IIb (5,6).
In 2004, Palmini et al (1) classified FCD according to the white
matter and cortical layer architecture as follows: Type I, presence
of heterotrophic neurons in the white matter, cortical layer
architecture alteration and giant neurons; Type IIa, presence of
heterotrophic neurons in the white matter, cortical layer
architecture alterations, giant neurons and dysmorphic cells; Type
IIb, presence of heterotrophic neurons in the white matter,
cortical layer architecture alterations, giant neurons, dysmorphic
cells and presence of balloons cells. In 2011, Blumcke et al
(7) modified the Palmini et
al (1) classification,
defining three types of FCD, known as Type I, II and III, where the
Type III was characterized by presence of abnormalities associated
with principal lesion, for example hippocampal sclerosis, glial or
glio-neuronal tumor, vascular malformation, trauma, ischemic injury
and encephalitis. However, at present, the mechanisms involved in
the pathogenesis of FCD have been poorly investigated, which is
primarily due to the limited number of cases and the lack of
suitable experimental models (5).
The exact etiology of FCD remains unknown; however,
it may be associated with clonal somatic mutations that, in certain
patients, affect the same signaling pathways (8). Previous studies have demonstrated an
increase in mechanistic target of rapamycin (mTOR) signaling in
patients with FCD based on the observation of phosphorylated
molecules, including S6 ribosomal proteins (8,9).
These alterations are primarily observed in FCD Type IIb, with
80–90% of balloon cells and giant neurons in the cerebral cortex
demonstrating increased mTOR signaling (9). Certain cases of FCD demonstrate
activation (phosphorylation) of molecules associated with the
phosphoinositide-3-dependent kinase (PI3K) and protein kinase B
(AKT) pathways in the dysplastic tissue (10). The phosphorylation of molecules
involved in the PI3K pathway in response to certain stimuli is
associated with a coordinated set of events that control cell
growth, cell cycle progression, cell migration and cell survival
(11).
The study of neurological and neuropsychiatric
disorders has been a longstanding challenge for researchers.
Despite significant investments in the field, pre-clinical models
suitable for studies concerning pathophysiology, mechanisms and
therapeutic targets, and for testing novel drugs, are scarce
(12). Although animal models are
valuable to elucidate disease mechanisms, develop specific markers
and identify genes associated with certain diseases, they have a
poor record when it comes to translating therapeutic discoveries
for human clinical application (13). The importance of the use of human
cells for the study of diseases is evident, given that a high
number of drugs that have demonstrated efficacy and safety when
tested in animal models fail in clinical trials, which is
attributed to differences between the species (14). Studies using post-mortem tissues
provide useful insight into cerebral structural alterations that
occur at molecular and cellular levels. Considering these surveys
and limitations, it is clear that the study of cerebral development
would benefit from using the patient's own cells (12).
The reprogramming of adult somatic cells at the
embryonic level is a promising approach for regenerative medicine,
which may also facilitate in vitro studies to gain an
improved understanding of complex genetic diseases. It is possible
to reprogram somatic cells by nuclear transfer into enucleated
oocytes or by cell fusion between somatic cells and embryonic
cells, co-culture of undifferentiated cells with somatic cells, and
adding genes that activate selective transcription factors
(15,16). In 2006, Takahashi and Yamanaka
(17) introduced a novel technique
for producing pluripotent cells by reprogramming mouse fibroblasts,
which was subsequently applied to human cells in 2007 (18). The cells were reprogrammed with the
addition of four genes, POU class 5 homeobox 1 (OCT4), sex
determining region Y-box 2 (SOX2), Kruppel-like factor 4
(KLF4) and c-MYC, using viral vectors. It is possible
to perform this reprogramming in various cell types. The cells
generated by this method are known as induced pluripotent stem
cells (iPSCs) and are similar to embryonic stem cells, with the
same self-renewal and differentiation potential characteristics for
cells of the three germ layers (17,19).
iPSCs differentiated into specific tissues are now
widely used for translational studies testing drugs in cells that
are difficult to obtain, including cardiomyocytes, neurons and
liver cells. The generation of iPSCs from cells of patients with
neurological diseases, and their tissue-specific differentiation,
serves as an invaluable source for testing and provides the
additional capacity to study the initial development and
progression of diseases associated with the central nervous system
(20). Cellular models exhibit
high relevance for the study of human diseases, providing excellent
conditions for understanding mechanisms and constituting an
effective tool for high-throughput experiments, even allowing for
the construction of platforms for screening novel drugs to treat
numerous human diseases (12).
Numerous studies have employed iPSCs for the study
of neurological diseases, including for multiple sclerosis
(21), cerebellar atrophy
(22), Alzheimer's disease
(23–26), Rett syndrome (27–30),
amyotrophic lateral sclerosis (31–34),
ataxia telangiectasia (35),
Dravet syndrome (36), familial
dysautonomia (37), fragile X
syndrome (38), Gaucher's disease
(39), Huntington's disease
(40,41), Lesch-Nyhan syndrome (42), microcephaly (43), Parkinson's disease (44–46)
and schizophrenia (47–49). The technology of cellular
reprogramming has highlighted the reality of the clinical
heterogeneity observed in patients from the lab bench to the
bedside (14). The use of iPSCs
derived from patients with specific neural diseases helps provide
information regarding embryonic neurogenesis, cortical formation
and pathophysiology. Therefore, the present study aimed to
establish a cellular model of refractory epilepsy by generating
iPSCs from fibroblasts obtained from patients with FCD.
Materials and methods
Ethics statement
The present study was reviewed and approved by the
Committee of Research Ethics of the Pontifical Catholic University
of Rio Grande do Sul (approval no. 17943213.9.0000.5336) through
the system Platafoma Brasil. Written informed consent was obtained
from patient 1 and from the parents of patient 2 (a minor) enrolled
in the present study, according to Brazil Resolution no.
466/12.
Patients
A total of 2 patients were enrolled in the present
study upon signing the ethical consent form, according to the
guidance of the Committee of Research Ethics. Patient 1 was a
45-year-old man with medically refractory seizures, whose
electroencephalogram monitoring demonstrated sharp waves in the
right frontal region and 3 seizures with onset in the same region
(Fig. 1A). Magnetic resonance
imaging revealed a small right frontal lesion with an increased
signal and blurring of the cortico-subcortical white matter
(Fig. 1B), which was resected
under acute electrocorticography. Histopathology (Fig. 1C and D) and immunohistochemistry
(Fig. 1E and F) revealed cortical
dyslamination and large, dysplastic neurons, with balloon cells,
which is compatible with a diagnosis of FCD Type IIb (International
League Against Epilepsy) (7).
