Introduction
Chronic airway infection with pseudomonas
aeruginosa (PA) represents a therapeutic challenge. Host immune
responses to PA often result in persistent airway inflammation and
immunopathological lung injury, characterized by polymorphonuclear
leukocyte infiltration (1).
Although the cause of PA-related airway inflammation remains
incompletely explored, it has been demonstratedthat Th17 responses
are associated with the neutrophil recruitment and activity in lung
defense against the infection. Significantly elevated levels of
interleukin (IL)-17A are reported in the sputum of patients with
cystic fibrosis who were colonized with PA at the time of pulmonary
exacerbation, and the levels declined with therapy directed against
PA (2,3). IL-23 mediates inflammatory responses
to mucoid PA lung infection, which induces IL-17 production and the
subsequent local production of cytokines and chemokines that are
critical to airway inflammation (4). IL-23 and the downstream cytokine
IL-17A are important molecules for proinflammatory gene expression
and are likely involved with the immunopathological injury in
chronic PA lung infection.
Th17 cells are a subset characterized by a unique
transcriptional program dependent on signal transducer and
activator of transcription 3 (STAT3) transduction pathways
(5). The Th17 transcription factor
RORγt induces the expression of IL-23 receptor through
STAT3-dependent mechanisms, rendering the differentiating cells
responsive to IL-23, which is an innate immune cell cytokine
essential for stabilization of the Th17 phenotype (6). When STAT3 is genetically ablated in
CD4+ cells, neither naturally occurring Th17 cells nor
Th17-dependent autoimmunity occurs (7). In PA lung infections, STAT3
activation has been demonstrated to be essential for the
translocation of nuclear factor-κB into the nucleus, which induced
elevated inflammatory cytokines (IL-6, tumor necrosis factor-α, and
IL-12) and increased superoxide release in the lung (8). These studies suggest that targeting
STAT3/Th17 pathway may be a potential therapeutic strategy for
controlling immunopathological injury during chronic PA lung
infection.
Suppressor of cytokine signaling (SOCS) proteins are
feedback inhibitors of the JAK/STAT pathway. The major function of
SOCS3 is inhibition of signaling by the IL-6 family of cytokines,
causing inhibition of STAT3 activation and Th17 generation
(9). Furthermore, SOCS3 expression
in T cells inhibits IL-23 signaling, which constrains Th17 cell
differentiation (10). In the
central nervous system, the STAT3/SOCS3 axis influences the T-cell
repertoire, with SOCS3 providing protection against autoimmune
diseases by blocking Th17 development (11). So far, in the field of chronic lung
infection, data regarding the effect of SOCS3 on STAT3/Th17 signal
pathway remains scarce.
In the present study, the authors investigated the
activation of the STAT3/Th17 signal pathway and the expression of
SOCS3 in the lung CD4+ T cells in a mouse model of
chronic PA lung infection. Following this, the SOCS3 gene was
lentivirally delivered into the CD4+ T cells isolated
from lung tissues of the mouse model and the effect of exogenous
SOCS3 on Th17-mediated neutrophil recruitment was investigated
in vitro.
Materials and methods
Animal model of chronic PA lung
infection
Specific pathogen-free female C57/BL6 mice (8–12
weeks of age, 18–22 g) were obtained from Shanghai SLAC Laboratory
Animal Co., Ltd. (Shanghai, China), and were kept under
environmentally controlled conditions with standard food and water.
Animal experimentations were approved by the Institutional Animal
Care and Use Committee of Shanghai Jiao Tong University School of
Medicine (Shanghai, China).
PA (ATCC© 27853) was purchased from China General
Microbiological Culture Collection Center (Beijing, China) and
loaded on agarose beads at a final concentration of
2×106 colony-forming units/50 µl phosphate-buffered
saline (PBS). The beads were measured to be 70 to 150 µm in
diameter by microscopic quantification. Mice were anesthetized with
intraperitoneal injection of 20 µl/g body weight of a stock
solution of 0.5 ml ketamine (100 mg/ml), 0.5 ml 1% xylazine
hydrochloride (equivalent to 10 mg/kg body weight) and 9 ml sterile
0.9% NaCl.
Mice were placed in the dorsal recumbent position.
The trachea was cannulated, and 50 µl PA-laden agarose beads were
introduced into the right lung. Mice were monitored, and allowed to
recover until euthanized by CO2 asphyxiation on d3, 5
and 7. Bacterial load was evaluated by quantitatively culturing
lung homogenates on sheep blood agar plates overnight at 37°C. Mice
in the control group were inoculated with sterile agarose beads. A
total of 72 C57BL/6 mice were randomly divided into a control group
(n=36) and a PA group (n=36). In each group, 12 mice were
euthanized at each time point. For PA group, the percentage of mice
positive for PA growth in homogenate cultures was 100% with the
bacterial load more than 1×105 colony forming units
(CFU)/g lung tissue at each time point.
Bronchoalveolar lavage cell
counts
The lungs were lavaged with 1.8 ml PBS.
Bronchoalveolar lavage (BAL) fluid was centrifuged at 300 × g. The
cell pellet from the BAL fluid was resuspended in 1 ml PBS, and the
cells were counted using a microscope counting chamber. The
percentage of neutrophils was determined by counting a minimum of
100 cells following staining with haematoxylin and eosin. A
pathologist, blinded to the experimental protocols, checked the
total and differential cell counts.
