Introduction
Parkinson's disease (PD) is the most common
neurodegenerative disorder that affects ~2% of the population aged
>65. Clinically, PD is characterized by slow movement, rigidity
and tremors (1). These classic
symptoms are primarily caused by the loss of dopamine (DA) in
neurons within the substantia nigra of the brain. The economic
burden of PD exceeded $14.4 billion in 2010 in the United States,
which equates to ~$22,800 per patient (2); the annual cost to Europe is ~€13.9
billion. At present, there are no effective treatments that prevent
or reverse DA degeneration and current therapies primarily aim to
alleviate the symptoms. When administered long-term, these
treatments may cause serious side effects including dyskinesias,
irritability and sleeplessness (3). Thus, extensive studies have
investigated the aetiology and physiopathology of PD, to develop
novel preventive or therapeutic strategies (4).
The aetiology of PD is complex, as PD may develop
from a combination of genetic and environmental factors (5). Genetic PD is relatively rare and
represents 10–15% of all PD cases. Determining the underlying
genetic mechanisms involved in PD is key in identifying novel
targets for the treatment of sporadic PD. Indeed, studies on
genetic factors of PD aid understanding of its pathophysiology
(6,7). For example, the identification of
mutations in the gene encoding human α-synuclein, SNCA, led
to the finding that α-synuclein aggregates are the primary
components of Lewy bodies, a pathological feature of sporadic and
genetic PD (8,9). Various causative genes have been
identified in rare cases of familial PD that exhibit autosomal
dominant or recessive patterns. Patients with recessive forms of PD
present with a range of clinical signs and symptoms, including
young-onset and onset with dystonia. However, the clinical symptoms
of dominant forms of PD are indistinguishable from sporadic PD,
suggesting that the underlying molecular signalling pathways are
similar. Studies have suggested that the genes involved in the
pathogenesis of dominant PD are associated with dysfunction of the
endosomal-lysosomal system, which is responsible for synaptic
vesicle and receptor recycling and protein degradation (10). Dysfunction of the
endosomal-lysosomal system induces PD via the accumulation of
α-synuclein (11); however, it may
additionally cause PD via an α-synuclein-independent pathway
(12). Previously, it was reported
that a point mutation (D620N) in vacuolar protein
sorting-associated protein 35 (VPS35), a gene that serves a
role in the endosomal-lysosomal system, is associated with
autosomal dominant PD (13,14).
VPS35 is a subunit of the retromer complex, which serves a
role in retrograde transport of cellular proteins from endosomes to
the trans-Golgi network (TGN) (15). The VPS35 D620 N mutation may
disrupt cellular functions via endosomal-lysosomal dysfunction,
autophagy impairment and mitochondrial dysfunction (16–19).
In addition to PD, VPS35 may be associated with Alzheimer's
disease in humans and animals. For example, the expression levels
of VPS35 are reduced in the hippocampi of patients with
Alzheimer's disease, and inhibition of VPS35 resulted in
elevated amyloid-β expression levels in animal models (20). Therefore, it remains uncertain how
the D620 N mutation in VPS35 is specifically associated with
PD.
α-synuclein serves a role in the pathogenesis of
genetic and sporadic PD. As α-synuclein is primarily degraded by
the endosomal-lysosome system, its role in VPS35-associated
PD has been extensively investigated; however, whether α-synuclein
is a neuropathological feature of VPS35-associated PD
remains unclear. Various studies have demonstrated an association
between α-synuclein and VPS35-linked PD (21,22).
For example, loss of VPS35 led to an increase in α-synuclein
accumulation and toxicity, whereas overexpression of VPS35
reversed this effect (17).
However, disruption of endosome-lysosomal function by mutations in
PD-associated genes may additionally induce loss of DA and
neurodegeneration via α-synuclein-independent mechanisms (22). In human brains from sporadic PD
patients and in animal models, VPS35 is not localised within
Lewy bodies and is not associated with α-synuclein. Overexpression
of VPS35 harbouring the D620N mutation in the substantia
nigra of rats may induce DA loss without formation of
α-synuclein-positive Lewy pathology (21). Collectively, these findings
suggested that α-synuclein-independent mechanisms may contribute to
the pathogenesis of VPS35-associated PD; however, the
association between α-synuclein and the D620N mutation within
VPS35 remains to be determined.
As numerous genes are conserved between species,
simple model organisms have been utilized as tools for examining
the underlying molecular mechanisms of neurodegenerative diseases.
The budding yeast, Saccharomyces cerevisiae (sc), is a
simple organism that has been widely used in the study of
neurodegenerative diseases. In yeast models, high expression levels
of α-synuclein are associated with key cellular defects implicated
in the aetiology of PD, including α-synuclein aggregation,
mitochondrial dysfunction and vesicle-trafficking dysfunction
(23,24).
To determine the underlying cellular mechanism by
which the VPS35 D620N mutation causes PD, cell growth and
endosomal-lysosomal function was examined in yeast cells with
various expression levels of sc VPS35 in the presence or
absence of non-toxic levels of α-synuclein. The results of the
present study revealed that high expression levels of sc
VPS35 D686N (the yeast equivalent of the human point
mutation D620N) were associated with α-synuclein-dependent toxicity
in yeast. In addition, sc VPS35 D686N promoted α-synuclein
aggregation whereas wild-type (WT) sc VPS35 reduced
α-synuclein aggregation. Fragmentation of vacuoles and dysfunction
of the lysosome may be the potential mechanism underlying the
synergistic effects of α-synuclein and sc VPS35.