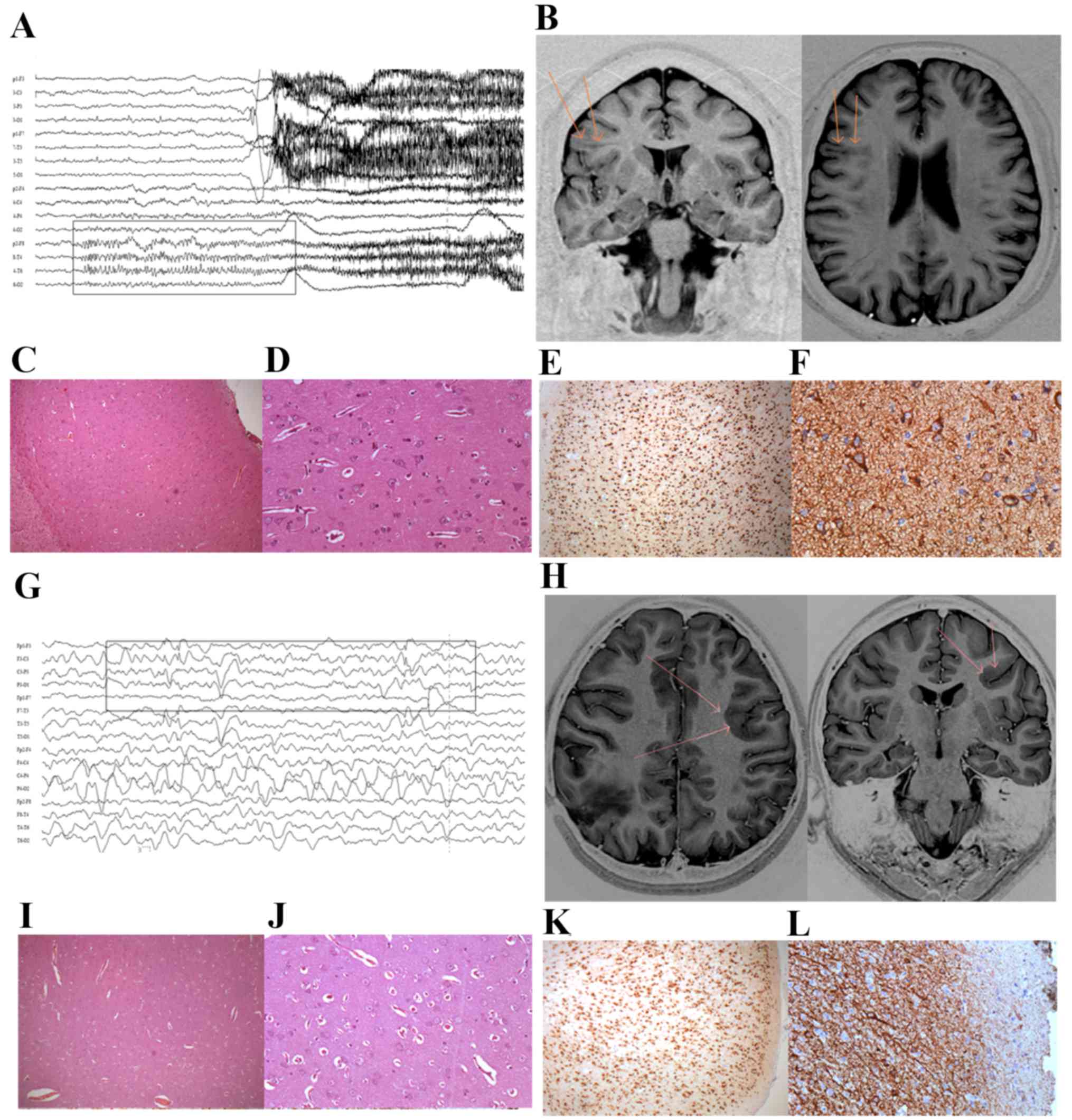 | Figure 1.Patient characteristics. For patient
1: (A) The EEG revealed rhythmic seizure discharge originating in
the right fronto-temporal region. (B) MR image indicating the area
of transmantle dysplasia in the right frontal lobe, with vagueness
and blurring of the cortico-subcortical interface (arrows). (C and
D) Histopathology of cortex morphology revealing delamination of
the layers and cortical disorganization, with dysmorphic neurons
and balloon cells under hematoxylin and eosin staining.
Magnification, ×200. Immunohistochemical staining of (E) NeuN,
revealing neurons with delamination of the cortical layers
(magnification, ×20) and (F) vimentin, marking balloon neurons.
Magnification, ×200. For patient 2: (G) The recorded EEG revealed
rhythmic seizure discharge originating in the left anterior
quadrant, with a maximum in the left frontal region. (H) MR image
revealing heterotopic subcortical and periventricular nodules in
the left frontal lobe, with vagueness and blurring of the
cortico-subcortical interface (arrows). (I and J) Histopathology of
cortex morphology revealing delamination of the layers and cortical
disorganization, with dysmorphic neurons and balloon cells under
hematoxylin and eosin staining. Magnification, ×20 and ×200.
Immunohistochemical staining of (K) NeuN, revealing neurons with
delamination of the cortical layers (magnification, ×20) and (L)
vimentin, marking balloon neurons (magnification, ×200). EEG,
electroencephalogram; MR, magnetic resonance; NeuN, RNA binding
protein, fox-1 homolog 3. |
Patient 2 was a 12-year-old girl who first started
experiencing seizures at the age of ~5 years, which were
characterized by sudden extension of the right arm and head drop.
The patient often had numerous seizures per day, despite attempts
to treat the seizures with several antiepileptic drug regimens. The
patient also had a cystic lesion with regular borders in the right
parietal region, which intermittently led to moderate intracranial
hypertension and was surgically targeted on several occasions.
However, this approach did not significantly improve seizure
control, which led to presurgical evaluation. The latter revealed
maximal interictal and ictal epileptic activity in the left frontal
region (Fig. 1G), where an
orbitofrontal dysplastic lesion was clearly observed by magnetic
resonance imaging (Fig. 1H). The
patient underwent resective surgery under acute
electrocorticography, and histopathology (Fig. 1I and J) and immunohistochemistry
(Fig. 1K and L) revealed a typical
pattern of FCD Type IIb (International League Against Epilepsy)
(7).
Production of fibroblasts from skin
biopsies
The human fibroblasts were obtained from residual
skin fragments from two patients that underwent surgical treatment
for medically refractory epilepsy (Epilepsy Surgery Program) at São
Lucas Hospital (Porto Alegre, Brazil) on November 2013 (patient 1)
and April 2015 (patient 2) at Pontifical Catholic University (Porto
Alegre, Brazil). The skin biopsies were cut into ~5 mm2
sections and the skin fragments were placed in a 60-mm Petri dish,
with the dermis facing the plate. The cells were cultured in
Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 20% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml
penicillin (Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml
streptomycin (Gibco; Thermo Fisher Scientific, Inc.), 100 µg/ml
gentamicin (Gibco; Thermo Fisher Scientific, Inc.) and 20 ng/ml
fibroblast growth factor (Thermo Fisher Scientific, Inc.). The skin
fragments were maintained at 37°C with 5% CO2 until
reaching >80% confluence, and a mycoplasma test (MycoAlert Plus;
Lonza Group, Ltd., Basel, Switzerland) was performed. Once the
cells were confirmed to be mycoplasma-free, they were maintained
under the same conditions up to the seventh passage. Cells in the
fourth, fifth, sixth and seventh passages were cryopreserved in
liquid nitrogen. In addition, fibroblasts morphological profile was
observed by the fifth-passage using an optical microscope Axiovert
25 (Carl Zeiss, Oberkochen, Germany) in bright field.