Histological examination and
immunohistochemistry
For histologic analysis, the right low lobe was
removed, fixed with 4% paraformaldehyde in PBS and embedded in
paraffin. The paraffin was sectioned and stained with haematoxylin
and eosin. The expression of phospho-STAT3 (p-STAT3) in the lung
tissues was analyzed by immunohistochemistry. Lung sections were
deparaffinized and stained with rabbit anti-mouse p-STAT3 antibody
(1:400; catalog no. 9145; Cell Signaling Technology, Inc., Danvers,
MA, USA) at 4°C overnight. The sections were incubated with
horseradish peroxidase-conjugated secondary antibody (1:1,000;
catalog no. 6721; Abcam, Cambridge, UK) at 37°C for 30 min. Then
the DAB substrate solution was applied to reveal the color of
antibody staining. Finally, sections were counterstained with
haematoxylin to visualize the nuclei.
Measurement of IL-17A and p-STAT3
concentrations in lung tissues by ELISA
The right middle lobes were weighed and homogenized
in 1 ml normal saline. Concentrations of IL-17A and p-STAT3 were
measured using commercially available ELISA kits (catalog nos.
M1700 and DYC4607B-2; R&D Systems, Inc., Minneapolis, MN, USA).
The samples were assayed in duplicate and values were expressed as
pg/mg lung tissue.
Mouse lung CD4+ T cells
isolation
The lungs were excised aseptically and homogenized
in 0.5 ml normal saline. Minced lungs were enzymatically digested,
using a mixture containing 6 Udispase II (catalog no. D4693;
Sigma-Aldrich, Merck KGaA, Darmstadt, Germany), 20 µg collagenase I
(catalog no. 1148089; United States Pharmacopeia, Rockville, MD,
USA). Following digestion, lung tissues were passed through a 40 µm
nylon mesh to obtain single-cell suspensions. CD4+ T
cells were isolated using a CD4+ T cell isolation kit
and Mini-MACS columns (MiltenyiBiotec, Inc., Auburn, CA, USA).
Following centrifugation at 300 × g, 4°C for 10 min and washing
with cold PBS, collected CD4+ T cells were dissolved in
culture medium consisting of RPMI 1640 (catalog no. 11875093;
Gibco; Thermo Fisher Scientific, Waltham, MA, USA) supplemented
with 25 mM HEPES, 2 mM glutamine, 10% fetal bovine serum,
penicillin (100 IU/ml) and streptomycin (100 IU/ml) at 37°C in a 5%
CO2 incubator.
Western blotting analysis of RORγt,
p-STAT3 and SOCS3 expression in lung CD4+ T cells
Proteins were extracted from lung CD4+ T
cells using a commercially available Mammalian Protein Extraction
Reagent (Pierce; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
The protein concentration was calculated using the bicinchoninic
acid protein assay (catalog no. 23227; Thermo Fisher Scientific,
Inc.). The extracts were diluted, mixed with protein loading buffer
and boiled for 5 min before loading onto an SDS-PAGE gel (with a
10% separating gel and a 5% stacking gel). Following
electrophoresis, proteins were transferred onto polyvinylidene
fluoride membranes, which were blocked with 5% skim milk for 1 h,
and blotted against rat anti-mouse RORγt monoclonal antibody
(1:1,000; catalog no. 14-6981-82; eBioscience, Inc., San Diego, CA,
USA), rabbit anti-mouse SOCS3 polyclonal antibody (1:500; catalog
no. sc-9023, Santa Cruz Biotechnology, Inc., Dallas, TX, USA),
rabbit anti-mouse p-STAT3 monoclonal antibody (1:2,000; catalog no.
9145, Cell Signaling Technology, Inc.), and rabbit anti-mouse
Histone H3 polyclonal antibody (1:1,000; catalog no.
sc-8654-R;Santa Cruz Biotechnology, Inc.) at 4°C overnight.
Membranes were washed and incubated with a horseradish
peroxidase-conjugated goat anti-rabbit IgG (1:5,000; catalog no.
111-035-144; Jackson ImmunoResearch Laboratories, West Grove, PA,
USA) or goat anti-rat IgG (1:5,000; catalog no. 97057;Abcam) at
room temperature for 1 h. Protein bands were visualized by enhanced
chemiluminescence substrate (catalog no. 32106; Thermo Fisher
Scientific, Inc.) and quantification was performed by densitometry
analysis in duplicate using Quantity One software version 4.6.3
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). The data were
normalized to Histone H3 expression in samples.
Reverse transcription-polymerase chain
reaction (RT-PCR) and RT-quantitative polymerase chain reaction
(RT-qPCR) analysis of SOCS3 mRNA in lung CD4+ T
cells
Lung CD4+ T cells were lysed in TRIzol
(catalog no. 15596026; Invitrogen, Thermo Fisher Scientific, Inc.)
and total RNA was extracted. The RT-PCR reaction for murine SOCS3
was carried out by standard methods using RNA LA PCR Kit (Takara
Bio, Inc., Otsu, Japan). The RT-qPCR reaction was conducted using
the GMR-Super™ Universal Master Mix Kit (Shanghai GenePharma Co.,
Ltd., Shanghai, China). The reaction was performed using the
Mx3000P qPCR system (Stratagene; Agilent Technologies, Inc., Santa
Clara, CA, USA). β-actin was used as an internal positive control
for normalization of data. The comparative threshold cycle method
was used to assess the relative mRNA levels of target genes
(12). The sequences of the
primers were as follows: Mouse SOCS3 sense,
5′-ACCTTCAGCTCCAAAAGCGAGTAC-3′ and anti-sense,
5′-CGCTCCAGTAGAATCCGCTCTC-3′; mouse β-actin sense,
5′-AAGATCAAGATCATTGCTCCTCC-3′ and anti-sense,
5′-GACTCATCGTACTCCTGCTTGC-3′.
