Introduction
Osteoarthritis (OA) is a disease affecting the
joints characterized by the thinning and disintegration of
cartilage, synovial inflammation and subchondral bone remodeling
(1). The primary feature of OA is
the gradual loss of articular cartilage (2). OA progression is complex and involves
numerous processes, including aggrecan and type II collagen
degradation due to increased cleavage by activated proteolytic
enzymes including matrix metalloproteinases (MMPs) and a
disintegrin and metalloproteinase with thrombospondin motifs
(ADAMTSs) (3). Focal loss of
articular cartilage in OA may be associated with alterations in the
subjacent bone via modified load transmission and/or direct
signaling between neighboring tissues (4).
Wnt signaling is critical for the regulation of
adult bone turnoveenhancing this signaling pathway has been
investigated as a potential therapeutic strategy for osteoporotic
and inflammatory bone loss, to induce bone production and inhibit
soluble antagonists (5). By
contrast, increased Wnt-β-catenin signaling has been demonstrated
to stimulate tissue degradation rather than formation in adult
cartilage (6). Increased levels of
β-catenin have been observed in chondrocytes at areas of cartilage
degeneration (7), and it is
upregulated in cartilage, particularly in the superficial cartilage
zone (8). This may result in
chondrocyte hypertrophy leading to cartilage damage (9). Furthermore, stimulation of
chondrocytes with Wnt-β-catenin has been demonstrated to increase
the expression levels of various factors involved in OA, including
runt-related transcription factor 2 (RUNX-2), MMP-13, ADAMTS-4, and
ADAMTS-5, facilitating cartilage matrix degradation (9).
Sclerostin (SOST), encoded by the Sost gene,
is a secreted cysteine-knot protein of the differential screening
selected gene abberative in neuroblastoma family, which acts as an
antagonistic ligand for the Wnt coreceptors, low-density
lipoprotein-related receptor (LRP)5 and LRP6, and an inhibitor of
the canonical Wnt/β-catenin signaling pathway (10). Although the alterations in SOST in
human osteoarthritic cartilage have been described (11), the complex role of SOST during OA
progression remains unclear. A previous study has demonstrated that
SOST is additionally expressed by chondrocytes in mineralized
cartilage (12). However, the
therapeutic effects of SOST in OA cartilage remain controversial.
Chan et al (13)
demonstrated in vitro that increased chondrocyte SOST may
protect against cartilage degradation in OA, and Bouaziz et
al (14) used SOST-knockout
mice to reveal that the loss of SOST promotes OA in mice via
β-catenin-dependent Wnt signaling pathways. However, Roudier et
al (15) used SOST-knockout
mice and an OA mouse model to demonstrate that SOST is expressed in
articular cartilage, but its loss does not affect cartilage
remodeling during aging or following mechanical injury. Whether
SOST protects cartilage from degradation via inhibiting
Wnt-β-catenin remains unknown. The present study therefore used
healthy and OA chondrocytes to investigate the complex role of SOST
in healthy and OA cartilage.
Materials and methods
Human samples
All human samples were obtained with the informed
consent of patients, and with approval from the Ningxia Medical
University Ethics Committee (Yinchuan, China). Human OA samples (OA
group: n=57; female, 42; male, 15; age, 61.6±6.8 years) were
obtained from patients undergoing total knee arthroplasty (TKA) for
OA. Healthy human specimens (healthy group: n=6; female, 2; male,
4; age, 24.7±5.9 years) were obtained from patients undergoing
lower extremity amputation due to destructive injury. X-ray films
of knees were used to determine whether patients had OA. Cartilage
from the medial condylar, encompassing the maximal cartilage
erosion focal area, was used for primary chondrocyte culture and
subsequent mRNA and protein extraction. Medial tibial plateaus,
encompassing the maximal cartilage erosion focal area, were
isolated from patients prior to fixing in 4% paraformaldehyde for
sectioning.
Histology
Knees were fixed in 4% paraformaldehyde for 24 h at
4°C, decalcified in 0.5 M ethylenediaminetetraacetic acid at room
temperature for 21 days and embedded in optimum cutting temperature
compound followed by paraffin. Serial 4-µm-thick sagittal sections
of the medial tibial plateau were obtained at three depths, at
50-µm intervals. Sections were stained with Safranin O and
cartilage degradation was determined using the modified Mankin
scoring system (16). Samples were
divided into three groups: Normal, mid-stage OA and end-stage OA.
Paraffin sections for SOST immunostaining were first dewaxed and
rehydrated, then antigen retrieval was performed with 0.1% trypsase
(Beijing Solarbio Science & Technology Co., Ltd.) and goat
serum (Boster Bio-Engineering Ltd. Co., Wuhan, China) was used as a
blocking antigen. Next, the slides were incubated with a primary
rabbit anti-SOST antibody (cat. no. ab63097; Abcam, Cambridge, MA,
USA) for 2 h. Subsequently, biotinylated goat anti-rabbit IgG
secondary antibody [cat. no. sv0002; Boster Bio-Engineering Ltd.
Co.; 1:100, diluted in PBS containing 1% bovine serum albumin
(GibcThermo Fisher Scientific, Inc., Waltham, MA, USA)] was applied
for 2 h at room temperature, followed by incubation for 1 h with
horseradish peroxidase-conjugated antibody (cat. no. sv0002; Boster
Bio-engineering Limited Company, Wuhan, China). Then the slides
were colored with 3,3-diaminobenzidin (DAB) and stained with
hematoxylin. Positive cells were stained yellow and counted under
the light microscope (Olympus CX31, Olympus Corporation, Tokyo,
Japan) on the tibial joint cartilage surface (magnification, ×25)
and expressed as a percentage of the total cells.
Ex vivo cartilage explant culture
Full-depth articular cartilage explants were
isolated from the medial femoral condyle of patients undergoing TKA
or amputation. Cartilage samples were cut into small pieces (~1
mm3) and digested individually with 0.25% trypsin
(Beijing Solarbio Science & Technology Co., Ltd., Beijing,
China) and collagenase II (Sigma-Aldrich; Merck Millipore,
Darmstadt, Germany) to isolate chondrocytes. As it is difficult to
isolate chondrocytes from end-stage OA patients, due to limited
cartilage on the bone surface, cartilage was not obtained from all
OA patients. In total, 42 samples of OA chondrocytes (n=42) and six
samples of healthy chondrocytes were cultured. All chondrocytes
were cultured in 20% fetal bovine serum (GibcThermo Fisher
Scientific, Inc., Waltham, MA, USA) in Dulbecco's modified Eagle's
medium (GibcThermo Fisher Scientific, Inc.) for 2 days to ensure
cell adhesion. Chondrocytes from healthy subjects were subcultured
once at a split ratio of 1:4 using trypsin, generating 24 culture
dishes of healthy chondrocytes (n=24). The medium was replaced
every 3 days and activation of the Wnt-β-catenin signaling pathway
was examined by incubating chondrocytes in the absence or presence
of 10 ng/ml interleukin-1-α (IL-1α; PeproTech, Inc., Rocky Hill,
NJ, USA) for 48 h. The effect of SOST was examined by addition of
250 ng/ml recombinant human SOST (PeproTech, Inc.) for 48 h in the
absence or presence of 10 ng/ml IL-1α. All chondrocytes were
subsequently prepared for mRNA and protein extractions.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
RNA was extracted from human chondrocytes using the
E.Z.N.A® Total RNA kit (Omega Bio-Tek, Inc., Norcross,
GA, USA), and quantified with a NanoDrop 2000 spectrophotometer
(Thermo Fisher Scientific, Inc.). Next, RT was performed using
RevertAid First Strand cDNA Synthesis kit (Thermo Fisher
Scientific, Inc.). The temperature protocol for RT was as follows:
25°C for 5 min; 42°C for 60 min, 70°C for 5 min for termination as
described in the kit manual. mRNA expression levels were quantified
by qPCR using a SYBR® Green Master mix (Thermo Fisher
Scientific, Inc.) and a LightCycler® 480 (Roche
Diagnostics, Basel, Switzerland). The temperature protocol for the
reaction was as follows: 95°C for 10 min, 40 cycles at 95°C for 15
sec, drawing melting curve at 65°C for 15 sec, 60°C for 1 min, 95°C
for 15 sec and 60°C for 15 sec. The primers used are presented in
Table I. Averaged quantification
cycle (Cq) values were normalized to the averaged Cq value of
β-actin. Adjusted average Cq values were used to calculate relative
expression vs. the control, using the 2−ΔΔCq method
(17).
 | Table I.Primer sequences used for polymerase
chain reaction analysis. |
Table I.