Materials and methods
Yeast strains and plasmid
construction
Saccharomyces cerevisiae strains BY4741,
BY4743 and Δ VPS35 were purchased from Invitrogen; Thermo
Fisher Scientific, Inc. (Waltham, MA, USA). The Q5®
High-Fidelity PCR kit and restriction enzymes below were purchased
from New England BioLabs, Inc. (Ipswich, MA, USA). Yeast vectors
pRS414, pRS415 and pYES2nt were received as gifts from Dr. Lu
Yongjun at the Sun Yat-sen University. The WT α-synuclein sequence
(NM_001146055.1) was cloned into the pRS414 yeast vector in the C
terminal region using SpeI and ClaI restriction
enzymes. The plasmid contained the GAL1 promoter and an enhanced
green fluorescent protein (eGFP) insert. To amplify the WT sc
α-synuclein sequence, polymerase chain reaction (PCR) was performed
with the following primer pair: Sense,
5′-GGACTAGTATGGATGTATTCATGAAAGG-3′ and antisense,
5′-CCATCGATAGCTTCAGGTTCGTAGTCTTG-3′. PCR cycling conditions were as
follows: an initial predenaturation step at 98°C for 1 min,
followed by 30 cycles of denaturation at 98°C for 10 sec, annealing
at 65°C for 20 sec and extension at 72°C for 30 sec. The sc
VPS35 sequence was amplified via PCR from yeast DNA and
inserted into the pRS415 plasmid between XbaI and
NotI restriction sites, and into the pYES2nt plasmid between
SacI and NotI restriction sites. The primer pair were
as follows: sense, 5′-ATGCGGACTCACC-3′ and antisense,
5′-TACATATATGACTTTGAAAC-3′. PCR cycling conditions were as follows:
an initial predenaturation step at 98°C for 1 min, followed by 30
cycles of denaturation at 98°C for 15 sec, annealing at 68°C for 20
sec and extension at 72°C for 1 min. The D686N mutation was
generated as previously described (25). In addition, sc VPS35 mutant
sequences were inserted into the GAL1 promoter and fused with the
mCherry tag in the C terminal of pRS415 and pYES2nt plasmids in the
same manner as with sc VPS35 WT.
Yeast cells were grown in non-selective medium at
30°C and transformed as previously described (21). Selective synthetic complete (SC)
medium (FunGenome Company, Beijing, China) contained 2% glucose or
2% galactose and lacked the nutrient corresponding to the marker
(tryptophan for pRS414 and uracil for pYES2nt).
Spotting tests were conducted to assess growth
differences. Yeast cells transfected with plasmids were grown
overnight to achieve an optical density (OD) at a wavelength of 600
nm (OD600) of >2.0. Cells were subsequently diluted to mid-log
phase (OD600, 0.5) and cultured for 4–6 h to achieve an OD600 of
1.0. Cells were normalized to equal densities, serially diluted
5-fold from an initial OD600 of 0.01, and spotted on plates
containing 2% glucose or 2% galactose. Following 2 days incubation
at 30°C, plates were imaged.
Fluorescence microscopy
Yeast cell cultures were grown in SC medium with
glucose to reach mid-log phase prior to replacement with medium
supplemented with galactose. Cells were visualized using a Leica
TCS SP5 II confocal microscope (Leica Microsystems GmbH, Wetzlar,
Germany) at ×63 magnification following a specific incubation
time.
Cells were stained with the FM® 1–43
lipophilic dye (T-35356; Thermo Fisher Scientific, Inc.) to observe
yeast vacuoles, as previously described (11).