Histologic analysis of dysplastic
tissue
Brain samples obtained via surgical resection were
immediately fixed in 10% buffered formaldehyde for 24 h at room
temperature, and the surgical specimens were processed and
paraffin-embedded. All specimens were cut into 5 µm sections with a
microtome (Leica Microsystems GmbH, Wetzlar, Germany) and stained
with hematoxylin and eosin, and additional slides were submitted to
automated 3,3-diaminobenzidine (DAB; Dako Autostainer Link 48;
Agilent Technologies, Inc., Santa Clara, CA, USA)
immunohistochemical staining using 5% FBS (Gibco; Thermo Fisher
Scientific, Inc.), 1% bovine serum albumin (BSA; Sigma-Aldrich;
Merck Millipore, Darmstadt, Germany) and 0,2% of Triton X-100
(Sigma-Aldrich; Merck Millipore) with blocking buffer for 1 h at
room temperature for anti-NeuN antibody (cat. no. ABN91; Merck
Millipore) and anti-vimentin (cat. no. GA630, Dako, Glostrup,
Denmark) diluted in blocking buffer (1:100). All reactions included
positive and negative external control samples on the same slide.
The slides were reviewed under a Zeiss Axiokop 40 microscope (Carl
Zeiss Group,). All images were documented in TIFF uncompressed
format with a Retiga 2000R color video camera (QImaging, Surrey,
Canada).
Generation of iPSCs
iPSCs were generated through exposure of fibroblasts
to viral vectors containing the genes OCT4, SOX2, KLF4 and c-MYC
using the CytoTune®-iPS 2.0 Sendai Reprogramming kit
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. The CytoTune 2.0 Sendai reprogramming vectors in this kit
are based on a modified, non-transmissible form of Sendai virus,
which has the Fusion protein (F) deleted, rendering the virus
incapable of producing infectious particles from infected cells.
Fibroblasts were cultured in 6-well plates and when ~70% confluence
was reached they were used for transfection. The number of viral
particles used was calculated according to the multiplicity of
infection (MOI) equation (1.5×106 cells at MOI=5-5-5-3; i.e., hOct4
MOI=5, hSox2 MOI=5, hc-Myc MOI=5, hKlf4 MOI=3). The specific amount
of virus was diluted in 1 ml DMEM/F12 culture medium supplemented
with 20% Knockout Serum Replacement (Gibco; Thermo Fisher
Scientific, Inc.), 1X non-essential amino acids of DMEM (Gibco;
Thermo Fisher Scientific, Inc.), 1X Glutamax (Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml of penicillin (Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml streptomycin (Gibco; Thermo Fisher
Scientific, Inc.) and 100 µg/ml of gentamicin (Gibco; Thermo Fisher
Scientific, Inc.). The fibroblasts were exposed to the medium
containing the virus and incubated at 37°C with 5% CO2
for 24 h. The cells were washed with PBS and cultured under the
same conditions for 6 days. On day 7 following transfection, the
cell cultures were treated with trypsin/ethylenediaminetetraacetic
acid and transferred to a culture dish containing BD Matrigel
hESC-qualified Matrix (BD Biosciences, Franklin Lakes, NJ, USA).
Following culture for 24 h, the culture medium was replaced with
embryonic cell mTeSR medium (Stemcell Technologies, Inc.,
Vancouver, Canada). Cell clones were manually removed ~20 days
later and transferred to new plates containing BD Matrigel
hESC-qualified Matrix. Following three subcultures, the clones were
characterized by immunostaining with antibodies against homeobox
protein NANOG (cat. no. MABD24A4, Merck Millipore) FITC conjugate,
SOX2 (cat. no. MAB4423C3, Merck Millipore) Cy3 conjugate, OCT4
(cat. no. MAB4419A4, Merck Millipore) FITC conjugate, TRA1-60 (cat.
no. MAB4360C3, Merck Millipore) Cy3 conjugate and TRA1-81 (cat. no.
MAB4381C3, Merck Millipore) Cy3 conjugate following the addition of
4% paraformaldehyde. The culture was incubated for 1 h at room
temperature with blocking buffer [5% FBS, 1% of BSA and 0.2% of
Triton X-100 (Sigma-Aldrich; Merck Millipore)] for NANOG, SOX2 and
OCT4 antibodies. The same blocking buffer without Triton was used
when cell surface proteins were analyzed (TRA1-60 and TRA-81
antibodies). Cells were incubated for 2 h at room temperature,
washed twice with PBS, and stained with
4′,6-diamidino-2-phenilindol (DAPI; Sigma-Aldrich; Merck
Millipore). The images were captured using confocal microscopy
Zeiss LSM-5 exciter (Carl Zeiss Group).
Analysis of the AKT and mTOR
pathway
The brain tissue was fixed with 4% buffered
formaldehyde for 24 h at room temperature, embedded in paraffin and
sliced into 5 µm sections. The primary antibodies against AKT (cat.
no. mAb2920; Cell Signaling Technology, Inc., Danvers, MA USA),
phosphorylated-AKT (cat. no. mAb4060; Cell Signaling Technology,
Inc.), mTOR (cat. no. mAb2983, Cell Signaling Technology, Inc.) and
phosphorylated-mTOR (cat. no. mAb2976, Cell Signaling Technology,
Inc.), diluted 1:100 with blocking buffer, were used. The slides
were incubated at 4°C for 12 h followed by further incubation with
a fluorescein isothiocyanate-conjugated secondary antibody diluted
1:100 (cat. no. A11029, Invitrogen; Thermo Fisher Scientific, Inc.)
for 2 h at room temperature. The slides were washed with PBS and
0.01% DAPI was added for nuclear staining. Analysis was performed
using a confocal microscope Zeiss LSM-5 (Carl Zeiss Group). For
quantitative analysis, 10 visual fields were randomly selected
using a ×20 objective lens, with a minimum of 20 DAPI-positive
cells. The images were quantified by area marker parameter using
Image-Pro Plus 7 software (Media Cybernetics, Inc., Rockville, MD,
USA). The quantification area was analyzed using one-way analysis
of variance followed by the Tukey post hoc test. Analyses were
performed using GraphPad Prism 5.0 software (GraphPad Software,
Inc., La Jolla, CA, USA).