Primer sequences used for polymerase
chain reaction analysis.
| Gene | Accession no. | Tm (°C) | Product size
(bp) | Forward | Reverse |
|---|
| SOST | NM_002427.3 | 60 | 126 |
TTCTCCTTCGGGACCTCAAT |
TCTCTCACCTCTGCCCATTC |
| β-catenin | NM_005099.4 | 58 | 122 |
GCTTTGTGTCGTCTTGAACG |
TCAGCAATCCCTTTCTCACC |
| LRP5 | AB257751.1 | 58 | 140 |
TTCATCTACTGGACCGACTGG |
TTGGTTCCGACGACCTTG |
| LRP6 | AB257752.1 | 59 | 114 |
ATCAAGCACCAAAGGCACT |
GTGGAAGGGCTGTTAGAAGAA |
| RUNX-2 | NM_001024630.3 | 60 | 100 |
GACGAGGCAAGAGTTTCACC |
GGTTCCCGAGGTCCATCTAC |
| MMP-13 | NM_001012329.1 | 60 | 129 |
TATGACTATGCGTGGCTGGA |
CCATTTGTGGTGTGGGAAGT |
| ADAMTS-4 | NM_005099.4 | 59 | 126 |
GGCTGTGATCGCATCATTGG |
CACATTGTTGTATCCGTACCTGA |
| ADAMTS-5 | NM_007038.3 | 59 | 172 |
TGTGAAGAGACCTTTGGTTCC |
TTCTGTGATGGTGGCTGAAG |
| COL2A1 | X02420 | 60 | 195 |
CATTCATCCCACCCTCTCAC |
TTCCTGTCTCTGCCTTGACC |
| β-actin | NM_001101 | 59 | 204 |
TGACGTGGACATCCGCAAAG |
CTGGAAGGTGGACAGCGAGG-3 |
Western blot analysis
Chondrocyte proteins were extracted using a Whole
Cell Lysis assay kit (Nanjing KeyGen Biotech Co., Ltd., Nanjing,
China) and quantified using the bicinchoninic acid assay. Membranes
were probed with the following primary antibodies: anti-SOST
(1:1,000; cat. no. ab63097), anti-β-catenin (1:1,000; cat. no.
ab6302), anti-MMP-13 (1:2,000; cat. no. ab39012) and anti-ADAMTS-4
(1:1,000; cat. no. ab84792; Abcam). Protein concentration was
determined using the bicinchoninc acid protein assay kit (Boster
Bio-engineering Limited Company, Wuhan, China). Equal quantities of
protein (20 µg) were separated by 10% SDS-PAGE and electroblotted
onto a PVDF membrane (EMD Millipore, Billerica, MA, USA). The
membrane was blocked in PBS containing 0.1% Tween-20 (Beyotime
Institute of Biotechnology, Inc., Haimen, China) and 5% non-fat dry
milk. The membrane was then probed with primary antibodiesfor
overnight incubation at 4°C. Horseradish peroxidase-conjugated
rabbit anti-human secondary antibody (cat. no. BA1070; 1:2,000;
Boster Bio-engineering Limited Company) was then added for 1 h at
26°C. Finally, the protein bands were detected using ECL solution
(GE Healthcare Life Sciences, Shanghai, China) and images were
captured using a FluorChem imaging system (Alpha Innotech, San
Leandro, CA, USA). β-actin was used as a loading control.
Unfortunately, western blot analysis was not performed on healthy
group chondrocytes. Only six healthy cartilage samples were used,
which were not subcultured to the third generation or cultured for
a prolonged time in case of cytometaplasia and apoptosis.
Therefore, not enough cells were obtained for western blotting and
SOST, β-catenin, MMP-13 and ADAMTS-4 protein expression levels were
detected only in OA human chondrocytes.
Statistical analysis
Data are presented as the mean ± standard deviation,
and were compared using SPSS software (version 22.0; IBM SPSS,
Armonk, NY, USA). Two-way analysis of variance was used to compare
the percentage of SOST-positive cells between healthy and OA
cartilage groups. Alterations in the number of positively stained
cells, and the fold change in gene expression are presented
graphically as means with 95% confidence intervals. The protein
expression levels in different groups were compared using a
Student's t-test. As certain data, for example gene expression,
were not normally distributed, treatment effects were assessed
using the non-parametric Mann-Whitney U test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Expression of SOST is not permanently
increased in cartilage during OA progression
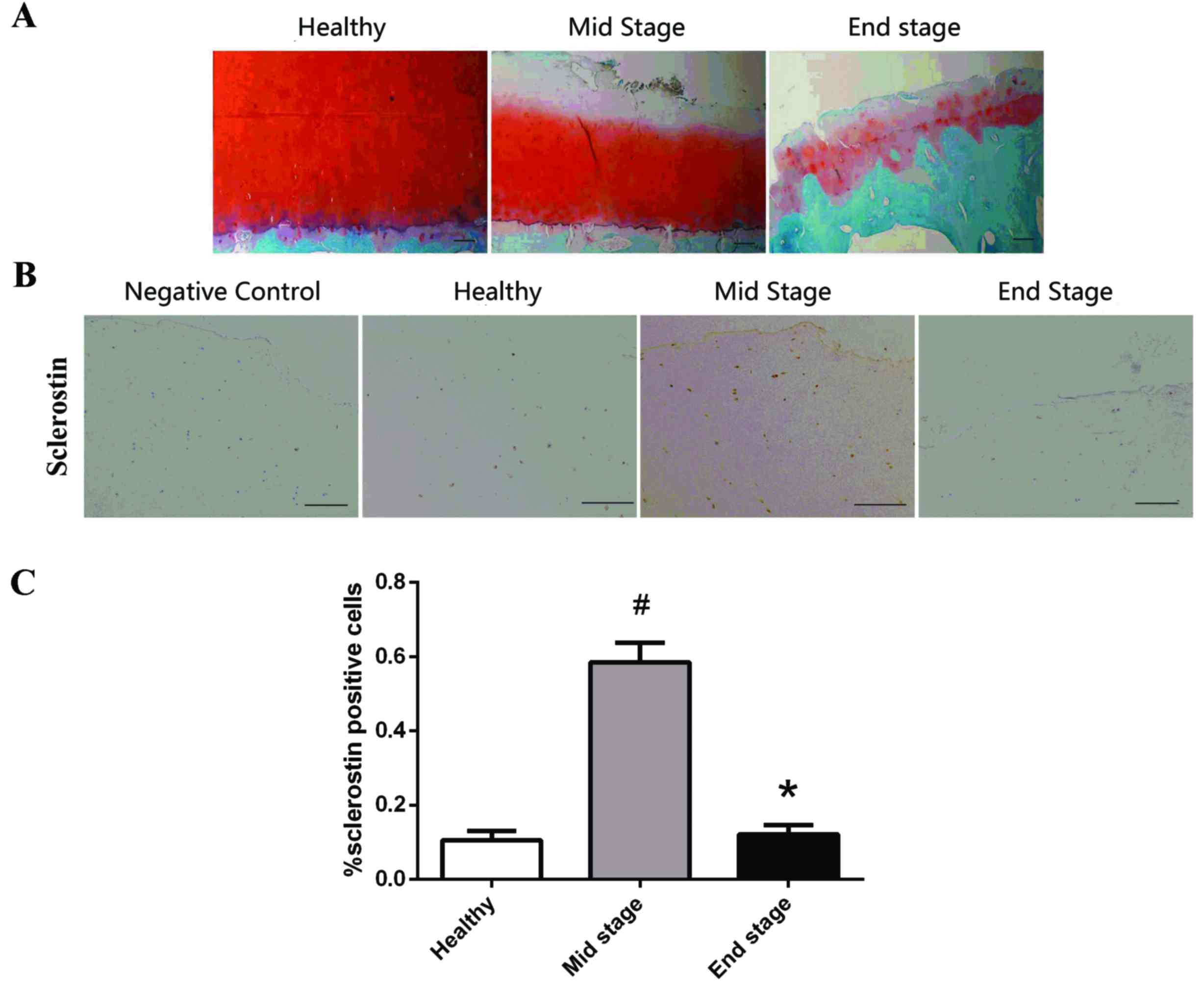
The Mankin score of the healthy group (n=6) was
1.3±1.2. Cartilage samples obtained from OA patients (n=57) were
divided into two groups, with scores for the mid-stage OA group
(n=37) being 7.7±0.9 and the end-stage OA group (n=20) being
13.0±1.2 (Fig. 1A).