For quantification of aggregation, ≥300 cells were
counted per strain in each experiment. For each strain, the number
of cells that exhibited cytoplasmic foci were reported as the total
number of cells with fluorescence and presented as a percentage.
Cells exhibiting only bright peripheral halos around the plasma
membrane, additional perivacuolar fluorescence or cytoplasmic
distribution were not considered to be aggregated and were excluded
from the count.
Promoter shut-off studies and drug
treatment
Yeast cells were cultured overnight in SC medium
with glucose and without tryptophan and uracil, and were
subsequently cultured in SC medium containing galactose for 12 h to
induce α-synuclein expression. Following pre-incubation in
galactose medium, cells were cultured in SC medium with glucose to
switch off the promoter for 2 h and observed under a fluorescence
microscope. A total of 1 mM phenylmethanesulfonyl fluoride (PMSF)
or ethanol (control; the solvent utilized to reconstitute PMSF)
were applied to yeast cells when the glucose medium was added.
Statistical analysis
Data are expressed as the mean ± standard error, of
three independent experiments. GraphPad Prism software version 6.0
(GraphPad Software, Inc., La Jolla, CA, USA) was used to perform
statistical analyses. A Brown-Forsythe test was used to confirm
that the group variances were statistically equal. A one-way
analysis of variance was performed to determine significant
differences between groups, followed by a Dunnett's multiple
comparison test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Overexpression of scVPS35 D686N does
not lead to toxicity in yeast
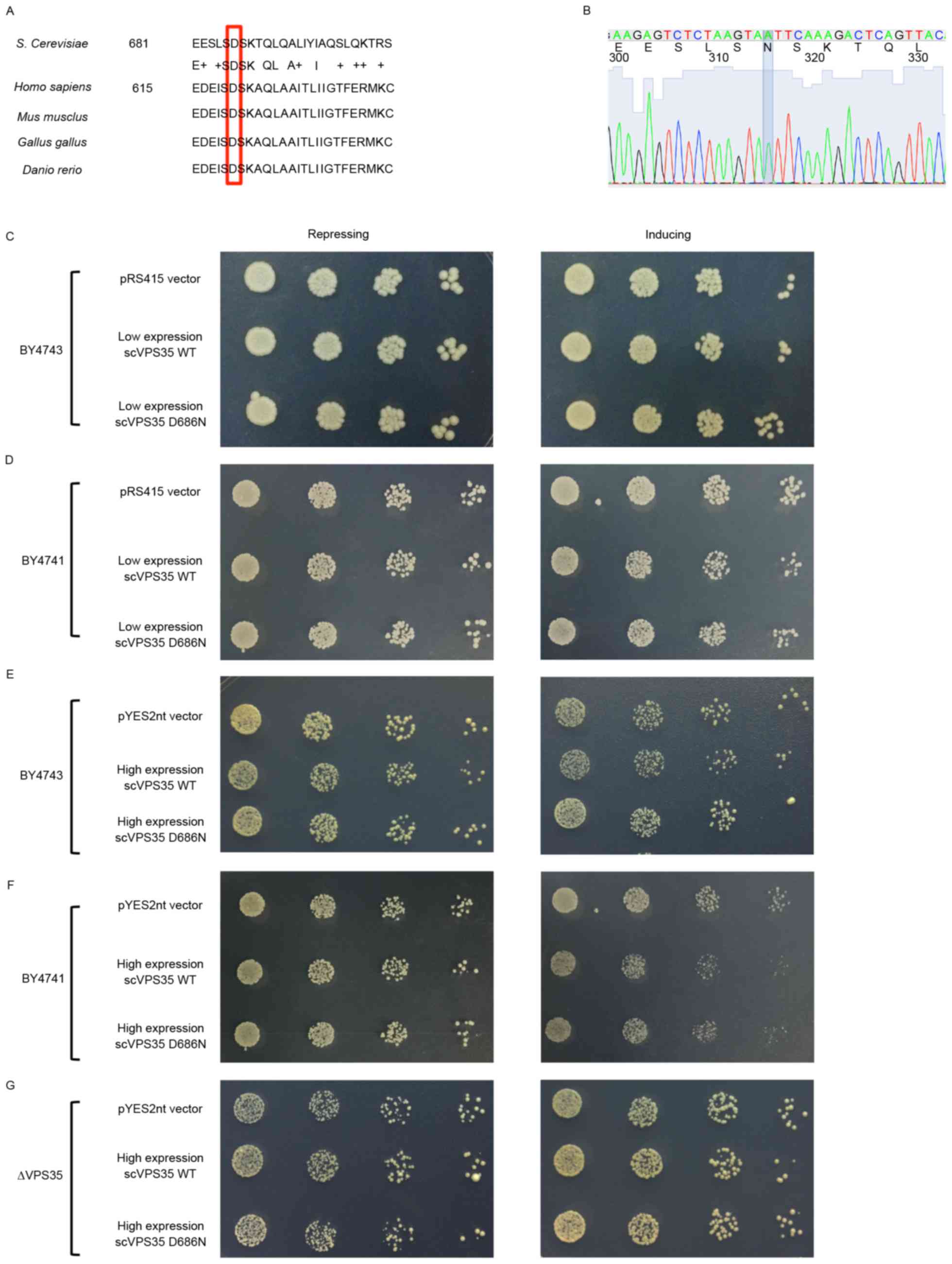
VPS35 is a highly conserved protein among
various species. Mouse, chicken, and zebrafish VPS35 share
≥90% similarity with humans. In addition, protein sequence
alignment revealed that the human VPS35 sequence is 52%
homologous to that of the budding yeast Saccharomyces
cerevisiae. Notably, the human PD-associated D620N mutation
site in VPS35 is conserved in yeast and corresponds to D686N
(Fig. 1A). To avoid possible
artificial dominant-negative impact by direct expression of human
genes in yeast, the human D620N mutation was mimicked by
introducing the analogous mutation D686N to examine the effects on
the biological functions in yeast. WT sc VPS35 was directly
cloned from yeast DNA and site 686 was subsequently mutated from
aspartic acid (D) to asparagine (N; Fig. 1B).
Cell growth is a key biological function of yeast.
To examine the potential role of VPS35 in cell growth, a
spotting assay was performed in yeast strains with or without sc
VPS35 expression. A spotting assay is a simple and effective
way to determine the effect of targeted genes on growth. Under the
control of the GAL1 inducible promoter, medium with galactose
significantly increases target gene expression, whereas medium
containing glucose represses gene expression. Therefore, the effect
of targeted genes on growth may be evaluated by assessing the
density of yeast in galactose medium. Under this system, yeast
strains with different expression levels of sc VPS35,
including a VPS35 deleted strain (Δ VPS35), in