Production of SOCS3 recombinant
lentiviral vectors
A fifth generation of the self-inactivating
lentiviral vector reporter was purchased from Shanghai GenePharma
Co., Ltd. The lentiviral vector system had four sections, including
the pGLV-EF1a-GFP plasmid, the pLV/helper-SL3 (gag/pol element)
plasmid, the pLV/helper-SL4 (pRev element) plasmid, and the
pLV/helper-SL5 (pVSV-G element) plasmid. The pGLV-EF1a-GFP plasmid
was constructed to encode the full length of the mouse SOCS3 gene
(NCBI reference sequence ID, NM_007707.3). PCR and DNA sequencing
confirmed the accurate insertion of the SOCS3 cDNA. The transient
transfection method was used to prepare the recombinant lentiviral
vectors. The plasmids were co-transfected into subconfluent 293T
cells in serum-free medium using the cationic liposome based
transfection reagent (Lipofectamine 2000; Invitrogen; Thermo Fisher
Scientific, Inc.). Following 8 h incubation, the medium was
completely exchanged. High-titer recombinant lentiviral vectors
with SOCS3 were harvested 48 h later. The viruses were precipitated
at 1,500 × g and resuspended with Opti-MEM (catalog no. 31985062;
Thermo Fisher Scientific, Inc.) medium.
SOCS3 overexpression in lung
CD4+ T cells by lentiviral infection in vitro
CD4+ T cells were isolated from mouse
lung tissues 3 d following inoculation of PA-laden agarose beads
using methods described above. The cells were cultured in
Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher
Scientific, Inc.) containing 10% horse serum, 1.5 g/l
NaHCO3 and 30 ng/ml IL-2 at 37°C in a 5% CO2
incubator. Cells were seeded on six-well plates at a density of
1×106 cells/well and incubated with SOCS3 recombinant
lentiviral vectors at a titer of 1×109 TU/ml for 72 and
96 h. ~90% of cells were infected as assessed by GFP staining. The
overexpression of SOCS3 gene expression in lung CD4+ T
cells was confirmed by RT-qPCR and western blotting. Cells
incubated with PBS were used as blank controls. Cells transfected
with the empty vector, which was not inserted with SOCS3, were used
as negative controls.
Western blotting and RT-qPCR analysis
in lung CD4+ T cells stimulated by IL-23
Following lentiviral infection, cells were seeded at
a concentration of 5×106 cells/well into a 96-well
culture plate and incubated with recombinant mouse IL-23 (10 ng/ml,
eBioscience, Inc.) for 24 h. The protein levels of p-STAT3 and
RORγt were analyzed by western blotting following the method
described above. The mRNA levels of STAT3 and RORγt in lung
CD4+ T cells were analyzed by RT-qPCR. The sequences of
primers were as follows: Mouse STAT3 sense,
5′-TGTCTCCACTTGTCTACCT-3′ and anti-sense, 5′-CAGCACCTTCACCGTTAT-3′;
mouse RORγt sense, 5′-TCTCTGCAAGACTCATCGACAAG-3′ and anti-sense,
5′-GCACAGGCTCCGGAGTTTT-3′. β-actin was used as an internal positive
control for normalization of data. The results were compared
between SOCS3 overexpressing group and control groups.
Measurement of IL-17A expression in
cell supernatant and flow cytometry analysis of IL-17A+
cells
Following IL-23 stimulation, protein levels of
IL-17A in supernatant of SOCS3 overexpressing cells were measured
by ELISA using commercially available kits (catalog no. M1700;
Quantikine ELISA; R&D Systems, Inc., Minneapolis, MN, USA). For
flow cytometry analysis, these cells were washed and incubated with
fixation/permeabilization solution for 1 h in the dark. Following a
blocking step with anti-mouse CD16/CD32 (1:1,000; catalog no.
14-0161-81; eBioscience, Inc.), allophycocyanin-conjugated IL-17A
antibody (1:200;catalog no. 17-7177; eBioscience, Inc.) was
incubated with prepared cells in a final volume of 100 µl flow
staining buffer at 4°C for 90 min. The percentage of
IL-17A+ cells within the CD4+ T-cell
population was determined using FACScan flow cytometer (BD
Biosciences, Franklin Lakes, NJ, USA) in the FL4 channel.
Background fluorescence was assessed using appropriate isotype- and
fluorochrome-matched control antibodies (eBioscience, Inc.).
Neutrophil isolation
Neutrophils were isolated from the peripheral blood
of healthy mice according to the manufacturer's instructions. In
brief, venous blood was drawn and neutrophils were isolated
immediately by mouse peripheral neutrophils separation medium
(Sangon Biotech Co., Ltd., Shanghai, China). Following lysis of the
erythrocytes, the neutrophils were harvested, washed twice and
resuspended in RPMI 1640 medium supplemented with 10% fetal bovine
serum at a cell concentration of 1×106/ml. The
percentage of viable neutrophils was assessed by morphology and the
trypan blue exclusion test. A population of >95% of viable
neutrophils was confirmed in the purified cells.
Chemotaxis analysis
Boyden chambers of 3 µm pore size (Corning
Incorporated, Corning, NY, USA) was used to assess chemotaxis for
neutrophils. MLE-12 cells (mouse lung epithelial cell lines, ATCC
CRL-2110; American Type Culture Collection, Manassas, VA, USA) were
plated in 24-well plates (4×105 per well) and incubated
with the three groups of lung CD4+ T cells (blank
control group, negative control group and SOCS3 overexpressing
group) for 48 h. The culture supernatant was harvested. Then, the
treated supernatant was added to the lower chambers, while
neutrophils were added to the top chambers for incubation for
another 90 min at 37°C in a humidified atmosphere with 5%
CO2. The filters were fixed with ethanol and stained
with DAPI to visualize the nuclei. The number of cells that had
migrated through the entire thickness of the filter was evaluated,
and the results were expressed as the chemotactic index, being the
number of cells that migrated towards the sample divided by the
number of cells that migrated towards the control medium.