Results
AKT/mTOR pathway analysis
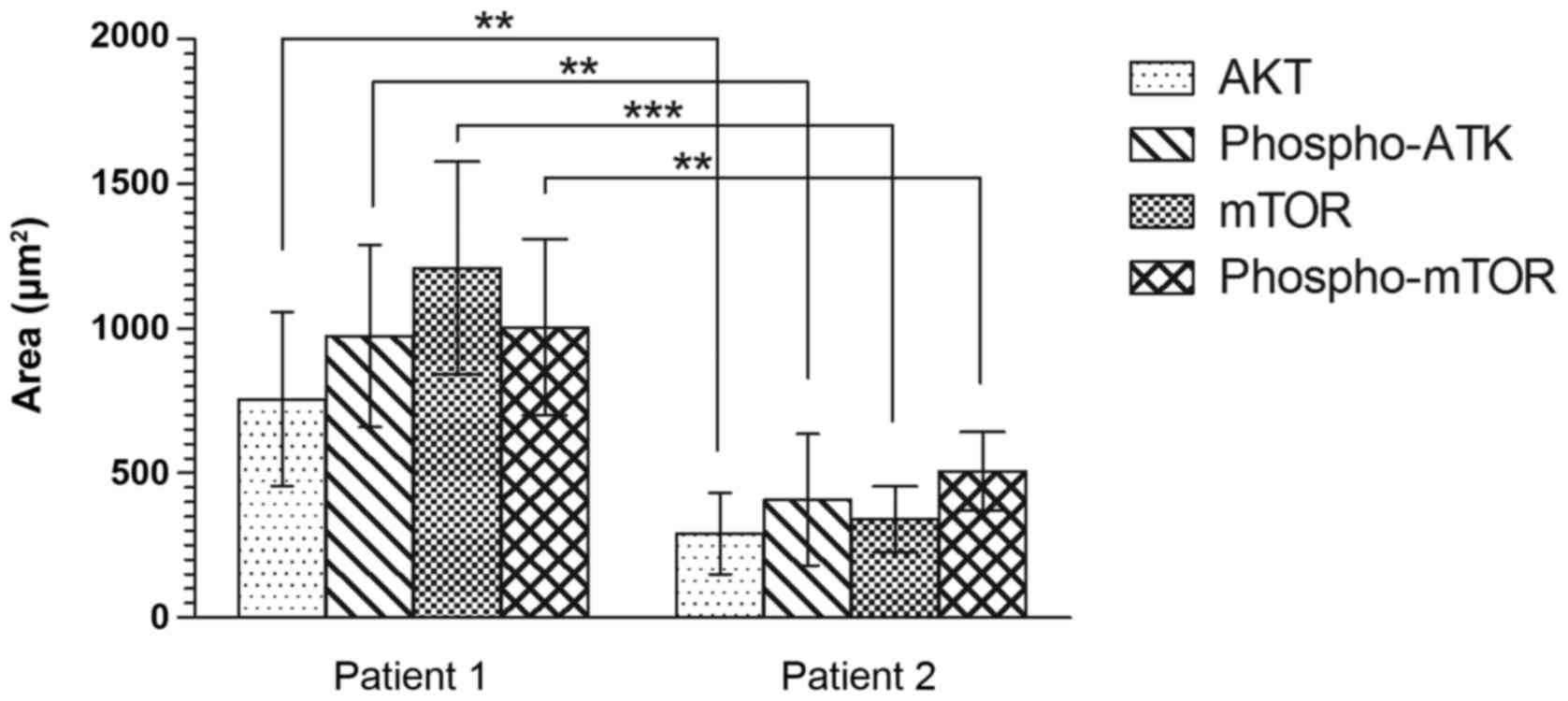
Analysis of the AKT/mTOR pathway in cerebral
dysplastic tissue revealed a statistically significant difference
between cerebral tissues in the two patients (Fig. 2). The quantified area stained with
anti-AKT was 756 and 291 µm2 in patients 1 and 2, respectively
(P=0.006; Fig. 2), and that of
phosphorylated-AKT staining was 974 and 408 µm2 for patients 1 and
2, respectively (P=0.004; Fig. 2).
In addition, the mTOR pathway analysis revealed a statistically
significant difference between the stained areas in the cerebral
tissues from the two patients (mTOR: Patient 1, 1,210 µm2; patient
2, 341 µm2; P=0.0003; and phosphorylated-mTOR: Patient 1, 1004 µm2;
patient 2, 507 µm2; P=0.004; Fig.
2).
iPSCs were generated from cellular
reprograming of fibroblasts
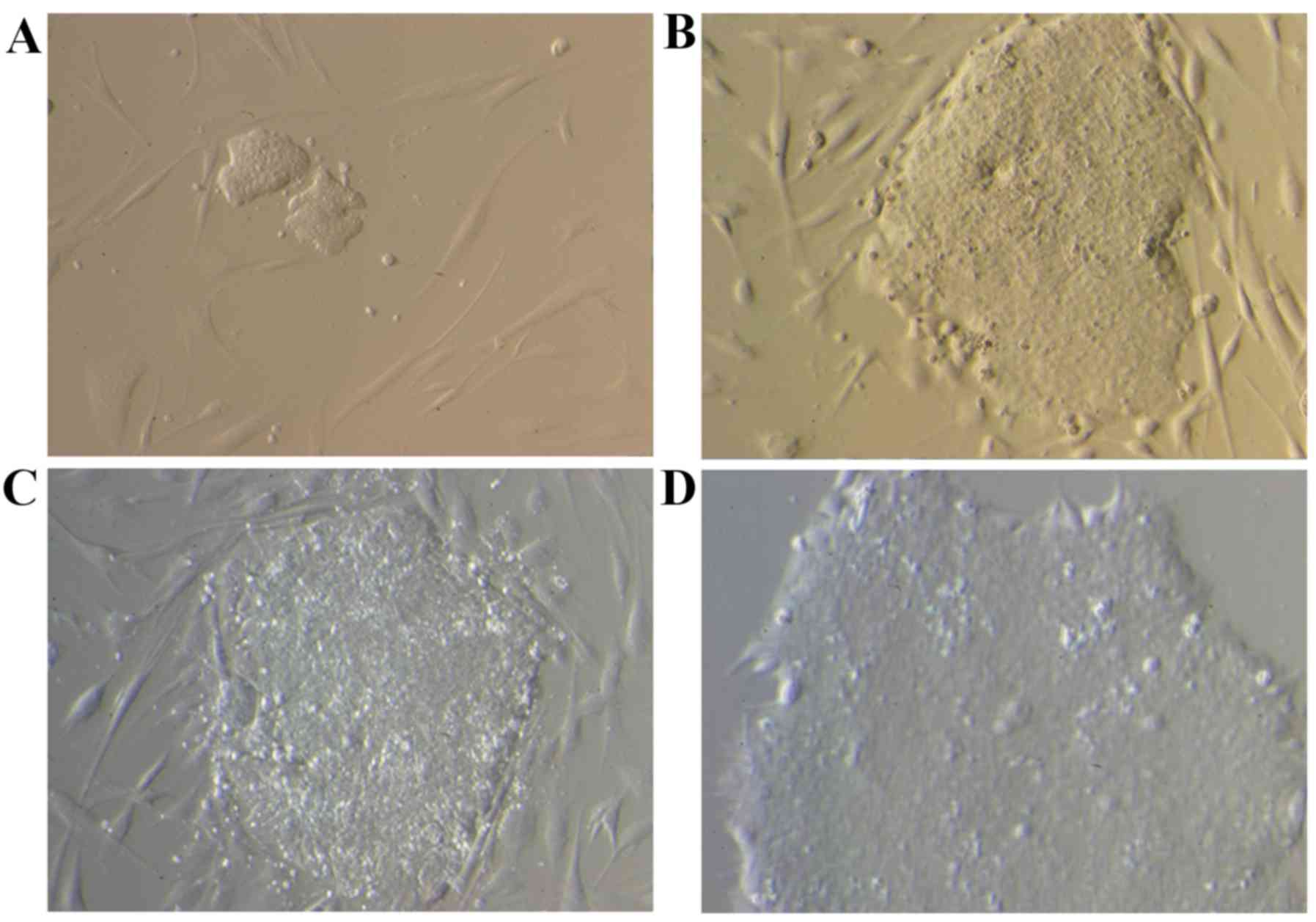
Clones with the morphological features of embryonic
cells were detected on the 13th day following viral transfection.