addition to BY4741 and BY4743 strains, were utilized to determine
whether sc VPS35 is essential for cell growth. Δ
VPS35, BY4741 and BY4743 strains contained zero, one or two
copies of sc VPS35, respectively. The cell density was
similar in these three strains (data not shown). Therefore,
endogenous sc VPS35 may not be essential for cell growth. To
examine the effect of the sc VPS35 D686N mutation on cell
growth, sc VPS35 WT and sc VPS35 containing the D686N
mutant were cloned into the low-copy plasmid pRS415 and transfected
into the yeast strains. Yeast with low expression levels of the sc
VPS35 WT or D686N mutant exhibited similar growth to the
pRS415 control vector, following incubation with repression and
induction medium in BY4741 and BY4743 strains (Fig. 1C and D). A high-copy plasmid,
pYES2nt, was utilized to address the potential threshold expression
levels of the sc VPS35 D686N mutant. As observed with the
low-copy plasmid, high expression levels of the sc VPS35 WT
and D686N mutant were equally as toxic in yeast strains as the
control (Fig. 1E, F and G).
Therefore, this data suggested that VPS35 D686N alone is not
sufficient for inhibition of growth, even at relatively high
expression levels.
High levels of scVPS35 D686N mutant
induce toxicity in an α-synuclein-dependent manner
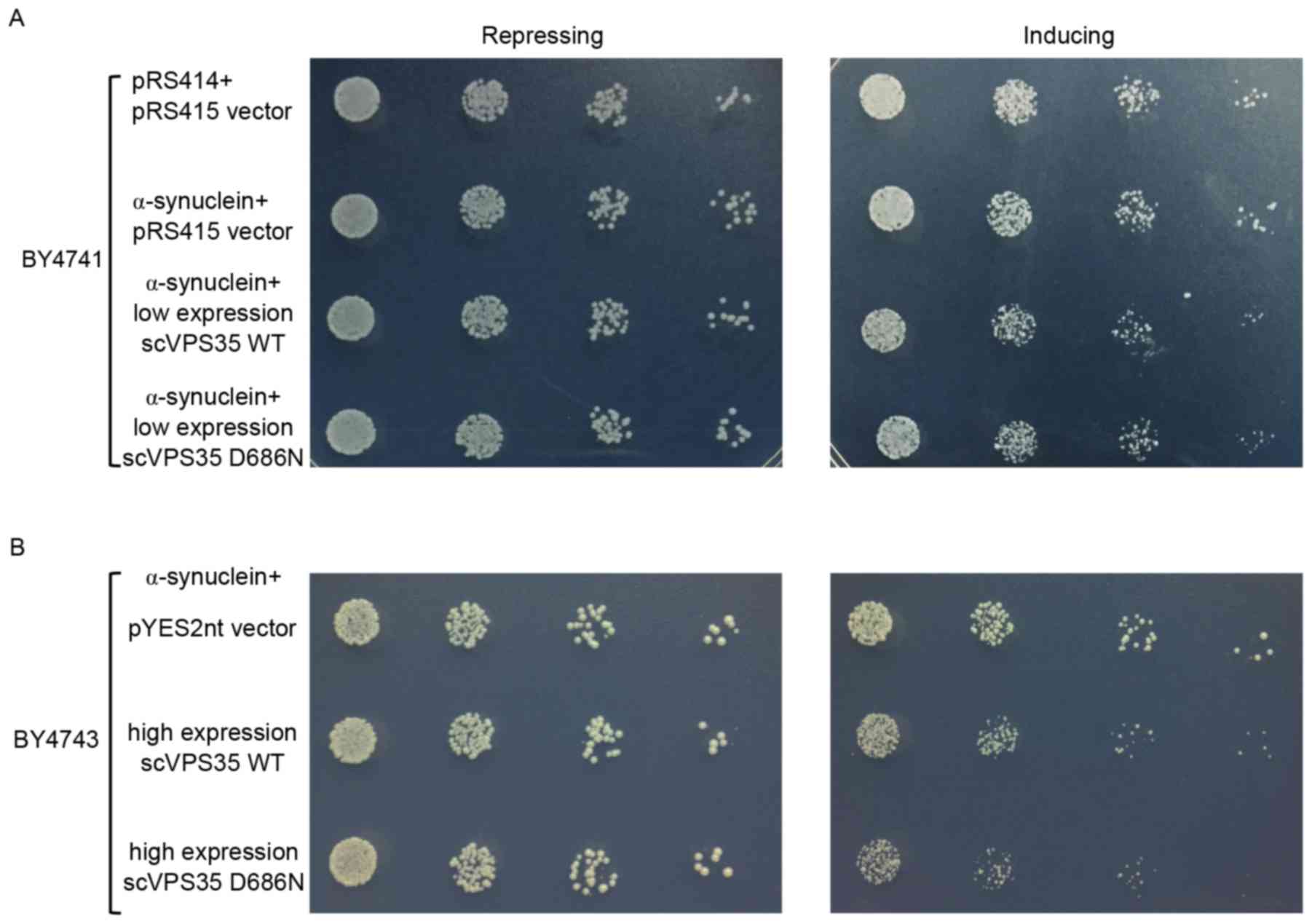
α-synuclein has a central role in the pathogenesis
of PD. Clinical and experimental studies have suggested that
overexpression of WT α-synuclein may lead to cell toxicity in a
dose-dependent manner (26). As
high expression of α-synuclein alone may lead to cell toxicity,
α-synuclein was expressed in yeast at non-toxic levels to determine
whether the interaction of VPS35 with physiological levels
of α-synuclein induces toxicity. Consistent with previous studies
(17,27), low expression levels of WT
α-synuclein in yeast were not toxic compared with the pRS415 vector
control (Fig. 2A). Co-expression
of sc VPS35 WT or D686N at low copy numbers with α-synuclein
did not affect cell growth compared with the vector control
(Fig. 2A). sc VPS35 WT and
D686N were subsequently cloned into the pYES2nt plasmid to enhance
their expression levels. High copy numbers of sc VPS35 D686N
or sc VPS35 WT were co-expressed with low copy numbers of
α-synuclein. Under induction, yeast cells showed relatively lower
growth rate due to carrying more protein compared with the control.
However, the interaction of sc VPS35 D686N with α-synuclein
suppressed cell growth significantly, compared with sc VPS35
WT (Fig. 2B). Collectively, this
data suggested that the negative effect of the VPS35 mutant
on yeast cell growth is dose- and α-synuclein-dependent.
scVPS35 D686N promotes fragmentation
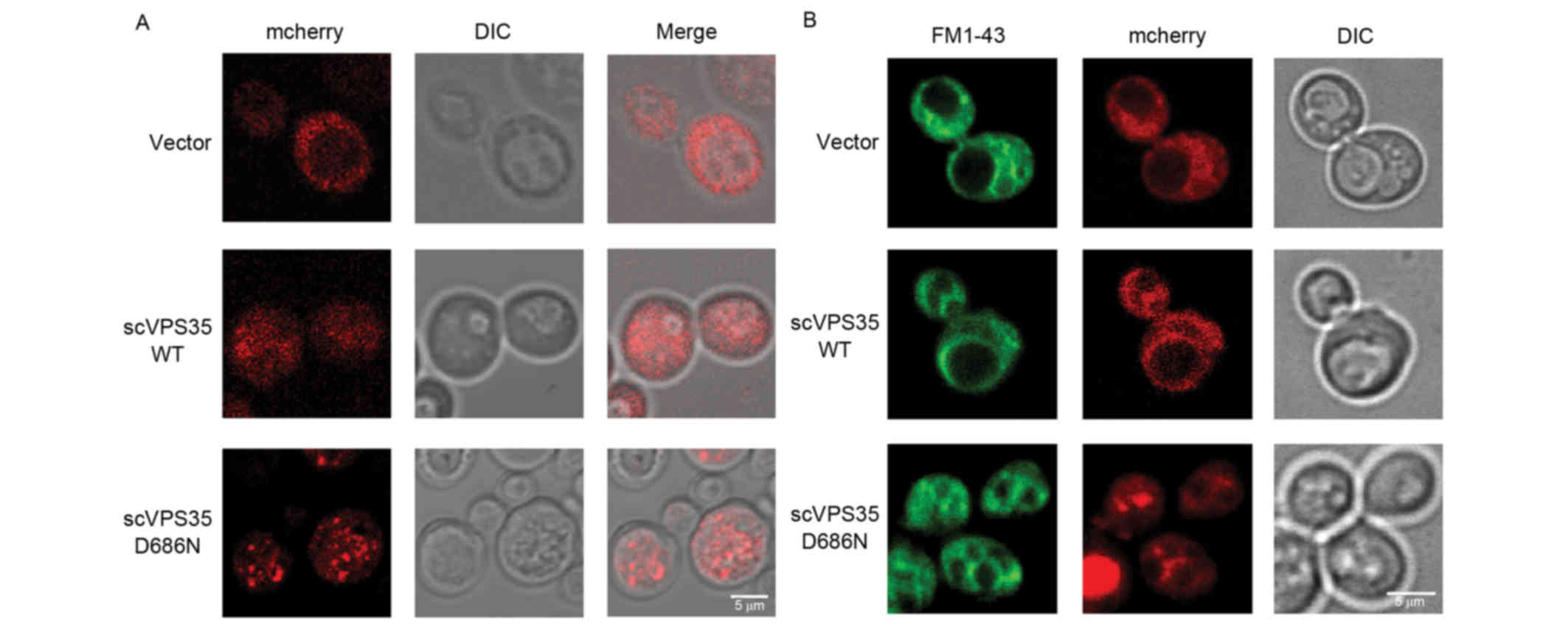
of vacuoles in yeast
The yeast vacuole is the equivalent of the mammalian
lysosome and serves numerous crucial functions in maintaining
cellular homeostasis, including protein degradation, removal of
harmful molecules and storage of organic molecules. To determine
whether the VPS35 mutant impairs vacuolar function, the
distribution of sc VPS35 in a pYES2nt plasmid tagged with
the mCherry fluorescent protein, was investigated in BY4741 cells.