Triplicate chambers were used in each experiment and five fields
were examined in each filter. CXCL1 and CXCL5 were measured in the
culture supernatant using commercially available ELISA kits
(catalog nos. MKC00B and MX000; R&D Systems, Inc., Minneapolis,
MN, USA) according to the manufacturer's protocol.
Statistical analysis
Data were summarized as means ± standard deviation.
Comparisons between groups were made with one-way analysis of
variance followed by the Tukey post hoc test, and a two-tailed
unpaired Student's t-test. Mann Whitney U test was used to analyze
the non-normal distribution data. P<0.05 was considered to
indicate a statistically significant difference. SAS version 8.1
statistical software (SAS Institute, Cary, NC, USA) was used for
data analysis.
Results
SOCS3 is upregulated in lung
CD4+ T cells following the activation of the STAT3/Th17
pathway during chronic PA lung infection
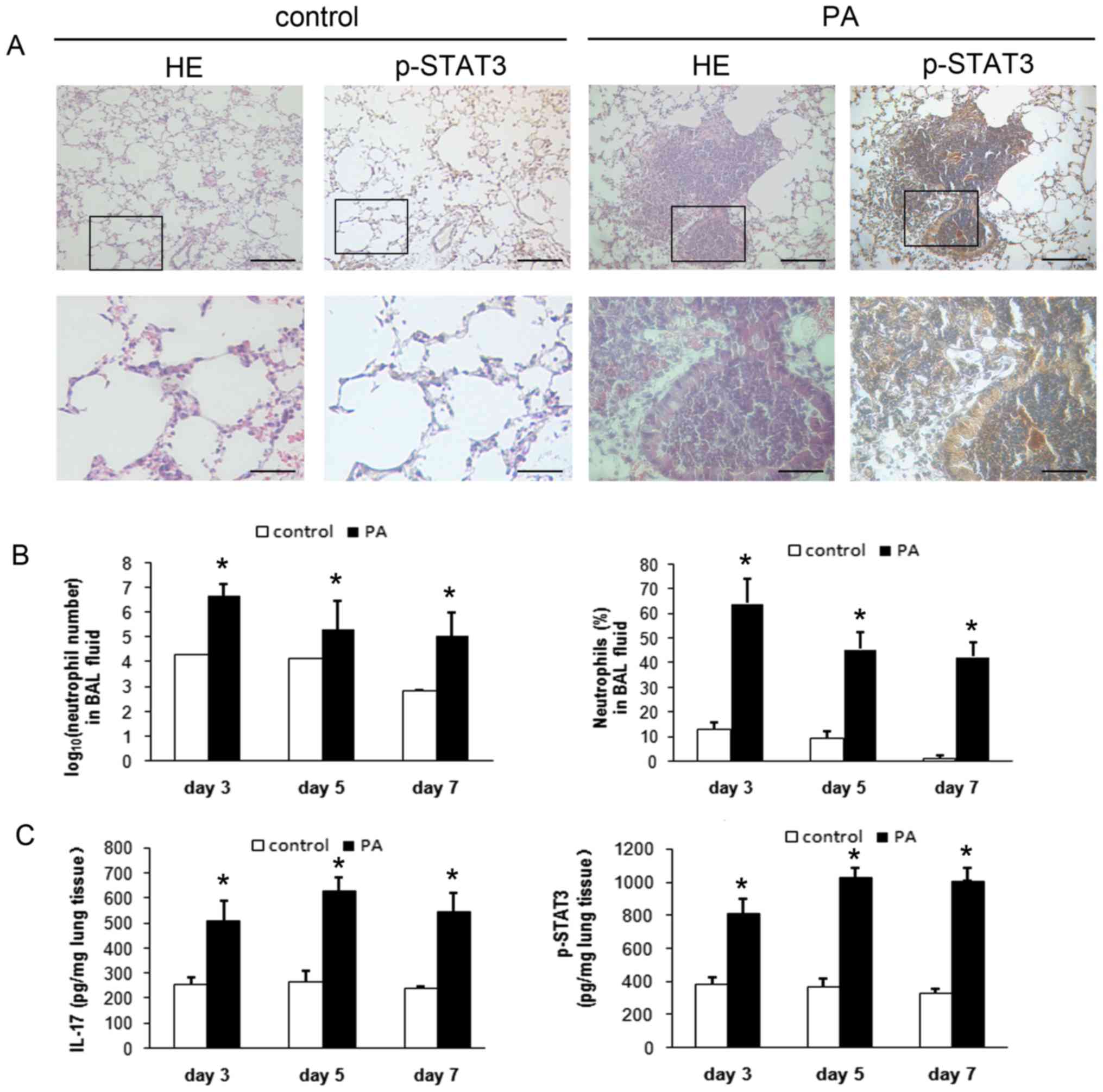
Lung tissue histology indicated that mice with
chronic PA lung infection had an intense neutrophilic infiltration
within and around the small- and medium-sized bronchi, and had
significantly higher expression of p-STAT3 in the lung tissues
compared with control mice (Fig.
1A). Both the number and percentage of recruited BAL
neutrophils were significantly higher in mice with chronic PA lung
infection (P<0.01; Fig. 1B).