The clones were manually selected and cultured over Matrigel at
~day 25. Small clones surrounded by a few fibroblasts were observed
on day 13 (Fig. 3A); however,
following 20 (Fig. 3B) and 25
(Fig. 3C) days of culture, larger
clones with the morphological features of embryonic cells were
detected, which still contained fibroblasts. Finally, following a
culture over a Matrigel support, a cell clone free of fibroblasts
was obtained (Fig. 3D).
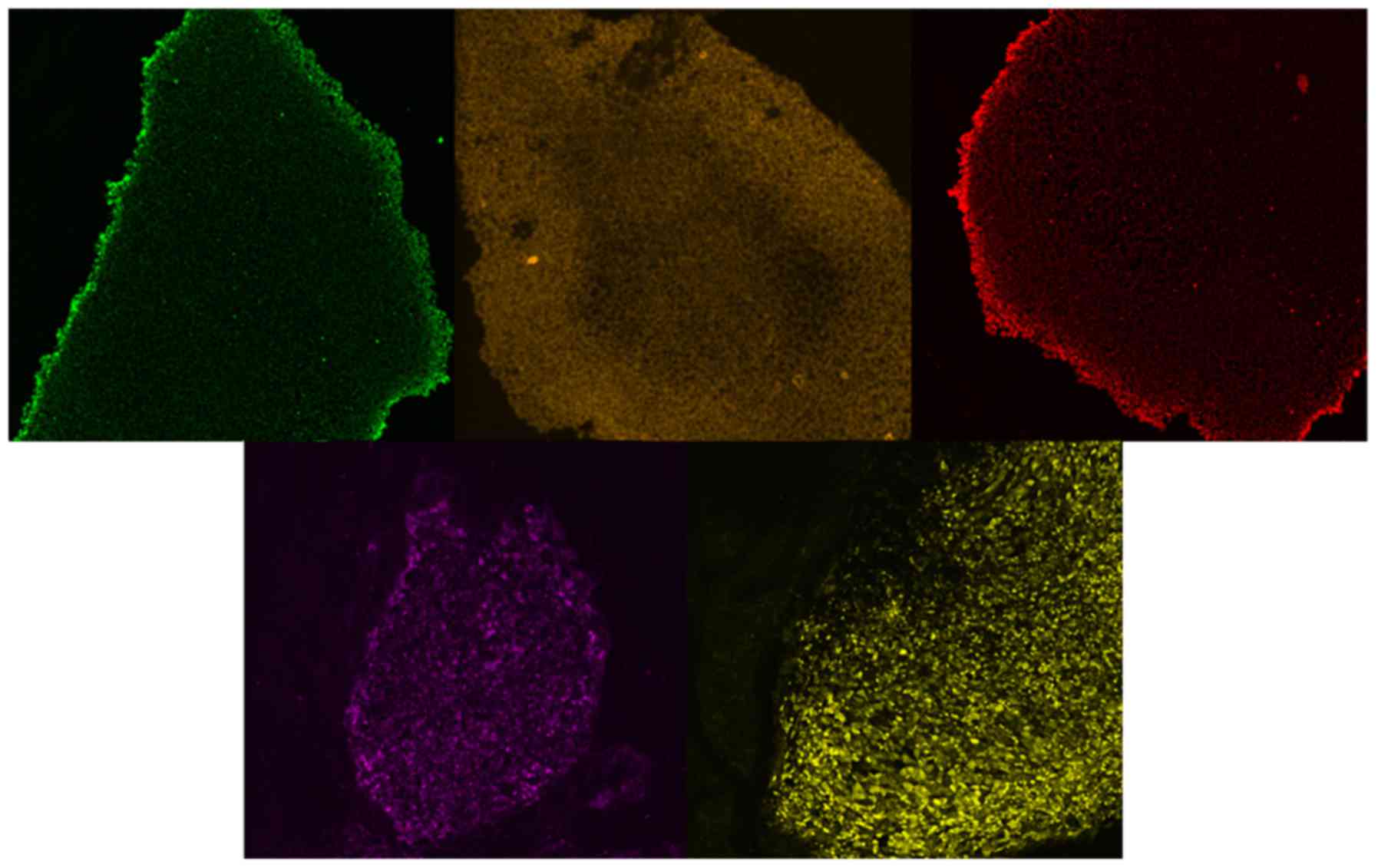
The features of embryonic cells were further
confirmed following 3 subcultures of the clones over Matrigel with
antibodies against the pluripotency markers Nanog, SOX2, OCT4,
TRA1-60 and TRA1-81 (Fig. 4). The
positive staining characteristics of the embryonic cells confirmed
the successful generation of iPSCs derived from fibroblasts from
patients with FCD.
Discussion
Several neural diseases remain poorly understood, in
particular those that affect the central nervous system, from the
course of embryonic development up to the onset of clinical signs.
These diseases represent a huge physical and social burden to
patients and families, with high financial costs to public health
systems. Although significant advances have been made in
understanding the genetic basis of these diseases, clinical
classification, patient care and effective treatments remain scarce
(14).