Immunohistochemical staining was performed to detect the expression
of SOST at different stages of OA (Fig. 1B). Few SOST-positive stained
chondrocytes were observed in the healthy and end-stage OA groups,
with focal localization in the calcified cartilage and deep
cartilage near the tidemark. However, the percentage of
SOST-positive chondrocytes in the mid-stage OA group was
significantly increased compared with the healthy (P=0.002) and
end-stage OA groups (P=0.001) (Fig.
1C). No differences were observed between the healthy and
end-stage OA groups (P=0.48); therefore, SOST was not permanently
increased during OA progression, instead increasing and
subsequently decreasing. A high number of positively stained
chondrocytes were observed in the calcified cartilage of the
healthy group; however, only a few were present in the deep
cartilage and none on the surface. Conversely, positively stained
chondrocytes were widely distributed in the surface, deep and
calcified cartilage in mid-stage OA cartilage, with low SOST
expression in the calcified cartilage in end-stage OA samples.
Wnt-β-catenin signaling is
overactivated in OA chondrocytes
β-catenin (P=0.011), LRP5 (P=0.023) and LRP6
(P=0.012) mRNA expression levels were upregulated in OA
chondrocytes, suggesting that the Wnt-β-catenin signaling pathway
was overactivated and may be important in cartilage degradation
(Fig. 2A). In addition, mRNA
expression levels of the cartilage catabolic factors, RUNX-2
(P=0.015), MMP-13 (P=0.015) and ADAMTS-4 (P=0.008), ADAMTS-5
(P=0.002) were upregulated, whereas the anabolic factor, COL2A1
(P=0.003) was downregulated, in the OA compared with the healthy
chondrocyte group, thus accelerating cartilage matrix breakdown
(Fig. 2A). Furthermore, SOST
(P=0.021) mRNA expression levels were increased in the OA group,
consistent with the results of the immunohistochemical analysis.
Increased SOST expression may be a reaction to the activation of
the Wnt-β-catenin signaling pathway and may inhibit the
overactivated signaling pathway to maintain the integrity and
normal structure of cartilage. If this is the case, SOST may be a
potential therapeutic to delay OA progression.
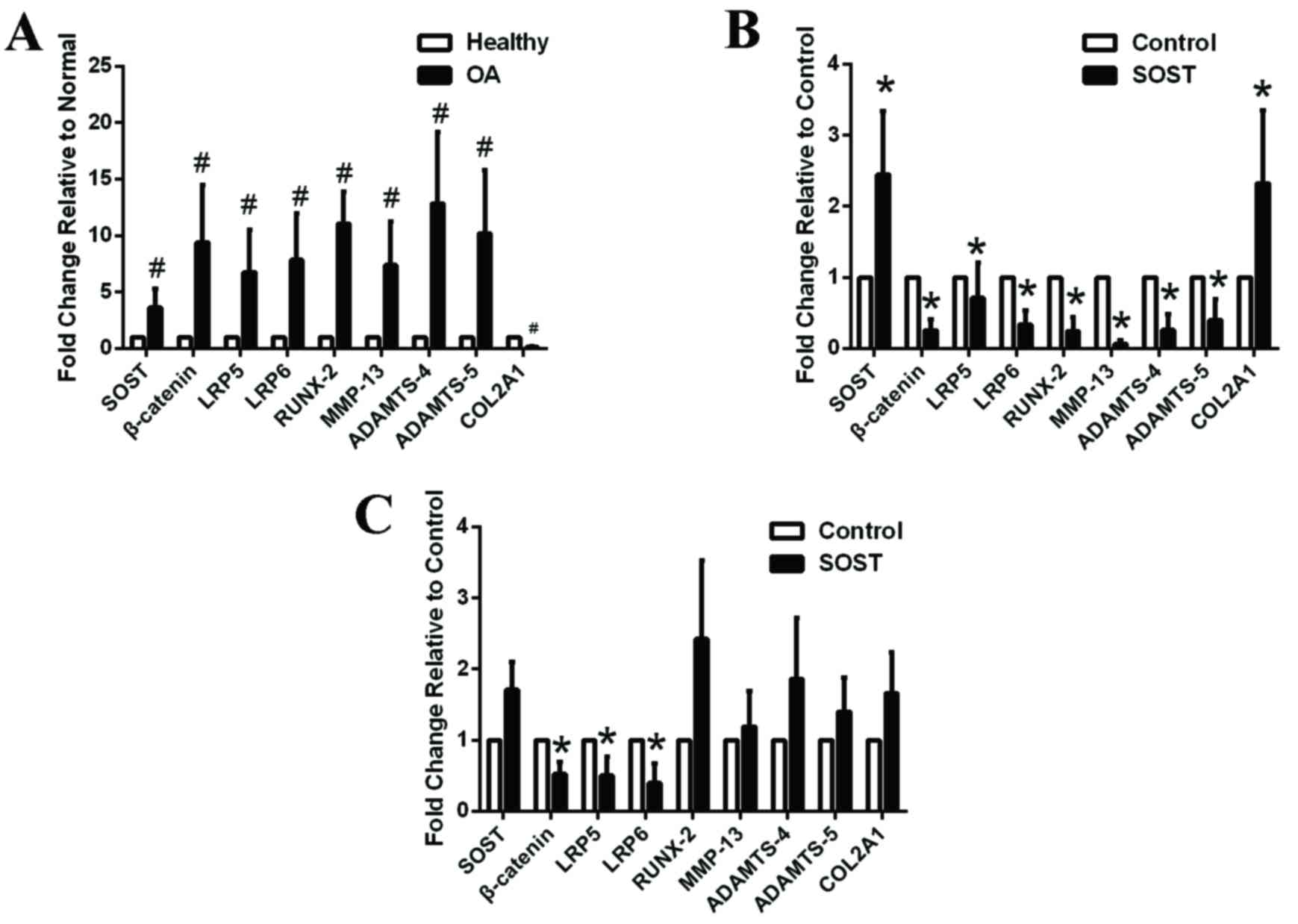 | Figure 2.Expression levels of Wnt-β-catenin
associated factors in healthy and OA chondrocytes incubated in the
absence or presence of SOST. (A) The Wnt-β-catenin signaling
pathway was overactivated in OA chondrocytes (n=6) compared with
healthy chondrocytes (n=6). (B) SOST treatment increased the mRNA
expression levels of the anabolic marker, COL2A1, but inhibited the
mRNA expression levels of the Wnt signaling-associated factors,
β-catenin, LRP5 and LRP6 and catabolic markers, RUNX-2, MMP-13 and
ADAMTS-4,5 in healthy chondrocytes (n=6). (C) SOST inhibited the
mRNA expression levels of the Wnt signaling-associated factors,
β-catenin, LRP5 and LRP6 in OA chondrocytes, but did not influence
downstream factors (n=6). (D) Western blot analysis of OA
chondrocytes incubated with SOST and/or IL-1α.