The concentrations of IL-17A and p-STAT3 in lung tissues were also
significantly higher in mice with chronic PA infection (P<0.01;
Fig. 1C).
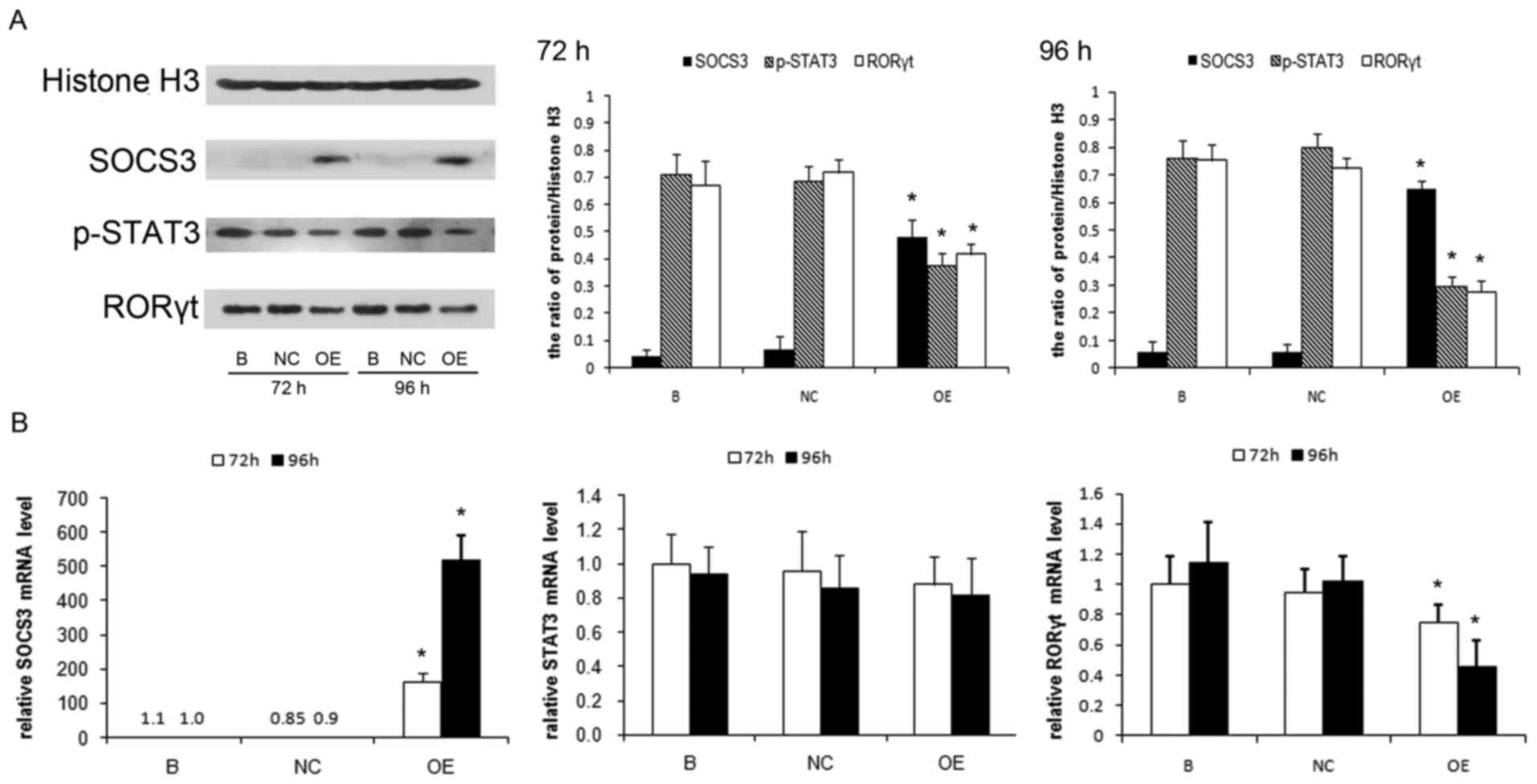
The authors examined RORγt, p-STAT3 and SOCS3
expression in the lung CD4+ T cells. The RORγt and
p-STAT3 protein were expressed more strongly in mice with chronic
PA infection than control mice on d5 and 7 (P<0.01). The SOCS3
protein was at low levels in control mice, while in mice with PA
infection, it gradually increased as chronic lung infection
developed (Fig. 2A). In addition
SOCS3 mRNA level in lung CD4+ T cells was examined using
RT-PCR and RT-qPCR analysis. SOCS3 mRNA level was demonstrated to
increase significantly in mice with chronic PA infection on d5 and
d7 (P<0.01; Fig. 2B).