VPS35 is involved in vesicle trafficking. Following a short
induction time of 4 h, sc VPS35 WT was distributed uniformly
throughout the cytoplasm with very few small puncta, which was
comparable to the mCherry vector control. By contrast, sc
VPS35 D686N exhibited large puncta, which is a
characteristic of a deficit in vesicle formation (Fig. 3A). This was consistent with the
characteristics of VPS35 in mammalian cells. As vesicle
formation is the first step of vesicle trafficking, the results of
the present study suggested that overexpression of sc VPS35
D686N disrupts vesicle trafficking.
To investigate the impact of sc VPS35 D686N
on the lysosome, the expression of VPS35 was extended to 12
h and FM 1–43 lipophilic dye was utilized to visualize the
morphology of yeast vacuoles. The vacuole is a dynamic organelle
whose morphology is responsive to varying intracellular conditions.
Normal cells contain one to two vacuoles with medium-sized lobes.
The morphology of vacuoles in cells treated with the sc
VPS35 WT plasmid was comparable to that of the vector
control. However, in yeast cells expressing sc VPS35 D686N,
vacuoles fragmented into multiple compartments, suggesting that the
cells were undergoing osmotic stress (Fig. 3B). The alteration in size and
compartment number corresponded to dysfunction of vacuoles and an
imbalance of the intracellular environment, which suggested that sc
VPS35 D68N overexpression causes defects in the lysosome
system.
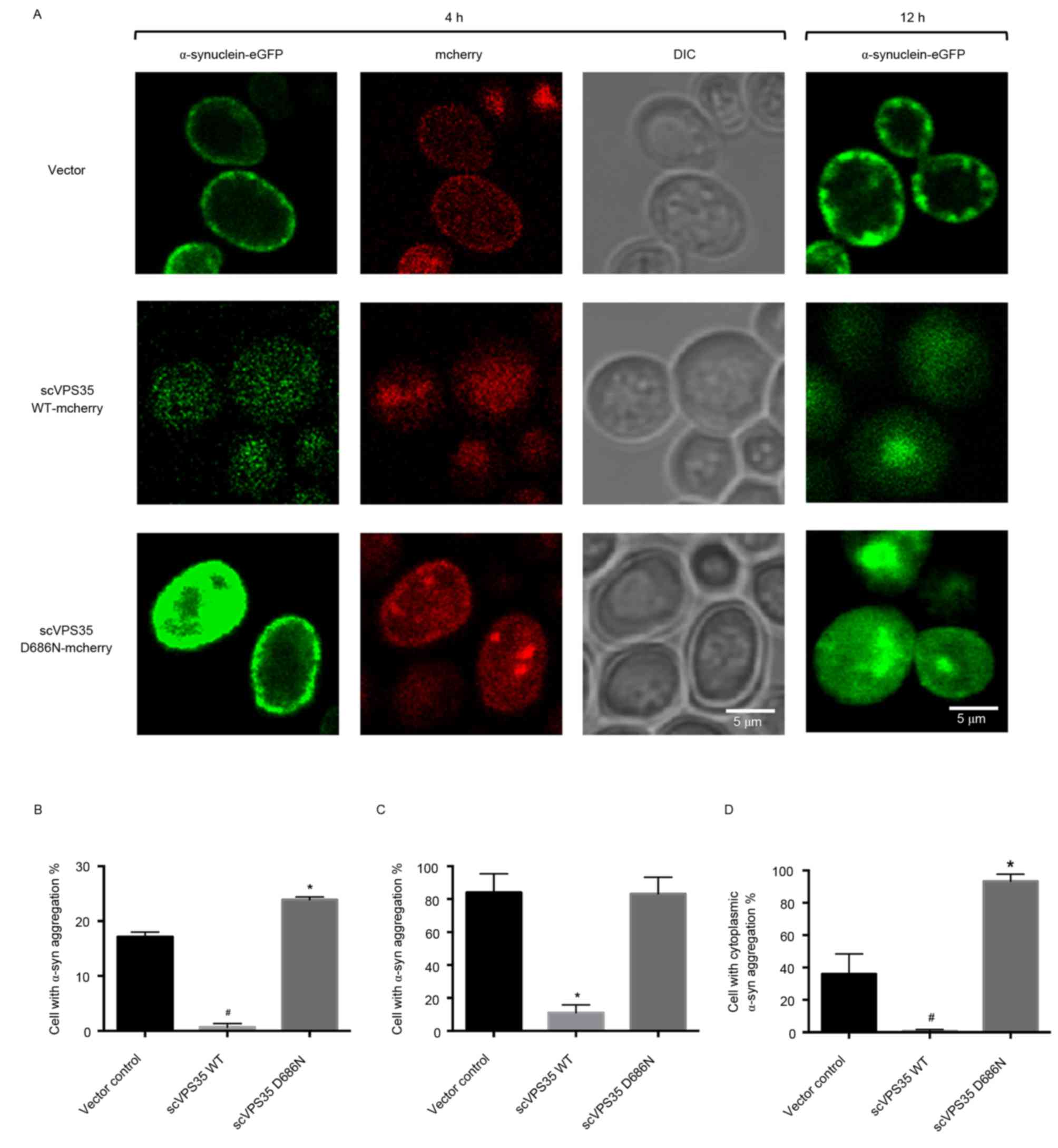
α-synuclein aggregation is enhanced by
expression of scVPS35 WT and is reduced by expression of scVPS35
WT
Tagging sc VPS35 and α-synuclein with the
fluorescent proteins mCherry and eGFP allowed visualization of
their interaction via fluorescent microscopy. Following 4 h of
induction, α-synuclein was localised primarily to the plasma
membrane in vector control cells with limited aggregation.
Transfection of cells with WT sc VPS35 resulted in reduced
expression levels of α-synuclein, which was uniformly distributed.