#P<0.05 vs. healthy chondrocyte *P<0.05 vs.
control. OA, osteoarthriti SOST, sclerostin; LRP, low-density
lipoprotein-related recepto; RUNX-2, runt-related transcription
factor 2; MMP, matrix metalloproteinas; ADAMTS, a disintegrin and
metalloproteinase with thrombospondin motif; COL2A1, collagen type
II alpha 1 chain. |
SOST inhibits the Wnt-β-catenin
signaling pathway in healthy and OA chondrocytes, with beneficial
effects observed in healthy chondrocytes only
SOST, as an inhibitor of the Wnt-β-catenin signaling
pathway, may decrease the expression of downstream factors,
including MMPs and ADAMTSs, to protect cartilage from degradation.
Healthy (Fig. 2B) and OA (Fig. 2C) chondrocytes were incubated with
250 ng/ml SOST for 48 h; this decreased β-catenin and LRP5/6 mRNA
expression levels in the two groups. β-catenin (healthy group,
P=0.012: OA group, P=0.022), LRP5 (healthy group, P=0.026; OA
group, 0.0016) and LRP6 (healthy group, P=0.015; OA group,
P=0.013). Therefore, SOST may inhibit the Wnt-β-catenin signaling
pathway by binding to LRP5/6 in chondrocytes. In addition, SOST
decreased RUNX-2 (P=0.015), MMP-13 (P=0.006) and ADAMTS-4
(P=0.013), ADAMTS-5 (P=0.015), and increased COL2A1 (P=0.002) mRNA
expression levels in healthy chondrocytes (Fig. 2B). This indicated that SOST may
assist in the maintenance of cartilage integrity and reduce
cartilage damage. Although SOST treatment decreased β-catenin and
LRP5/6 mRNA expression levels, it did not influence RUNX-2
(P=0.065), MMP13 (P=0.083), ADAMTS4 (P=0.074), ADAMTS-5 (P=0.063)
and COL2A1 (P=0.068) mRNA expression levels in OA chondrocytes
compared with the control group (Fig.
2C). This indicated that SOST may not have beneficial effects
on OA chondrocytes despite decreasing β-catenin expression levels
via binding to LRP5/6.
IL-1α may regulate cartilage
degradation via activation of the Wnt-β-catenin signaling pathway;
this may be inhibited by SOST
Chondrocytes were incubated with 10 ng/ml IL-1α for
48 h to simulate the inflammatory environment, determine the
activation effect on the Wnt-β-catenin signaling pathway and
quantify the effects of IL-1α on chondrocytes. Following treatment
with IL-1α in the absence or presence of SOST, RT-qPCR was
performed on healthy (Fig. 3) and
OA (Fig. 4) chondrocytes and
western blotting was performed on OA chondrocytes to detect protein
expression of SOST, β-catenin, ADAMTS-4 and MMP-13 (Fig. 5). The mRNA expression levels of
β-catenin (P=0.002), LRP5 (P=0.004) and LRP6 (P=0.014) in healthy
(Fig. 3) were increased following
treatment with IL-1α. Furthermore, mRNA expression levels of the
cartilage catabolic factors, RUNX-2, MMP13 and ADAMTS4/5 were
significantly increased, and the anabolic factor, COL2A1 was
decreased, following treatment with IL-1α in healthy (P-values were
0.015, 0.009, 0.022, 0.012; Fig.
3) but not OA (P-values were 0.62, 0.53, 0.71, 0.53; Fig. 4) chondrocytes. These results
indicated that IL-1α may accelerate the degradation of cartilage by
activating the Wnt-β-catenin signaling pathway in healthy but not
OA cartilage. Furthermore, chondrocytes exposed to IL-1α
demonstrated increased mRNA expression levels of SOST in the
healthy (P=0.002) but not the OA group (P=0.56). Chondrocytes were
treated with a combination of 250 ng/ml SOST and 10 ng/ml IL-1α to
investigate whether SOST inhibited Wnt-β-catenin following
overactivation. Compared with IL-1α only, IL-1α plus SOST treatment