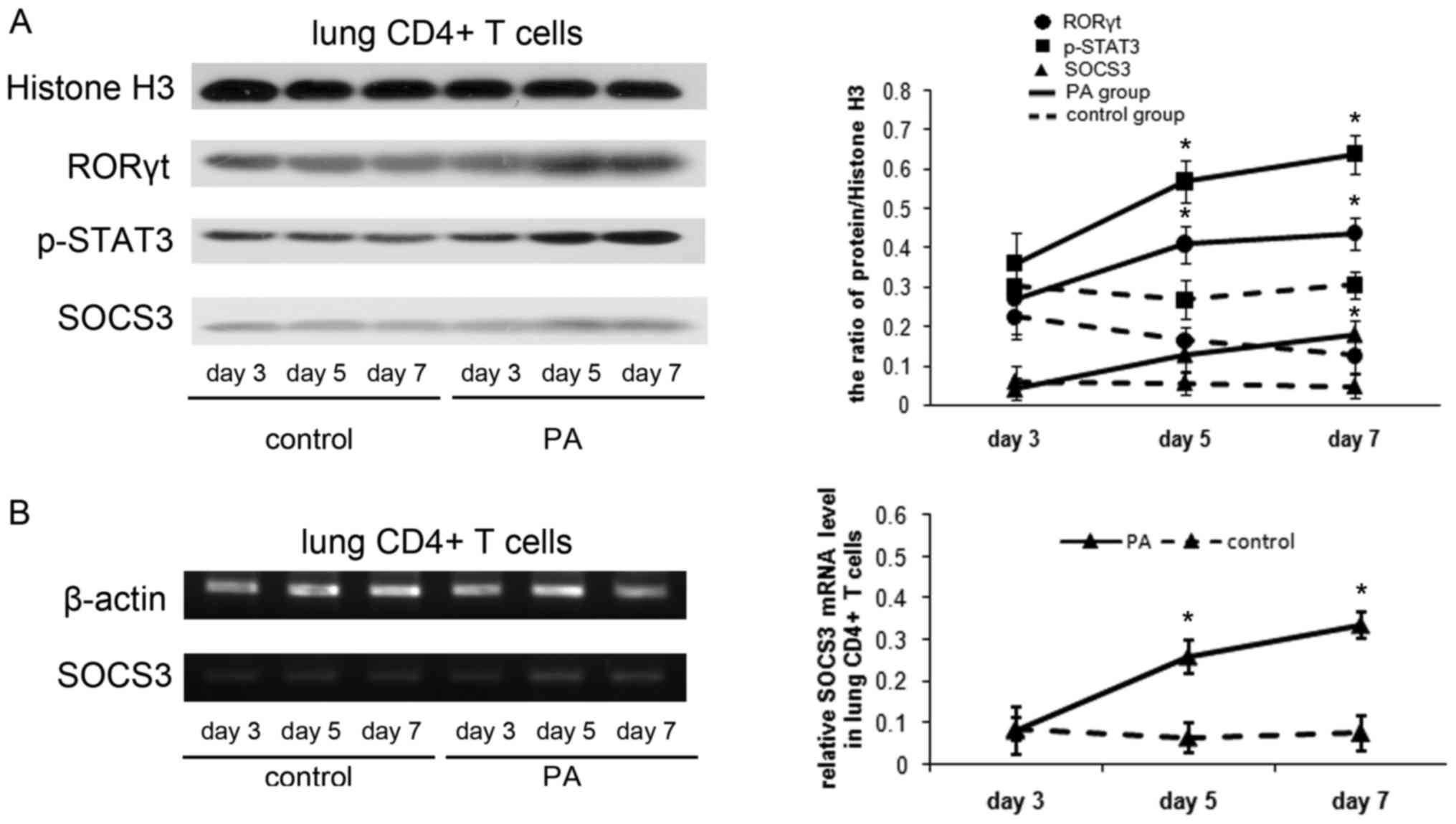 | Figure 2.Induction of SOCS3 following the
activation of STAT3/Th17 in a mouse model of chronic PA lung
infection. (A) Protein expression of RORγt, p-STAT3 and SOCS3 in
the lung CD4+ T cells by western blot analysis. Total cell proteins
were extracted from lung CD4+ T cells on days 3, 5 and 7, then were
immunoblotted with anti-RORγt, anti-p-STAT3 or anti-SOCS3
antibodies. Protein quantification was performed by densitometry
analysis using Quantity One software. (B) Reverse transcription-PCR
analysis of SOCS3 mRNA in the lung CD4+ T cells on days
3, 5 and 7. The relative expression of SOCS3 mRNA was reverse
transcription-quantitative PCR analysis. Values are presented as
the means ± standard deviation for 12 mice per group. *P<0.01
vs. control. B, blank control group; NC, negative control group;
OE, SOCS3 overexpression group. PA, Pseudomonas aeruginosa; RORγt,
retinoid-related orphan receptor γt; STAT3, signal transducer and
activator of transcription 3; p-STAT3, phospho-STAT3; SOCS3,
suppressor of cytokine signaling 3; Th17, T helper 17; PCR,
polymerase chain reaction. |
In vitro SOCS3 gene transfer
suppresses p-STAT3 expression and Th17 response in lung
CD4+ T cells
To investigate the significance of SOCS3 expression
in the regulation of STAT3/Th17 activation in lung CD4+
T cells during chronic PA infection, the authors overexpressed the
SOCS3 gene in lung CD4+ T cells isolated from the
chronic PA-infection mouse model. These cells were stimulated with
IL-23 and the p-STAT3 expression and Th17 response were compared
between SOCS3-overexpressing cells and control cells. The
overexpression of SOCS3 strongly suppressed the protein level of
p-STAT3 and RORγt in the lung CD4+ T cells (P<0.01;
Fig. 3A). The effect is more
obvious when the SOCS3 expression is higher after 96 h of
lentiviral infection compared with that of 72 h of infection
(P<0.05). The mRNA level of RORγt was significantly lower in the
SOCS3 overexpressing cells (P<0.01), but no significant change
of STAT3 mRNA level was observed between these groups (Fig. 3B).