Fortunately, the unquestionable advance of methods
for iPSC generation and their subsequent differentiation into
several tissue types have rendered these cells a standout cellular
model for diverse diseases, including those affecting the central
nervous system. This strategy allows for the investigation and
development of novel approaches to study the mechanisms of
embryonic neurodevelopment and pathological contexts specific for
each patient, considering their unique genetic backgrounds
(14). To the best of our
knowledge, the present study is the first to present a method for
the generation of a cell model to study the embryonic neurogenesis
of epilepsy refractory to drug treatment in the context of FCD.
Previous studies have demonstrated a link between
genetic alterations and various types of cortical malformation,
which may be specifically associated with the main stages of
central nervous system development (8). More than 100 genes have been
associated with various types of cortical malformation (50). The major genes identified,
including those that are involved in signaling pathways associated
with cerebral cortex malformation, are associated with apoptosis,
cell proliferation, cytoskeletal structure, cell migration and
neurodifferentiation. Alterations in signaling and/or other
regulatory pathways may have a variable impact on not only the
pattern of brain cortical malformation but also on the site
affected (8).
The diagnoses of certain cortical defects, including
megalencephaly, polymicrogyria, hemimegalencephaly and cortical
dysplasia, are generated following the observation of typical
features in clinical imaging. Pathological alterations associated
with these disorders include a wide range of abnormalities,
including those typically associated with FCD. A growing number of
gene alterations have been associated with polymicrogyria and
hemimegalencephaly, in particular in cases with more severe
phenotypes. Megalencephaly with polymicrogyria has been associated
with a mutation in PIK3CA and PIK3R2 genes and isolated
hemimegalencephaly has been associated with mosaic mutations in the
PI3K, AKT and mTOR pathways. However, unlike these malformations,
the etiology of FCD remains unknown (8–10).
Normal PI3K/AKT signaling integrates fundamental
physiological responses for healthy aging and longevity. Previous
studies have demonstrated that downregulation of the PI3K/AKT
signaling pathway may be associated with the lifespan of particular
species (51,52). Alterations in the PI3K/AKT/mTOR
signaling pathway are involved in age-related diseases, including
heart and neurological conditions. Increased activation of this
pathway is considered a feature of early onset Alzheimer's disease,
but is also associated with normal aging processes in healthy
subjects (53). The brain tissue
from the patients investigated in the present study revealed a
difference regarding both pathways. A significant increase in AKT,
phosphorylated-AKT, mTOR and phosphorylated-mTOR expression was
observed in the older patient (patient 1; 45 years old) compared
with in the younger patient (patient 2; 12 years old). Indeed, it
has previously been reported that balloon and giant cells in the
brain tissue from patients with FCD express markers of mature,
undifferentiated neuronal cells and glial cells (5). However, it will only be possible to
confirm this hypothesis by investigating the brain tissue from
patients of the same age, although the effect of genetic background
should not be ignored.
It has been hypothesized that clonal somatic
mutations are shared among patients with FCD. Previous studies have
observed an increase in mTOR signaling in 80–90% of the balloon
cells present in the cortex of patients with FDC Type IIb. Certain
cases of FCD Type IIb also exhibit increased PI3K and AKT activity
(8–10). Increased signaling of the
PI3K/AKT/mTOR pathway was demonstrated in FCD Types IIa and IIb
without genetic mutation, which was attributed to other mechanisms
associated with other common diseases (54).
iPSCs are obtained from somatic cells by means of
distinct techniques, including chemical induction or gene
transfection. Using the premise introduced by Takahashi and
Yamanaka (17), at least 4 genes
that confer pluripotency should be included for this strategy to be
successful. Retroviral vectors require the integration of
transfected genes into the host genome in order to be expressed
along with the other host genes. Adenoviral, adeno-associated virus
and plasmid vectors do not require integration, but may integrate
and disrupt the host genome. The viral vectors used in the present
study are not integrative and do not influence the genome of the
host cell (55,56). This property is an additional
advantage to the embryonic features already mentioned, including
pluripotency.
The consensus method currently used to characterize
pluripotent cells begins with observing the clone morphology. iPSC
clones generated from human cells have a distinct morphology,
containing large nuclei and well-organized colonies with clearly
defined edges (57). In addition,
adequate clones should be well organized and tightly adhered,
without areas of differentiation. In the present study, the
pluripotency of the clones was confirmed by positive expression of
Nanog, SOX2, OCT4, TRA1-60 and TRA1-81. There is no minimum
criterion required for iPSC characterization. However, the presence
of certain markers is essential to confirm the pluripotency, as
well as the maintenance of the undifferentiated condition (58).
Gaining a global understanding of the development of
normal brain function depends on extensive knowledge concerning
brain formation, connection patterns between neurons and brain
regions, and the synaptic communications present in these
connections. Studies with iPSCs from patients with FCD will enable
investigations of different neurodevelopmental stages, and allow
the gathering of molecular and clinical evidence from observations
of the affected adult tissue. Furthermore, the generated iPSCs will
motivate in vitro studies to determine the processes
involved in embryonic neurogenesis and to elucidate potential
changes that may be associated with the abnormal development of the
cerebral cortex, leading to FCD.
The present study provides a useful tool that may
help to understand embryonic brain development associated with the
development of FCD, a disease with an unclear genesis. Insight into
other diseases may also be achieved using the same approach.
Acknowledgements
The authors would like to thank the National Council
for Scientific and Technological Development (grant no.
457384/2013-1) and the Coordination of Improvement of Higher Level
Personnel (grant no. 380095/2014-9) for their financial
support.
Glossary
Abbreviations
Abbreviations:
|
FCD
|
focal cortical dysplasia
|
|
mTOR
|
mechanistic target of rapamycin
|
|
PI3K
|
phosphoinositide-3-dependent
kinase
|
|
AKT
|
protein kinase B
|
|
iPSC
|
induced pluripotent stem cell
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
References
|
1
|
Palmini A, Najm I, Avanzini G, Babb T,
Guerrini R, Foldvary-Schaefer N, Jackson G, Lüders HO, Prayson R,
Spreafico R and Vinters HV: Terminology and classification of the
cortical dysplasias. Neurology. 62:S2–8. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Arai A, Saito T, Hanai S, Sukigara S,
Nabatame S, Otsuki T, Nakagawa E, Takahashi A, Kaneko Y, Kaido T,
et al: Abnormal maturation and differentiation of neocortical
neurons in epileptogenic cortical malformation: Unique distribution
of layer-specific marker cells of focal cortical dysplasia and
hemimegalencephaly. Brain Res. 1470:89–97. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Prayson RA, Spreafico R and Vinters HV:
Pathologic characteristics of the cortical dysplasias. Neurosurg
Clin N Am. 1317–25. (vii)2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guerrini R, Dobyns WB and Barkovich AJ:
Abnormal development of the human cerebral cortex: Genetics,
functional consequences and treatment options. Trends Neurosci.