By contrast, expression of sc VPS35 D686N resulted in
enhanced expression levels of α-synuclein, which was localised to
the plasma membrane (Fig. 4A). In
addition, the percentage of α-synuclein aggregation was greater in
cells expressing sc VPS35 D686N compared with the vector
control (23.88±0.52 vs. 17.14±0.89%; P<0.05; Fig. 4B). Induction for 12 h resulted in
formation of α-synuclein aggregates at the membrane in control
cells, and larger α-synuclein aggregates in the cytoplasm of yeast
cells expressing sc VPS35 D686N. However, cells transfected
with sc VPS35 WT had reduced expression levels of
α-synuclein compared with the control. Quantification analysis
revealed that the percentage of control cells bearing aggregates
was not significantly different to cells expressing sc VPS35
D686N (84.03±11.47 vs. 83.30±10.01%; Fig. 4C). However, compared with the
control, the percentage of cells containing cytoplasmic aggregates
was significantly enhanced following expression of sc VPS35
D686N (35.98±12.45 vs. 93.30±4.40%; P<0.05; Fig. 4D). These results suggested that sc
VPS35 WT reduces α-synuclein aggregation and the sc
VPS35 D686N mutant enhances α-synuclein aggregation.
scVPS35 D686N mutant increases
α-synuclein aggregation in a lysosome-dependent manner
As the lysosome serves a key role in the clearance
of α-synuclein aggregation, it was hypothesized that the sc
VPS35 mutant may interfere with the degradation of
α-synuclein via the lysosome system. The GAL1 promoter shut-off
system is an effective tool to evaluate the efficiency of protein
clearance, and enables investigation of α-synuclein clearance and
the effect of sc VPS35 on cytoplasmic fluorescent foci.
Promoter shut-off was achieved by culturing cells in
glucose-containing medium. Following 12 h of pre-incubation in
galactose-containing medium, cells harbouring α-synuclein-eGFP
exhibited aggregates. Promoter shut-off for 2 h resulted in a
significant reduction in aggregate formation. The vacuolar protease
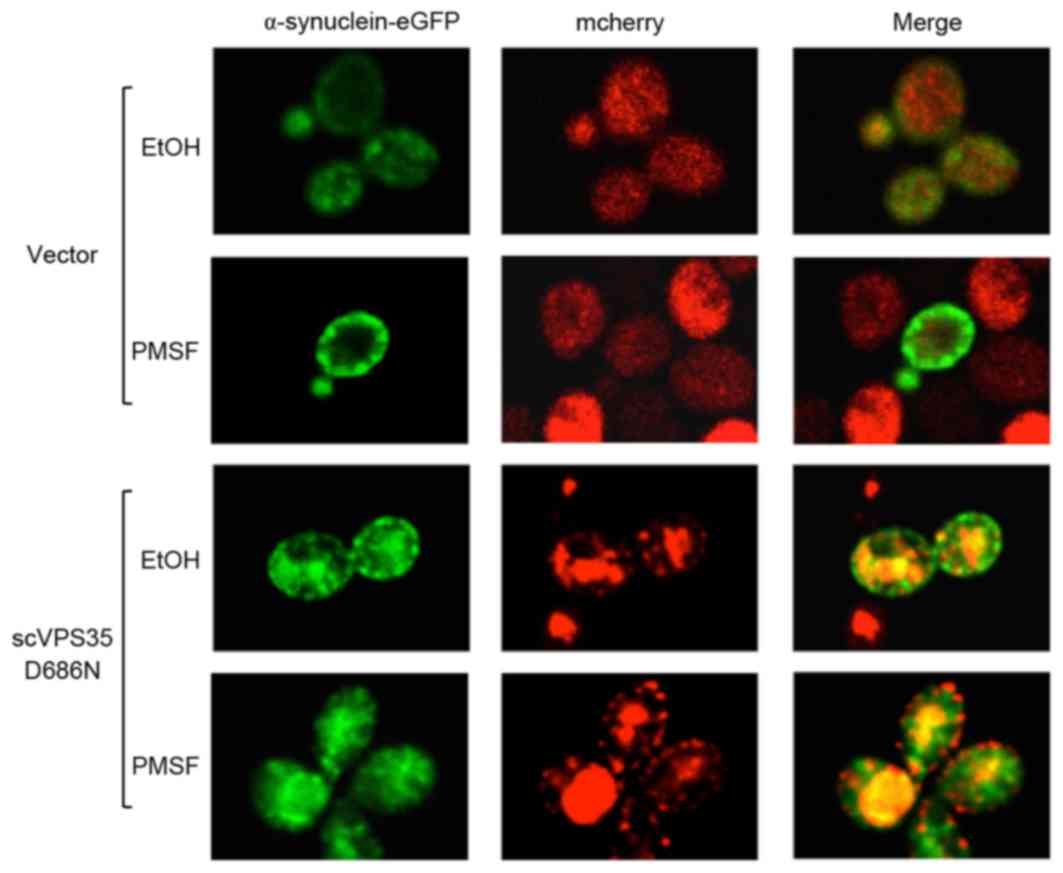
inhibitor PMSF was utilized to investigate α-synuclein aggregation
in yeast cells expressing the WT or mutant VPS35. PMSF
blocked the lysosome system and clearance of α-synuclein
aggregation was prevented following promoter shut-off. Aggregation
was primarily localized to the plasma membrane; however, aggregates
were additionally localized to cytoplasmic vesicular inclusions.
The fluorescence intensity of α-synuclein was maintained; however,
there were fewer aggregated and co-localized vacuoles in the
cytoplasm of yeast cells expressing sc VPS35 D686N following
PMSF treatment compared with cells treated with ethanol (Fig. 5). The alteration in the
distribution of aggregated α-synuclein in the cell suggested that
scVSP35 D686N may affect clearance of α-synuclein prior to the
lysosome, and direct inhibition of lysosome function by PMSF may
lead to accumulation of α-synuclein in the cytoplasm. Therefore,
the data of the present study suggested that impaired
endosome-to-Golgi retrieval of proteins may contribute to VSP35
D686N mutant-mediated α-synuclein aggregation.
Discussion
In the present study, the role of the VPS35
D686N mutant was examined in yeast. It was demonstrated that
deletion of VPS35, and expression of WT or mutant
VPS35, did not suppress cell growth. Therefore, VPS35
may not be necessary to maintain yeast homeostasis. However, when
high expression levels of VPS35 D686N were combined with
non-toxic levels of α-synuclein, cell growth was inhibited,
suggesting that α-synuclein is involved in VPS35-associated
PD. In addition, vacuoles, which are the equivalents of lysosomes
in mammalian cells, were compromised in cells expressing
VPS35 D686N, and α-synuclein aggregation was evident in
yeast cells expressing high levels of VPS35 D686N and low
levels of α-synuclein. Together with previous studies (17,28),
the data of the present study supported the hypothesis that
impairment of lysosomal degradation of α-synuclein is responsible
for the phenotypes in VPS35-associated PD.