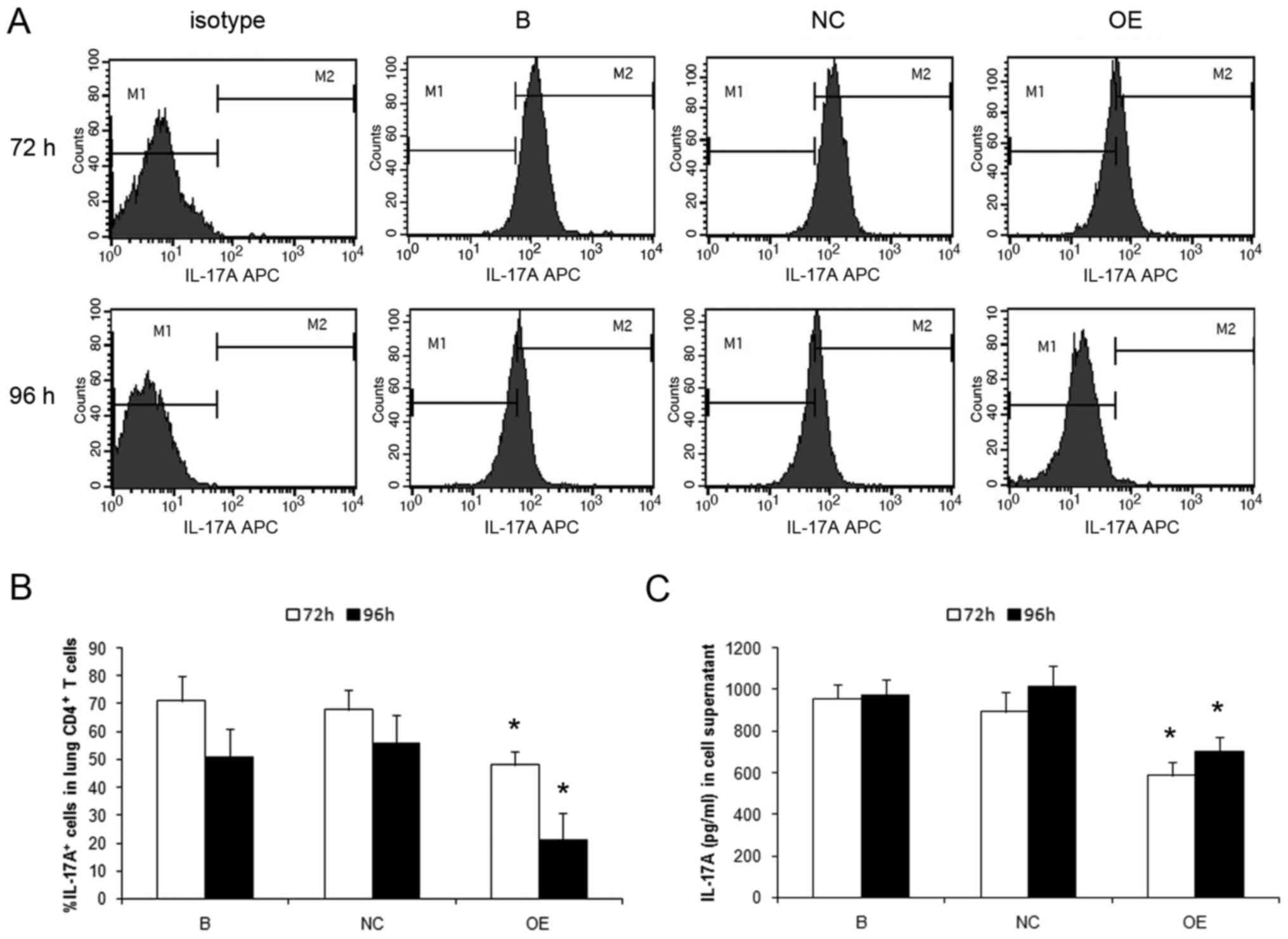
Flow cytometry analysis indicated that the
percentage of IL-17A+ cells was significantly lower in
SOCS3-overexpressing cells compared with that of control cells
(P<0.01; Fig. 4A and B). The
level of IL-17A was analyzed in the cell supernatant by ELISA.
IL-23 induced IL-17A production was significantly decreased in the
supernatant of SOCS3-overexpressing cells, but not in that of
control cells (P<0.01; Fig.
4C).