31:154–162. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kabat J and Król P: Focal cortical
dysplasia-review. Pol J Radiol. 77:35–43. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Taylor DC, Falconer MA, Bruton CJ and
Corsellis JA: Focal dysplasia of the cerebral cortex in epilepsy. J
Neurol Neurosurg Psychiatry. 34:369–387. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Blümcke I, Thom M, Aronica E, Armstrong
DD, Vinters HV, Palmini A, Jacques TS, Avanzini G, Barkovich AJ,
Battaglia G, et al: The clinicopathologic spectrum of focal
cortical dysplasias: A consensus classification proposed by an ad
hoc Task Force of the ILAE Diagnostic Methods Commission.
Epilepsia. 52:158–174. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kuzniecky R: Epilepsy and malformations of
cortical development: New developments. Curr Opin Neurol.
28:151–157. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hsu PP, Kang SA, Rameseder J, Zhang Y,
Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, et al:
The mTOR-regulated phosphoproteome reveals a mechanism of
mTORC1-mediated inhibition of growth factor signaling. Science.
332:1317–1322. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou J, Blundell J, Ogawa S, Kwon CH,
Zhang W, Sinton C, Powell CM and Parada LF: Pharmacological
inhibition of mTORC1 suppresses anatomical, cellular, and
behavioral abnormalities in neural-specific Pten knock-out mice. J
Neurosci. 29:1773–1783. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dolmetsch R and Geschwind DH: The human
brain in a dish: The promise of iPSC-derived neurons. Cell.
145:831–834. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dragunow M: The adult human brain in
preclinical drug development. Nat Rev Drug Discov. 7:659–666. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ichida JK and Kiskinis E: Probing
disorders of the nervous system using reprogramming approaches.
EMBO J. 34:1456–1477. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Takahashi K, Okita K, Nakagawa M and
Yamanaka S: Induction of pluripotent stem cells from fibroblast
cultures. Nat Protoc. 2:3081–3089. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Marinowic DR, Domingues MF, Machado DC and
DaCosta JC: The expression of pluripotency genes and neuronal
markers after neurodifferentiation in fibroblasts co-cultured with
human umbilical cord blood mononuclear cells. In Vitro Cell Dev
Biol Anim. 51:26–35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Takahashi K and Yamanaka S: Induction of
pluripotent stem cells from mouse embryonic and adult fibroblast
cultures by defined factors. Cell. 126:663–676. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Takahashi K, Tanabe K, Ohnuki M, Narita M,
Ichisaka T, Tomoda K and Yamanaka S: Induction of pluripotent stem
cells from adult human fibroblasts by defined factors. Cell.
131:861–872. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fries KM, Blieden T, Looney RJ, Sempowski
GD, Silvera MR, Willis RA and Phipps RP: Evidence of fibroblast
heterogeneity and the role of fibroblast subpopulations in
fibrosis. Clin Immunol Immunopathol. 72:283–292. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Parent JM and Anderson SA: Reprogramming
patient-derived cells to study the epilepsies. Nat Neurosci.
18:360–366. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Song B, Sun G, Herszfeld D, Sylvain A,
Campanale NV, Hirst CE, Caine S, Parkington HC, Tonta MA, Coleman
HA, et al: Neural differentiation of patient specific iPS cells as
a novel approach to study the pathophysiology of multiple
sclerosis. Stem Cell Res. 8:259–273. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Luo Y, Fan Y, Zhou B, Xu Z, Chen Y and Sun
X: Generation of induced pluripotent stem cells from skin
fibroblasts of a patient with olivopontocerebellar atrophy. Tohoku
J Exp Med. 226:151–159. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Israel MA, Yuan SH, Bardy C, Reyna SM, Mu
Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, et al:
Probing sporadic and familial Alzheimer's disease using induced
pluripotent stem cells. Nature. 482:216–220. 2012.PubMed/NCBI
|
|
24
|
Yagi T, Ito D, Okada Y, Akamatsu W, Nihei
Y, Yoshizaki T, Yamanaka S, Okano H and Suzuki N: Modeling familial
Alzheimer's disease with induced pluripotent stem cells. Hum Mol
Genet. 20:4530–4539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hossini AM, Megges M, Prigione A, Lichtner
B, Toliat MR, Wruck W, Schröter F, Nuernberg P, Kroll H,
Makrantonaki E, et al: Induced pluripotent stem cell-derived
neuronal cells from a sporadic Alzheimer's disease donor as a model
for investigating AD-associated gene regulatory networks. BMC
Genomics. 16:842015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Duan L, Bhattacharyya BJ, Belmadani A, Pan
L, Miller RJ and Kessler JA: Stem cell derived basal forebrain
cholinergic neurons from Alzheimer's disease patients are more
susceptible to cell death. Mol Neurodegener. 9:32014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Marchetto MC, Carromeu C, Acab A, Yu D,
Yeo GW, Mu Y, Chen G, Gage FH and Muotri AR: A model for neural
development and treatment of Rett syndrome using human induced
pluripotent stem cells. Cell. 143:527–539. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Williams EC, Zhong X, Mohamed A, Li R, Liu
Y, Dong Q, Ananiev GE, Mok JC, Lin BR, Lu J, et al: Mutant
astrocytes differentiated from Rett syndrome patients-specific
iPSCs have adverse effects on wild-type neurons. Hum Mol Genet.
23:2968–2980. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Djuric U, Cheung AY, Zhang W, Mok RS, Lai
W, Piekna A, Hendry JA, Ross PJ, Pasceri P, Kim DS, et al: MECP2e1
isoform mutation affects the form and function of neurons derived
from Rett syndrome patient iPS cells. Neurobiol Dis. 76:37–45.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livide G, Patriarchi T, Amenduni M,
Amabile S, Yasui D, Calcagno E, Lo Rizzo C, De Falco G, Ulivieri C,
Ariani F, et al: GluD1 is a common altered player in neuronal
differentiation from both MECP2-mutated and CDKL5-mutated iPS
cells. Eur J Hum Genet. 23:195–201. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sareen D, O'Rourke JG, Meera P, Muhammad
AK, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian
A, et al: Targeting RNA foci in iPSC-derived motor neurons from ALS
patients with a C9ORF72 repeat expansion. Sci Transl Med.
5:208ra1492013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wainger BJ, Kiskinis E, Mellin C, Wiskow
O, Han SS, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, et
al: Intrinsic membrane hyperexcitability of amyotrophic lateral
sclerosis patient-derived motor neurons. Cell Rep. 7:1–11. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kiskinis E, Sandoe J, Williams LA,
Boulting GL, Moccia R, Wainger BJ, Han S, Peng T, Thams S,
Mikkilineni S, et al: Pathways disrupted in human ALS motor neurons
identified through genetic correction of mutant SOD1. Cell Stem
Cell. 14:781–795. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Devlin AC, Burr K, Borooah S, Foster D,
Cleary EM, Geti I, Vallier L, Shaw CE, Chandran S and Miles GB:
Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS
mutations are dysfunctional despite maintaining viability. Nat
Commun. 6:59992015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee P, Martin NT, Nakamura K, Azghadi S,
Amiri M, Ben-David U, Perlman S, Gatti RA, Hu H and Lowry WE: SMRT
compounds abrogate cellular phenotypes of ataxia telangiectasia in
neural derivatives of patient-specific hiPSCs. Nat Commun.