decreased β-catenin and LRP5/6 mRNA expression levels in healthy
(P=0.002, 0.012, 0.012) and OA chondrocytes (P=0.022, 0.002, 0.015)
and decreased RUNX-2, MMP-13 and ADAMTS4/5, and increased COL2A1 in
healthy (P=0.023, 0.009, 0.013, 0.016; Fig. 3) but not OA (P=0.68, 0.57, 0.55;
Fig. 4) chondrocytes. SOST
therefore inhibited overactivation of the Wnt-β-catenin signaling
pathway in healthy and OA chondrocytehowever, it did not decrease
the expression of downstream catabolic factors to induce
‘anti-catabolic’ effects on OA chondrocytes.
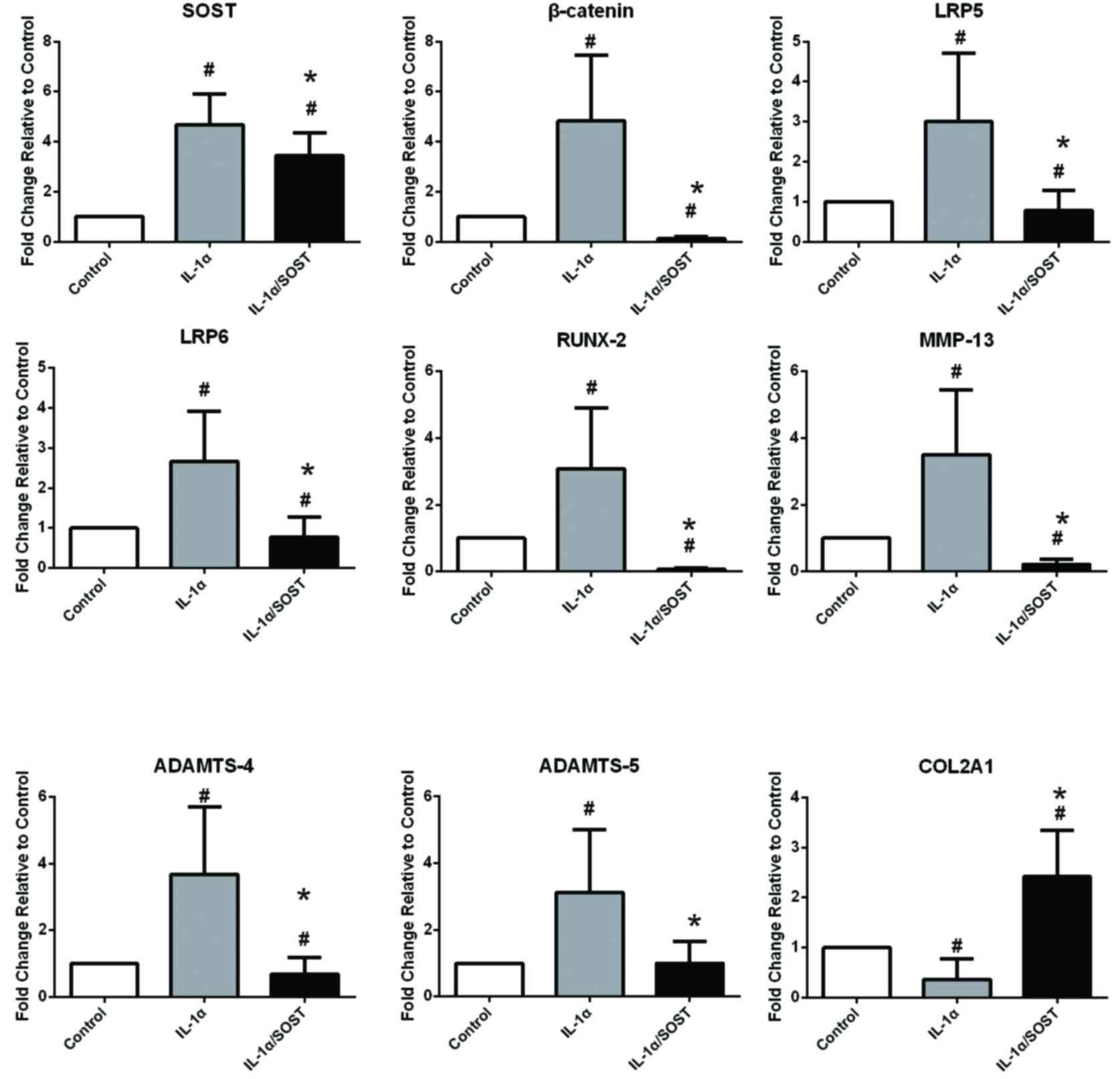 | Figure 3.mRNA expression levels of
Wnt-β-catenin-associated genes in healthy chondrocytes incubated
with IL-1α in the absence or presence of SOST. mRNA expression
levels of the Wnt-β-catenin-associated factors, β-catenin and
LRP5/6 and the catabolic markers, RUNX-2, MMP-13 and ADAMTS-4,5
were increased, whereas the anabolic marker, COL2A1 was decreased
following IL-1α treatment in healthy chondrocytes. These effects
were inhibited by SOST (n=6). The increasing of ADAMTS-5 by IL-1α
was inhibited by SOST, but has no difference with the control group
after the decreasing. #P<0.05 vs. control; *P<0.05
vs. IL-1α. IL-1α, interleukin-1-α; SOST, sclerostin; LRP,
low-density lipoprotein-related recepto; RUNX-2, runt-related
transcription factor 2; MMP, matrix metalloproteinas; ADAMTS, a
disintegrin and metalloproteinase with thrombospondin motif;
COL2A1, collagen type II alpha 1 chain. |
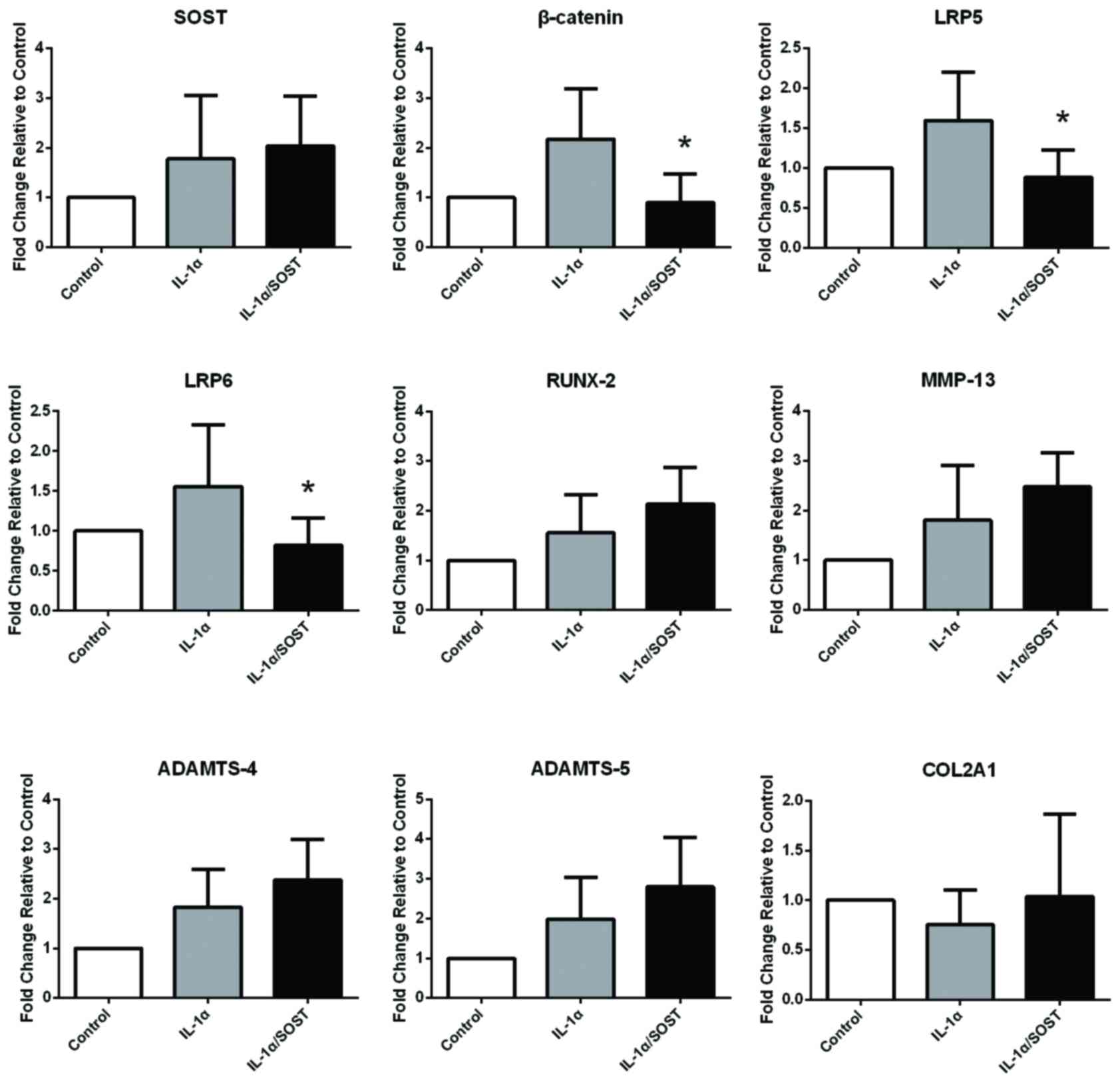 | Figure 4.mRNA expression levels of
Wnt-β-catenin-associated genes in OA chondrocytes incubated with
IL-1α in the absence or presence of SOST. mRNA expression levels of
the Wnt-β-catenin-associated factors, β-catenin and LRP5/6 were not
influenced by IL-1α in OA chondrocytehowever, the expression of
these factors was inhibited by SOST. Catabolic and anabolic markers
were not influenced by IL-1α alone or with SOST (n=6). *P<0.05
vs. IL-1α. OA, osteoarthriti; IL-1α, interleukin-1-α; SOST,
sclerostin; LRP, low-density lipoprotein-related recepto; RUNX-2,
runt-related transcription factor 2; MMP, matrix metalloproteinas;
ADAMTS, a disintegrin and metalloproteinase with thrombospondin
motif; COL2A1, collagen type II alpha 1 chain. |
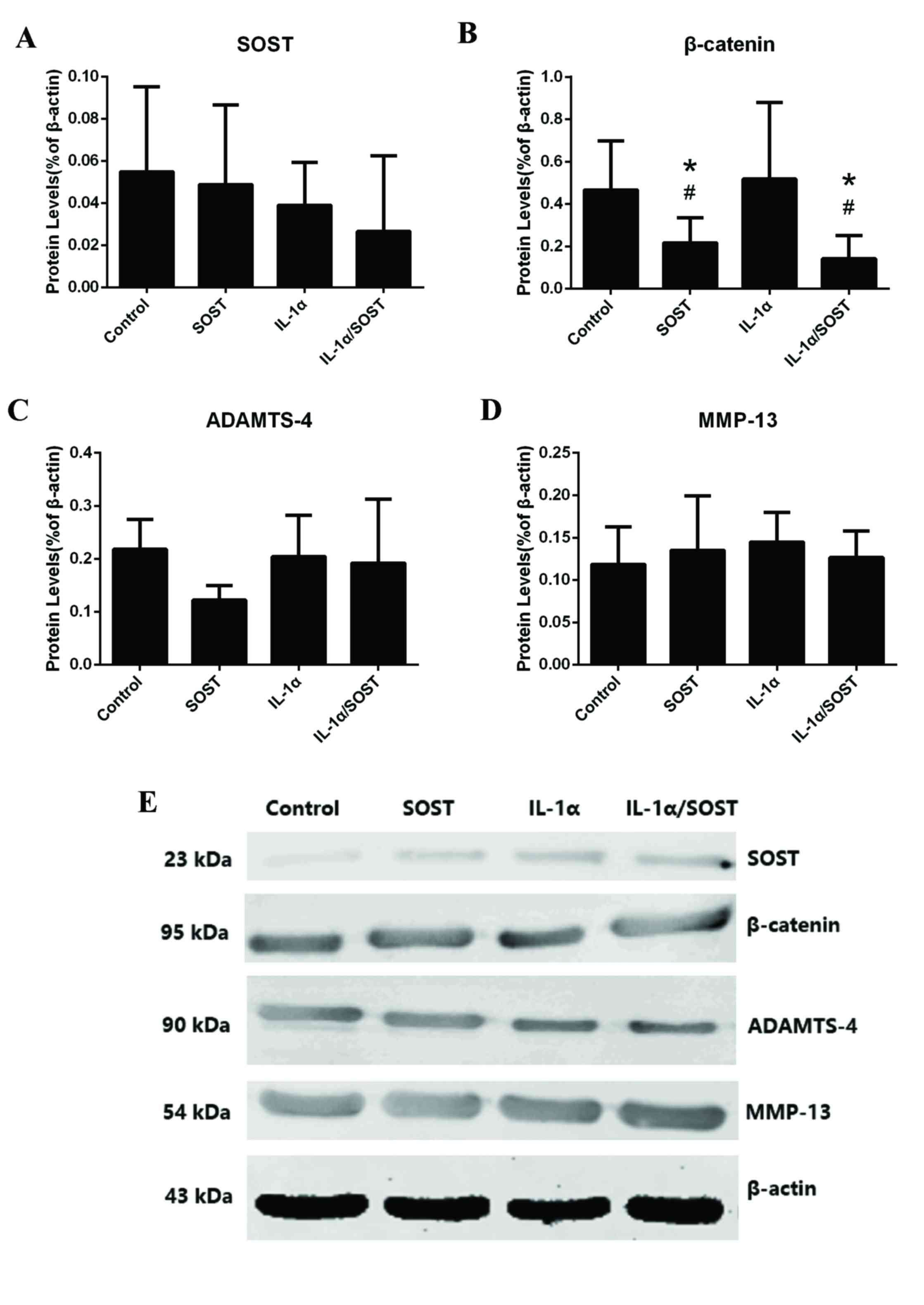 | Figure 5.Protein expression levels of
Wnt-β-catenin-associated genes in OA chondrocytes incubated with
IL-1α in the absence or presence of SOST. (A) Protein levels of
SOST in OA chondrocytes was not influenced by IL-1α (P=0.76), SOST
(P=0.43) and IL-1α/SOST (P=0.17; n=6) compared with control group
(n=6). (B) Wnt-β-catenin-associated factors, β-catenin were
influenced by SOST (P=0.035) and IL-1α/SOST (P=0.024) in OA
chondrocytes, but not influenced by IL-1α (P=0.15). (C) Protein
levels of ADAMTS-4 in OA chondrocytes was not influenced by IL-1α
(P=0.56), SOST (P=0.23) and IL-1α/SOST (P=0.31; n=6) compared with
control group (n=6). (D) Protein levels of MMP-13 in OA
chondrocytes were not influenced by IL-1α (P=0.13), SOST (P=0.21)
and IL-1α/SOST (P=0.37; n=6) compared with control group (n=6). (E)
Western blot analysis of OA chondrocytes incubated with SOST and/or
IL-1α. #P<0.05 vs. control; *P<0.05 vs.
IL-1α/SOST. OA, osteoarthriti; SOST, sclerostin; LRP, low-density
lipoprotein-related recepto; RUNX-2, runt-related transcription
factor 2; MMP, matrix metalloproteinas; ADAMTS, a disintegrin and
metalloproteinase with thrombospondin motif; COL2A1, collagen type
II alpha 1 chain. |