The endosome is a component of the endocytic
membrane transport pathway that primarily mediates cellular
transport of endocytic proteins. Once proteins enter into endocytic
system, they are transferred to lysosomes for degradation or
recycled back to the plasma membrane for reuse. There are two
primary pathways for the reuse of endocytic proteins, including the
recycling endosome-mediated endosome-to-plasma membrane pathway and
the endosome-to-TGN retrograde pathway. In the TGN pathway, the
retromer serves a critical role in shifting proteins from endosomes
back to the TGN, resulting in escape of lysosomal degradation.
VPS35 is a subunit of the retromer complex and functions to
sort cellular cargo within the retromer complex (29). Although mutations in VPS35
have been associated with a late-onset, autosomal dominant form of
PD, the underlying mechanism by which VPS35 mutants
contribute to PD is not fully understood. Loss and gain of function
have been proposed to be involved in the pathogenesis of
VPS35-associated PD. As genetic manipulation in yeast is
relatively simple, the present study generated yeast cells with
varying expression levels of VPS35, and the subsequent
effect on cell growth was investigated. In agreement with previous
studies, deletion of sc VPS35 and varying expression levels
of WT sc VPS35 did not inhibit cell growth (21,27).
This suggested that VPS35 may not be involved in the
maintenance of yeast homeostasis. Similarly, varying the expression
levels of the VPS35 mutant did not affect cell growth.
However, large puncta, a sign of a compromised lysosome, was
detected in cells transfected with the mutant VPS35,
indicating that the VPS35 mutant may induce lysosome
dysfunction. However, the non-toxic effect of the VPS35
mutant observed in the present study was not in agreement with
previous reports, which demonstrated that the VPS35 mutant
induced toxicity (30,31). This discrepancy may be due to the
yeast model expressing insufficient levels of mutant VPS35
to induce toxicity. An alternative explanation may be the lack of
endogenous α-synuclein in yeast cells. In addition, as α-synuclein
is degraded by the endosome-lysosome pathway, accumulation of
α-synuclein may impair this pathway. Therefore, the interaction
with α-synuclein may be necessary for other cellular pathways,
which may synergistically lead to neurodegeneration.
Endogenous expression levels of the α-synuclein gene
and protein are associated with the disease phenotype, and have
been investigated in yeast (11).
Consistent with clinical observations (17,32),
the present study demonstrated that at low to moderate expression
levels, α-synuclein was localized to the membrane and did not
induce toxicity, whereas at high levels, α-synuclein formed protein
aggregation and caused toxicity. Previous studies have demonstrated
that VPS35 deficiency induces toxicity, whereas
overexpression of WT VPS35 reduces α-synuclein toxicity
(18), suggesting that α-synuclein
is involved in VPS35-associated PD. However, the majority of
studies were conducted in animals with high expression levels of
α-synuclein, and it is important to assess the impact of the
VPS35 mutant in models that express α-synuclein at low
levels, which corresponds with physiological endogenous expression
in sporadic PD. As the majority of sporadic PD patients do not
exhibit point mutations in genes that are associated with high
expression of α-synuclein, yeast strains expressing low levels of
α-synuclein were utilized to investigate the effect of the
interaction between VPS35 and a physiological level of
α-synuclein. The results of the present study revealed that the
interaction between low expression levels of α-synuclein and high
expression levels of the VPS35 mutant impaired cell growth.
Additionally, α-synuclein aggregation was evident. This suggested
that high expression levels of the VPS35 mutant may be
required to trigger α-synuclein-dependent toxicity. However,
whether this is the case in PD patients requires further
investigation. Notably, α-synuclein was less aggregated in cells
transfected with WT VPS35, and high expression levels of WT
VPS35 did not affect the morphology of the lysosome. These
results suggested that overexpression of WT VPS35 may have a
protective function in α-synuclein pathology.
As high expression levels of the sc VPS35
mutant caused morphological alteration of lysosomes, the effect of
the sc VPS35 mutant on clearance of α-synuclein was
investigated via a GAL1 promoter shut-off system. Under control of
the shut-off system, α-synuclein accumulation was evident in sc
VPS35 mutant yeast cells; however, this was not observed in
sc VPS35 WT yeast cells, suggesting that the VPS35
mutant impaired the degradation of α-synuclein via the
endosome-lysosome pathway.
In conclusion, the present data suggested that a
high expression level of the analogous PD mutation D686N in
VPS35 disrupted α-synuclein homeostasis in yeast. These
results, in addition to previous studies (21,27),
suggested that a mutual mechanism may be responsible for the
interaction between α-synuclein and mutant VPS35. However,
the VPS35 mutant may negatively modulate the clearance of
α-synuclein and trigger toxicity; accumulation of α-synuclein may
impair vacuolar function and vesicular trafficking to exacerbate
VPS35 mutant-mediated cell sensitivity. Collectively, the
data suggested that α-synuclein and scVPS35 is interlinked via the
endosomal-lysosome pathway. The present study thus implicates
endosome-lysosome as a promising therapeutic target for PD.
Acknowledgements
The present study was supported by the National Key
Clinical Department, Guangdong Key Laboratory For Diagnosis and
Treatment of Major Neurological Diseases (grant no.
2014B030301035), the National Natural Science Foundation of China
(grant no. 81371255), the National Science and Technology Support
Program (grant no. 2015BAI07B01) and the Guangdong Science and
Technology Project (grant nos. 2013B051000018, 2014A030304018,
2014B040404053 and 2015B050501003). We appreciate the gifts of
yeast vectors pRS414, pRS415 and pYES2nt from Dr. Lu Yongjun at the
Sun Yat-sen University.
References
|
1
|
Jankovic J: Parkinson's disease: Clinical
features and diagnosis. J Neurol Neurosurg Psychiatry. 79:368–376.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kowal SL, Dall TM, Chakrabarti R, Storm MV
and Jain A: The current and projected economic burden of
Parkinson's disease in the United States. Mov Disord. 28:311–318.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
LeWitt PA and Fahn S: Levodopa therapy for
Parkinson disease: A look backward and forward. Neurology. 86:(14
Suppl 1). S3–S12. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Trinh J and Farrer M: Advances in the
genetics of Parkinson disease. Nat Rev Neurol. 9:445–454. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Warner TT and Schapira AH: Genetic and
environmental factors in the cause of Parkinson's disease. Ann
Neurol. 53:(Suppl 3). S16–S25. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mouradian MM: Recent advances in the
genetics and pathogenesis of Parkinson disease. Neurology.