4:18242013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jiao J, Yang Y, Shi Y, Chen J, Gao R, Fan
Y, Yao H, Liao W, Sun XF and Gao S: Modeling Dravet syndrome using
induced pluripotent stem cells (iPSCs) and directly converted
neurons. Hum Mol Genet. 22:4241–4252. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee G, Papapetrou EP, Kim H, Chambers SM,
Tomishima MJ, Fasano CA, Ganat YM, Menon J, Shimizu F, Viale A, et
al: Modelling pathogenesis and treatment of familial dysautonomia
using patient-specific iPSCs. Nature. 461:402–406. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Doers ME, Musser MT, Nichol R, Berndt ER,
Baker M, Gomez TM, Zhang SC, Abbeduto L and Bhattacharyya A:
iPSC-derived forebrain neurons from FXS individuals show defects in
initial neurite outgrowth. Stem Cells Dev. 23:1777–1787. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tiscornia G, Vivas EL, Matalonga L,
Berniakovich I, Barragán Monasterio M, Eguizábal C, Gort L,
González F, Mellet C Ortiz, García F, ernández JM, et al:
Neuronopathic Gaucher's disease: Induced pluripotent stem cells for
disease modelling and testing chaperone activity of small
compounds. Hum Mol Genet. 22:633–645. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Guo X, Disatnik MH, Monbureau M, Shamloo
M, Mochly-Rosen D and Qi X: Inhibition of mitochondrial
fragmentation diminishes Huntington's disease-associated
neurodegeneration. J Clin Invest. 123:5371–5388. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yao Y, Cui X, Al-Ramahi I, Sun X, Li B,
Hou J, Difiglia M, Palacino J, Wu ZY, Ma L, et al: A
striatal-enriched intronic GPCR modulates huntingtin levels and
toxicity. Elife. 4:2015. View Article : Google Scholar
|
|
42
|
Mekhoubad S, Bock C, De Boer AS, Kiskinis
E, Meissner A and Eggan K: Erosion of dosage compensation impacts
human iPSC disease modeling. Cell Stem Cell. 10:595–609. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lancaster MA, Renner M, Martin CA, Wenzel
D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP and
Knoblich JA: Cerebral organoids model human brain development and
microcephaly. Nature. 501:373–379. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Reinhardt P, Schmid B, Burbulla LF,
Schöndorf DC, Wagner L, Glatza M, Höing S, Hargus G, Heck SA,
Dhingra A, et al: Genetic correction of a LRRK2 mutation in human
iPSCs links parkinsonian neurodegeneration to ERK-dependent changes
in gene expression. Cell Stem Cell. 12:354–367. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chung CY, Khurana V, Auluck PK, Tardiff
DF, Mazzulli JR, Soldner F, Baru V, Lou Y, Freyzon Y, Cho S, et al:
Identification and rescue of α-synuclein toxicity in Parkinson
patient-derived neurons. Science. 342:983–987. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sanders LH, Laganière J, Cooper O, Mak SK,
Vu BJ, Huang YA, Paschon DE, Vangipuram M, Sundararajan R, Urnov
FD, et al: LRRK2 mutations cause mitochondrial DNA damage in
iPSC-derived neural cells from Parkinson's disease patients:
Reversal by gene correction. Neurobiol Dis. 62:381–386. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Robicsek O, Karry R, Petit I,
Salman-Kesner N, Müller FJ, Klein E, Aberdam D and Ben-Shachar D:
Abnormal neuronal differentiation and mitochondrial dysfunction in
hair follicle-derived induced pluripotent stem cells of
schizophrenia patients. Mol Psychiatry. 18:1067–1076. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yoon KJ, Nguyen HN, Ursini G, Zhang F, Kim
NS, Wen Z, Makri G, Nauen D, Shin JH, Park Y, et al: Modeling a
genetic risk for schizophrenia in iPSCs and mice reveals neural
stem cell deficits associated with adherens junctions and polarity.
Cell Stem Cell. 15:79–91. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bda S Paulsen, de Moraes Maciel R, Galina
A, da Silveira M Souza, Souza C dosSantos, Drummond H, Pozzatto E
Nascimento, Silva H Jr, Chicaybam L, Massuda R, et al: Altered
oxygen metabolism associated to neurogenesis of induced pluripotent
stem cells derived from a schizophrenic patient. Cell Transplant.
21:1547–1559. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Guerrini R and Dobyns WB: Malformations of
cortical development: Clinical features and genetic causes. Lancet
Neurol. 13:710–726. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Johnson TE: Caenorhabditis elegans 2007:
The premier model for the study of aging. Exp Gerontol. 43:1–4.
2008.PubMed/NCBI
|
|
52
|
Kenyon CJ: The genetics of ageing. Nature.
464:504–512. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
O'Neill C: PI3-kinase/Akt/mTOR signaling:
Impaired on/off switches in aging, cognitive decline and
Alzheimer's disease. Exp Gerontol. 48:647–653. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Jansen LA, Mirzaa GM, Ishak GE, O'Roak BJ,
Hiatt JB, Roden WH, Gunter SA, Christian SL, Collins S, Adams C, et
al: PI3K/AKT pathway mutations cause a spectrum of brain
malformations from megalencephaly to focal cortical dysplasia.
Brain. 138:1613–1628. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Fusaki N, Ban H, Nishiyama A, Saeki K and
Hasegawa M: Efficient induction of transgene-free human pluripotent
stem cells using a vector based on Sendai virus, an RNA virus that
does not integrate into the host genome. Proc Jpn Acad Ser B Phys
Biol Sci. 85:348–362. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Li HO, Zhu YF, Asakawa M, Kuma H, Hirata
T, Ueda Y, Lee YS, Fukumura M, Iida A, Kato A, et al: A cytoplasmic
RNA vector derived from nontransmissible Sendai virus with
efficient gene transfer and expression. J Virol. 74:6564–6569.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
International Stem Cell Banking
Initiative, ; Andrews PW, Arias-Diaz J, Auerbach J, Alvarez M,
Ahrlund-Richter L, Baker D, Benvenisty N, Ben-Josef D, Blin G, et
al: Consensus guidance for banking and supply of human embryonic
stem cell lines for research purposes. Stem Cell Rev. 5:301–314.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Asprer JS and Lakshmipathy U: Current
methods and challenges in the comprehensive characterization of
human pluripotent stem cells. Stem Cell Rev. 11:357–372. 2015.
View Article : Google Scholar : PubMed/NCBI
|