Discussion
The etiology and progression of OA remain to be
fully elucidated owing to the complexity of the diseastherefore,
the role of SOST in cartilage and subchondral bone during
degradation requires further investigation. Whether SOST protects
cartilage from degradation remains controversial. SOST is typically
expressed in osteocytes, particularly mature osteocytes, which are
surrounded by mineralized matrix (18,19).
However, later studies have suggested that SOST is expressed by
hypertrophic chondrocytes in calcified cartilage around the growth
plate (20–23). Full-depth articular cartilage
explants from the trochlear groove of ovine knee joints were used
to examine the activation of IL-1α in the Wnt-β-catenin signaling
pathway; chondrocytes were revealed to express SOST, which is
regulated by IL-1 (24). Increased
SOST in human chondrocytes may inhibit the Wnt-β-catenin signaling
pathway and downstream MMPs and ADAMTS in vivo, thus
protecting cartilage from degradation (13). Bouaziz et al (14) demonstrated using destabilization of
the medial meniscus (DMM) mice that SOST is only secreted by
calcified matrix-embedded cells, and is increased during the
development of OA but decreased in end-stage OA. In addition, this
study demonstrated using SOST-knockout mice that loss of SOST
accelerates degradation of cartilage in OA. However, Roudier et
al (15) revealed using human
articular cartilage that SOST is increased in OA cartilage, and
that loss of SOST in the joint enhanced the bone mass of
subchondral bonhowever, there was no difference between the
cartilage of SOST-knockout mice and DMM mice. Furthermore,
increased SOST in cartilage did not affect cartilage remodeling
during aging or following mechanical injury.
The present study demonstrated that SOST was
expressed only in the focal area of calcified cartilage and deep
cartilage adjacent to the tidemark in the medial tibial plateau of
healthy and end-stage OA human articular cartilage. The expression
of SOST was significantly increased in mid-stage OA and the
positively stained chondrocytes were closer to the surface of
cartilage compared with the healthy and end-stage OA groups. This
is consistent with the study by Bouaziz et al (14) using DMM mice. SOST was not
continuously increased in cartilage during the development of OA,
instead first increasing and subsequently decreasing, which is in
contrast to previous studies (13,25,26).
In early-stage OA, SOST in the cartilage may be secreted by
osteocytes in the subchondral bone as a result of stimuli,
including mechanical loading, but is not secreted by chondrocytes.
Furthermore, in the present study almost every hypertrophic
chondrocyte was positively stained. SOST therefore may not be
secreted by healthy chondrocytes but by hypertrophic ones. In
early-stage OA, chondrocytes in the cartilage did not express SOST;
therefore, it must be secreted by osteocytes in the subchondral
bone, from which it penetrates into cartilage via microchannels or
vessels. Therefore, SOST-positive chondrocytes were observed only
in the calcified cartilage. In mid-stage OA, the chondrocytes
became hypertrophic and began to secrete SOST, resulting in a
significant increase in SOST expression compared with early-stage
OA (27). In end-stage OA, SOST
expression was significantly reduced compared with mid-stage OA.
This is not consistent with some previous studies and further
investigation is required to confirm these results.