58:179–185. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gasser T: Update on the genetics of
Parkinson's disease. Mov Disord. 22:(Suppl 17). S343–S350. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Polymeropoulos MH, Lavedan C, Leroy E, Ide
SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et
al: Mutation in the alpha-synuclein gene identified in families
with Parkinson's disease. Science. 276:2045–2047. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Baba M, Nakajo S, Tu PH, Tomita T, Nakaya
K, Lee VM, Trojanowski JQ and Iwatsubo T: Aggregation of
alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and
dementia with Lewy bodies. Am J Pathol. 152:879–884.
1998.PubMed/NCBI
|
|
10
|
Tofaris GK: Lysosome-dependent pathways as
a unifying theme in Parkinson's disease. Mov Disord. 27:1364–1369.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee HJ, Khoshaghideh F, Patel S and Lee
SJ: Clearance of alpha-synuclein oligomeric intermediates via the
lysosomal degradation pathway. J Neurosci. 24:1888–1896. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chu Y, Dodiya H, Aebischer P, Olanow CW
and Kordower JH: Alterations in lysosomal and proteasomal markers
in Parkinson's disease: Relationship to alpha-synuclein inclusions.
Neurobiol Dis. 35:385–398. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vilarino-Guell C, Wider C, Ross OA,
Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA,
Wilhoite GJ, Bacon JA, et al: VPS35 mutations in Parkinson disease.
Am J Hum Genet. 89:162–167. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zimprich A, Benet-Pagès A, Struhal W, Graf
E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC,
Lichtner P, et al: A mutation in VPS35, encoding a subunit of the
retromer complex, causes late-onset Parkinson disease. Am J Hum
Genet. 89:168–175. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Follett J, Norwood SJ, Hamilton NA, Mohan
M, Kovtun O, Tay S, Zhe Y, Wood SA, Mellick GD, Silburn PA, et al:
The Vps35 D620N mutation linked to Parkinson's disease disrupts the
cargo sorting function of retromer. Traffic. 15:230–244. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zavodszky E, Seaman MN, Moreau K,
Jimenez-Sanchez M, Breusegem SY, Harbour ME and Rubinsztein DC:
Mutation in VPS35 associated with Parkinson's disease impairs WASH
complex association and inhibits autophagy. Nat Commun. 5:38282014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Miura E, Hasegawa T, Konno M, Suzuki M,
Sugeno N, Fujikake N, Geisler S, Tabuchi M, Oshima R, Kikuchi A, et
al: VPS35 dysfunction impairs lysosomal degradation of α-synuclein
and exacerbates neurotoxicity in a Drosophila model of Parkinson's
disease. Neurobiol Dis. 71:1S–13S. 2014. View Article : Google Scholar
|
|
18
|
Tang FL, Liu W, Hu JX, Erion JR, Ye J, Mei
L and Xiong WC: VPS35 deficiency or mutation causes dopaminergic
neuronal loss by impairing mitochondrial fusion and function. Cell
Rep. 12:1631–1643. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang W, Wang X, Fujioka H, Hoppel C, Whone
AL, Caldwell MA, Cullen PJ, Liu J and Zhu X: Parkinson's
disease-associated mutant VPS35 causes mitochondrial dysfunction by
recycling DLP1 complexes. Nat Med. 22:54–63. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wen L, Tang FL, Hong Y, Luo SW, Wang CL,
He W, Shen C, Jung JU, Xiong F, Lee DH, et al: VPS35
haploinsufficiency increases Alzheimer's disease neuropathology. J
Cell Biol. 195:765–779. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dhungel N, Eleuteri S, Li LB, Kramer NJ,
Chartron JW, Spencer B, Kosberg K, Fields JA, Stafa K, Adame A, et
al: Parkinson's disease genes VPS35 and EIF4G1 interact genetically
and converge on α-synuclein. Neuron,. 85:76–87. 2015. View Article : Google Scholar
|
|
22
|
Tang FL, Erion JR, Tian Y, Liu W, Yin DM,
Ye J, Tang B, Mei L and Xiong WC: VPS35 in dopamine neurons is
required for Endosome-to-Golgi retrieval of Lamp2a, a receptor of
chaperone-mediated autophagy that is critical for α-synuclein
degradation and prevention of pathogenesis of Parkinson's disease.
J Neurosci. 35:10613–10628. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Outeiro TF and Lindquist S: Yeast cells
provide insight into alpha-synuclein biology and pathobiology.
Science. 302:1772–1775. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Franssens V, Boelen E, Anandhakumar J,
Vanhelmont T, Büttner S and Winderickx J: Yeast unfolds the road
map toward alpha-synuclein-induced cell death. Cell Death Differ.
17:746–753. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Edelheit O, Hanukoglu A and Hanukoglu I:
Simple and efficient site-directed mutagenesis using two
single-primer reactions in parallel to generate mutants for protein
structure-function studies. BMC Biotechnol. 9:612009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Anandhan A, Rodriguez-Rocha H, Bohovych I,
Griggs AM, Zavala-Flores L, Reyes-Reyes EM, Seravalli J, Stanciu
LA, Lee J, Rochet JC, et al: Overexpression of alpha-synuclein at
non-toxic levels increases dopaminergic cell death induced by
copper exposure via modulation of protein degradation pathways.
Neurobiol Dis. 81:76–92. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Auluck PK, Caraveo G and Lindquist S:
α-Synuclein: Membrane interactions and toxicity in Parkinson's
disease. Annu Rev Cell Dev Biol. 26:211–233. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li D, Shi JJ, Mao CJ, Liu S, Wang JD, Chen
J, Wang F, Yang YP, Hu WD, Hu LF and Liu CF: Alteration of dynein
function affects α-synuclein degradation via the
autophagosome-lysosome pathway. Int J Mol Sci. 14:24242–24254.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bonifacino JS and Hurley JH: Retromer.
Curr Opin Cell Biol. 20:427–436. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tsika E, Glauser L, Moser R, Fiser A,
Daniel G, Sheerin UM, Lees A, Troncoso JC, Lewis PA, Bandopadhyay
R, et al: Parkinson's disease-linked mutations in VPS35 induce
dopaminergic neurodegeneration. Hum Mol Genet. 23:4621–4638. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang HS, Toh J, Ho P, Tio M, Zhao Y and
Tan EK: In vivo evidence of pathogenicity of VPS35 mutations in the
Drosophila. Mol Brain. 7:732014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sahay S, Ghosh D, Singh PK and Maji SK:
Alteration of structure and aggregation of a-synuclein by familial
Parkinson's disease associated mutations. Curr Protein Pept Sci.
Mar 14–2016.(Epub ahead of print). PubMed/NCBI
|