In the present study, the Wnt-β-catenin signaling
pathway was overactivated in OA chondrocytes, and matrix breakdown
factors including MMPs and ADAMTSs were upregulated. Inhibiting the
Wnt-β-catenin signaling pathway may decrease these mediators and
therefore be a potential therapeutic approach for the treatment of
OA. SOST has been identified to inhibit the Wnt-β-catenin signaling
pathway by binding to LRP5/6 in healthy and OA chondrocytehowever,
it decreased expression levels of cartilage catabolic factors only
in healthy chondrocytes, with no beneficial effects observed on OA
chondrocytes. This finding is inconsistent with previous studies in
animal OA models (13,26). It may be that SOST expression is
affected by mechanical loading in osteoblasts in subchondral bone,
and alterations of load bearing on cartilage may stimulate signal
transduction between subchondral bone and cartilage. Due to the
weight-bearing diversity across different areas of cartilage within
the joint, SOST expression and its effect may vary between areas
(28). As there is a marked
difference in walking and limb alignment between humans and
rodents, mechanical loading in the same area of cartilage within
the joint may cause different consequences of gene expression,
regulation and transduction of factors in the Wnt-β-catenin
signaling pathway (29). In
previous studies using rodent OA models, different experimental
results may be due to a number of reasons. For example, using
different joint cartilage regions for quantification due to the
small scale and blurred boundaries of different parts of rodent
joints, ignoring species specificity, in particular mechanical
loading within the joint, and interactions of different signaling
pathways. To avoid these errors, the present study used cartilage
from the medial femoral condyle for chondrocyte isolation and
primary culture to determine the effects of SOST on cartilage, and
the medial tibial plateau for paraffin sectioning and
immunohistochemistry to measure SOST expression in cartilage at
different stages of OA. However, only six samples of healthy human
cartilage were obtained. To generate sufficient chondrocyte
numbers, cells were subcultured at a ratio of 1:4 to obtain 24
bottles of chondrocytes in the healthy group. Due to differing
conditions in vivo and in vitro, chondrocytes were
passaged only to the first generation in case of cytometaplasia and
apoptosis. Consequently, not enough chondrocytes were obtained for
western blot analysis, which may lead to inaccurate results within
the present study. SOST may therefore only be a precautionary
measure to prevent cartilage degradation, and not a potential
therapeutic strategy to restore integrity or healthy cartilage
structure during OA progression.
In addition, the route of injecting SOST may have a
marked impact on whether SOST may be a potential therapeutic in OA.
Intraperitoneal injection of SOST as a systemic drug delivery
method would be convenient and effective. However, there is a very
limited blood supply within cartilage tissue, although certain
microchannels and micrangium penetrate calcified cartilage and the
tidemark to deliver mediators between cartilage and subchondral
bone. Whether SOST is small enough to access these channels remains
unknown; therefore, the concentration of SOST in cartilage may not
be enough to elicit an effect. Furthermore, SOST has been reported
to inhibit osteoblast differentiation, proliferation and activity,
resulting in reduced osteoblastic bone formation (30), which may lead to decreased
subchondral bone stiffness and increased mechanical stress in
cartilage. Intra-articular injection may have an advantage in
maintaining the concentration of SOST; however, whether SOST
permeates cartilage into the calcified region and subchondral bone
remains unknown. It is currently difficult to state which
drug-delivery method is more suitable.
To determine whether SOST inhibited overactivation
of the Wnt-β-catenin signaling pathway in healthy and OA
chondrocytes, chondrocytes were incubated with IL-1α to activate
Wnt signaling. IL-1α treatment increased mRNA expression levels of
β-catenin and downstream catabolic factors, including MMP-13 and
ADAMTS-4/5, in healthy chondrocytes, and SOST inhibited this
phenomenon. Notably, IL-1α and SOST did not influence OA
chondrocytes. This may be due to the reaction to the simulation of
inflammatory factors of hypertrophic chondrocytes in OA not being
as sensitive as healthy chondrocytehealthy chondrocytes may react
to inflammatory stimuli and thus contribute to the maintenance of
the normal structure and integrity of the cartilage, whereas
hypertrophic chondrocytes may have lost this ability.
However, the results of the present study obtained
by comparing SOST expression at different stages of OA and the
incubation of chondrocytes with SOST ex vivo may not be
accurate. All structures within the joint are affected and may
interact during the progression of OA, including the cartilage,
subchondral bone and synovium. Chondrocytes and cells in the bone
may react independently to identical environmental stimuli, leading
to altered cell phenotypes in OA. Alternatively, cellular
alterations in one cell type may have an affect on another cell
type. Previous in vitro and in vivo studies have
demonstrated that chondrocytes and osteoblasts influence each other
(31,32). Extensive research has demonstrated
that cartilage and subchondral bone are not separate in the
progression of OA, therefore the progression of the disease should
be considered in the context of the interaction of these two
compartments (33,34). Recently, molecular crosstalk
between osteoblasts/osteocytes and chondrocytes has been
demonstrated, revealing a previously unappreciated complexity
(35). The dense subchondral
vasculature in close proximity to the cartilage and the
microchannels that infiltrate the subchondral mineralization region
has been revealed to allow communication between bone and cartilage
(36). Uncalcified cartilage may
be observed dipping through the calcified cartilage into the bone
and marrow spaces, which may provide a molecular diffusion pathway
with potential nutritional, metabolic and biomechanical effects. In
addition, as this interface is involved in OA, these areas may
enable trafficking of humoral mediators between tissues (37). In the present study, no crosstalk
was observed between subchondral bone and cartilage as it is
challenging to simulate the physical and chemical environments
within the joint. As alterations in one compartment may influence
the other in OA, the normalization of cells in one compartment may
have beneficial effects on the other. The two compartments may
subsequently influence each other to create a more favorable cycle,
thus delaying the progression of OA.
Although the present study revealed that SOST
benefited healthy but not OA chondrocytes, this does not mean that
SOST may not influence chondrocytes, owing to the complexity of
mediator translation between cartilage and subchondral bone. One
possibility is that in the presence of SOST or as a result of
pharmacologic inhibition, a compensatory molecule, for example
another Wnt signaling inhibitor, is upregulated in the
cartilagalternatively, the crosstalk between cartilage and
subchondral bone may mask the effect of SOST inhibition. Further
studies are required to clarify the complex role of the Wnt
signaling pathway in OA and the interaction of SOST in the two
compartments, which may contribute to an improved understanding of
the pathogenesis and future therapies of human OA.
In conclusion, the results of the present study
demonstrated that SOST is secreted by chondrocytes at various
stages of OA and is not permanently increased in cartilage, first
being increased and subsequently decreased. Furthermore, SOST
inhibited the Wnt-β-catenin signaling pathway in healthy and OA
chondrocytehowever, SOST downregulated catabolic factors to benefit
only healthy chondrocytes. SOST may prevent cartilage degradation;
however, it may be that SOST cannot restore the integrity or normal
structure of cartilage during OA progression.
References
|
1
|
Bijlsma JW, Berenbaum F and Lafeber FP:
Osteoarthritis: An update with relevance for clinical practice.
Lancet. 377:2115–2126. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Felson DT: Developments in the clinical
understanding of osteoarthritis. Arthritis Res Ther. 11:2032009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Welgus HG: Stromelysin: Structure and
function. Agents Actions Suppl. 35:61–67. 1991.PubMed/NCBI
|
|
4
|
Karsdal MA, Leeming DJ, Dam EB, Henriksen
K, Alexandersen P, Pastoureau P, Altman RD and Christiansen C:
Should subchondral bone turnover be targeted when treating
osteoarthritis? Osteoarthritis Cartilage. 16:638–646. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hoeppner LH, Secreto FJ and Westendorf JJ:
Wnt signaling as a therapeutic target for bone diseases. Expert
Opin Ther Targets. 13:485–496. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yuasa T, Otani T, Koike T, Iwamoto M and
Enomoto-Iwamoto M: Wnt/beta-catenin signaling stimulates matrix
catabolic genes and activity in articular chondrocytes: Its
possible role in joint degeneration. Lab Invest. 88:264–274. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu M, Tang D, Wu Q, Hao S, Chen M, Xie C,
Rosier RN, O'Keefe RJ, Zuscik M and Chen D: Activation of
beta-catenin signaling in articular chondrocytes leads to
osteoarthritis-like phenotype in adult beta-catenin conditional
activation mice. J Bone Miner Res. 24:12–21. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Blom AB, Brockbank SM, van Lent PL, van
Beuningen HM, Geurts J, Takahashi N, van der Kraan PM, van de Loo
FA, Schreurs BW, Clements K, et al: Involvement of the Wnt
signaling pathway in experimental and human osteoarthritis:
Prominent role of Wnt-induced signaling protein 1. Arthritis Rheum.
60:501–512. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tamamura Y, Otani T, Kanatani N, Koyama E,
Kitagaki J, Komori T, Yamada Y, Costantini F, Wakisaka S, Pacifici
M, et al: Developmental regulation of Wnt/beta-catenin signals is
required for growth plate assembly, cartilage integrity, and
endochondral ossification. J Biol Chem. 280:19185–19195. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
van Bezooijen RL, Roelen BA, Visser A, van
der Wee-Pals L, de Wilt E, Karperien M, Hamersma H, Papapoulos SE,
ten Dijke P and Löwik CW: Sclerostin is an osteocyte-expressed
negative regulator of bone formation, but not a classical BMP
antagonist. J Exp Med. 199:805–814. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang
J, Harris SE and Wu D: Sclerostin binds to LRP5/6 and antagonizes
canonical Wnt signalin. J Biol Chem. 280:19883–19887. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Karlsson C, Dehne T, Lindahl A, Brittberg
M, Pruss A, Sittinger M and Ringe J: Genome-wide expression
profiling reveals new candidate genes associated with
osteoarthritis. Osteoarthritis Cartilage. 18:581–592. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chan BY, Fuller ES, Russell AK, Smith SM,
Smith MM, Jackson MT, Cake MA, Read RA, Bateman JF, Sambrook PN and
Little CB: Increased chondrocyte sclerostin may protect against
cartilage degradation in osteoarthritis. Osteoarthritis Cartilage.
19:874–885. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bouaziz W, Funck-Brentano T, Lin H, Marty
C, Ea HK, Hay E and Cohen-Solal M: Loss of sclerostin promotes
osteoarthritis in mice via β-catenin-dependent and -independent Wnt
pathways. Arthritis Res Ther. 17:242015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Roudier M, Li X, Niu QT, Pacheco E,
Pretorius JK, Graham K, Yoon BR, Gong J, Warmington K, Ke HZ, et
al: Sclerostin is expressed in articular cartilage but loss or
inhibition does not affect cartilage remodeling during aging or
following mechanical injury. Arthritis Rheum. 65:721–731. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Moody HR, Heard BJ, Frank CB, Shrive NG
and Oloyede AO: Investigating the potential value of individual
parameters of histological grading systems in a sheep model of
cartilage damage: The Modified Mankin method. J Anat. 221:47–54.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Poole KE, van Bezooijen RL, Loveridge N,
Hamersma H, Papapoulos SE, Löwik CW and Reeve J: Sclerostin is a
delayed secreted product of osteocytes that inhibits bone
formation. FASEB J. 19:1842–1844. 2005.PubMed/NCBI
|
|
19
|
Irie K, Ejiri S, Sakakura Y, Shibui T and
Yajima T: Matrix mineralization as a trigger for osteocyte
maturation. J Histochem Cytochem. 56:561–567. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
van Bezooijen RL, Bronckers AL, Gortzak
RA, Hogendoorn PC, van der Wee-Pals L, Balemans W, Oostenbroek HJ,
Van Hul W, Hamersma H, Dikkers FG, et al: Sclerostin in mineralized
matrices and van Buchem disease. J Dent Res. 88:569–574. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Winkler DG, Sutherland MK, Geoghegan JC,
Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR,
Staehling-Hampton K, et al: Osteocyte control of bone formation via
sclerostin, a novel BMP antagonist. EMBO J. 22:6267–6276. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ellies DL, Viviano B, McCarthy J, Rey JP,
Itasaki N, Saunders S and Krumlauf R: Bone density ligand,
Sclerostin, directly interacts with LRP5 but not LRP5G171 V to
modulate Wnt activity. J Bone Miner Res. 21:1738–1749. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lin C, Jiang X, Dai Z, Guo X, Weng T, Wang
J, Li Y, Feng G, Gao X and He L: Sclerostin mediates bone response
to mechanical unloading through antagonizing Wnt/beta-catenin
signaling. J Bone Miner Res. 24:1651–1661. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hwang SG, Yu SS, Ryu JH, Jeon HB, Yoo YJ,
Eom SH and Chun JS: Regulation of beta-catenin signaling and
maintenance of chondrocyte differentiation by ubiquitin-independent
proteosomal degradation of alpha-catenin. J Biol Chem.
280:12758–12765. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Goldring MB and Goldring SR: Articular
cartilage and subchondral bone in the pathogenesis of
osteoarthritis. Ann N Y Acad Sci. 1192:230–237. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lewiecki EM: Role of sclerostin in bone
and cartilage and its potential as a therapeutic target in bone
diseases. Ther Adv Musculoskelet Dis. 6:48–57. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Findlay DM and Atkins GJ:
Osteoblast-chondrocyte interactions in osteoarthritis. Curr
Osteoporos Rep. 12:127–134. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bove SE, Calcaterra SL, Brooker RM, Huber
CM, Guzman RE, Juneau PL, Schrier DJ and Kilgore KS: Weight bearing
as a measure of disease progression and efficacy of
anti-inflammatory compounds in a model of monosodium
iodoacetate-induced osteoarthritis. Osteoarthritis Cartilage.
11:821–830. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Teichtahl AJ, Davies-Tuck ML, Wluka AE,
Jones G and Cicuttini FM: Change in knee angle influences the rate
of medial tibial cartilage volume loss in knee osteoarthritis.
Osteoarthritis Cartilage. 17:8–111. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Baron R and Rawadi G: Targeting the
Wnt/beta-catenin pathway to regulate bone formation in the adult
skeleton. Endocrinology. 148:2635–2643. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mohan G, Perilli E, Kuliwaba JS, Humphries
JM, Parkinson IH and Fazzalari NL: Application of in vivo
micro-computed tomography in the temporal characterisation of
subchondral bone architecture in a rat model of low-dose monosodium
iodoacetate-induced osteoarthritis. Arthritis Res Ther.
13:R2102011. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Findlay DM and Atkins GJ:
Osteoblast-chondrocyte interactions in osteoarthritis. Current
osteoporosis reports. 12:127–134. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Amin AK, Huntley JS, Simpson AH and Hall
AC: Chondrocyte survival in articular cartilage: The influence of
subchondral bone in a bovine model. J Bone Joint Surg Br.
91:691–699. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sanchez C, Deberg MA, Piccardi N, Msika P,
Reginster JY and Henrotin YE: Osteoblasts from the sclerotic
subchondral bone downregulate aggrecan but upregulate
metalloproteinases expression by chondrocytes. This effect is
mimicked by interleukin-6, −1beta and oncostatin M pre-treated
non-sclerotic osteoblasts. Osteoarthritis Cartilage. 13:979–987.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang B, Zhou X, Price C, Li W, Pan J and
Wang L: Quantifying load-induced solute transport and solute-matrix
interaction within the osteocyte lacunar-canalicular system. J Bone
Miner Res. 28:1075–1086. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Imhof H, Sulzbacher I, Grampp S, Czerny C,
Youssefzadeh S and Kainberger F: Subchondral bone and cartilage
disease: A rediscovered functional unit. Invest Radiol. 35:581–588.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Botter SM, van Osch GJ, Clockaerts S,
Waarsing JH, Weinans H and van Leeuwen JP: Osteoarthritis induction
leads to early and temporal subchondral plate porosity in the
tibial plateau of mice: An in vivo microfocal computed tomography
study. Arthritis Rheum. 63:2690–2699. 2011. View Article : Google Scholar : PubMed/NCBI
|