In vitro SOCS3 gene overexpression in
lung CD4+ T cells suppresses neutrophil recruitment
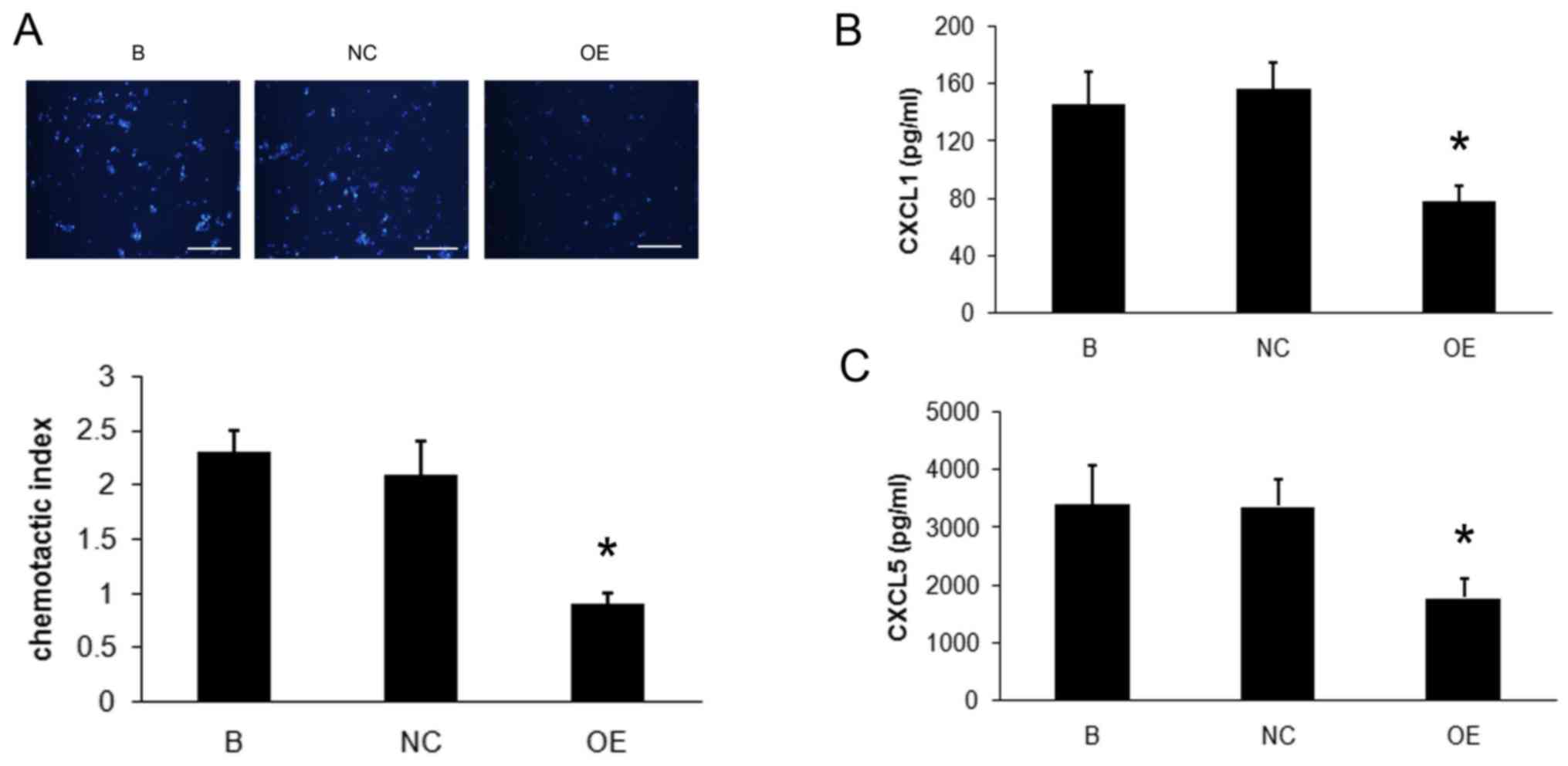
The mouse lung epithelial cell line, MLE-12, was
incubated with lung CD4+ T cells that overexpressed the
SOCS3 gene. Using a chemotaxis assay it was demonstrated that,
compared with those incubated with control cells, the MLE-12 cells
incubated with SOCS3-overexpressing lung CD4+ T cells
induced significantly smaller number of migrating neutrophils
isolated from peripheral blood of healthy mice (P<0.01, Fig. 5A). The expression of neutrophil
chemoattractants CXCL1 and CXCL5 secreted by MLE-12 cells was
examined. The expressions of CXCL1 and CXCL5 were significantly
lower in the supernatant of MLE-12 cells incubated with
SOCS3-overexpressing cells (P<0.01; Fig. 5B and C).
Discussion
The immunological injury induced by chronic PA lung
infection remains a challenge for both clinicians and scientists.
So far, therapeutic interventions to control the sustained
neutrophilic airway inflammation are not ideal. The aim of the
present study was to investigate the role served by a
cytokine-signaling negative regulator, SOCS3, in the neutrophilic
airway inflammation of chronic PA lung infection. The results
indicated that SOCS3 was upregulated following the STAT3/Th17
activation in a mouse model of chronic PA lung infection. In
vitro exogenous SOCS3 gene transfer in lung CD4+ T
cells decreased p-STAT3 expression and Th17 response, and
suppressed the neutrophil recruitment induced by lung epithelial
cells. These results suggested that SOCS3 gene therapy maybe a
potential way for immunotherapy to treat neutrophillic airway
inflammation in chronic PA lung infection.
It was reported previously that the integration of
IL-17A into the IL-6/STAT3 signaling axis mediates lung
inflammation, and that SOCS3, the feedback inhibitor of the
JAK/STAT3 pathway, was increased in lungs during chronic
inflammation (13). In the field
of chronic PA lung infection, however, the role of SOCS3 in the
regulation of STAT3/IL-17A pathway has been scarcely reported.
Here, it was reported that the levels of p-STAT3 expression and
Th17 response were higher in the mouse model of chronic PA lung
infection than those in control mice, and SOCS3 protein and mRNA
levels increased following the protein levels of p-STAT3 and RORγt
became significantly higher at d5. These results suggested that
STAT3 activation and enhanced Th17 responses were related to the
sustained neutrophillic airway inflammation in chronic PA lung
infection, and SOCS3 may function as a negative feedback regulator
of p-STAT3 to control the Th17-mediated inflammation.
Although SOCS3 expression was demonstrated to be
upregulated following STAT3 activation in the mouse model of
chronic PA lung infection, a strong activation of STAT3 and Th17
responses was still observed, even in the presence of high SOCS3
mRNA expression. This may be explained by the possibility that the
SOCS3 expression in chronic airway inflammation may not reach a
high enough level to shut off STAT3 activation completely. As
reported previously, SOCS3 can be rapidly degraded by the
ubiquitin-proteasome system (14),
and has indicated to be phosphorylated in response to cytokine
stimulation (15). Thus, to
guarantee a high level of SOCS3 expression, additional expression
of exogenous SOCS3 by gene transfer could be an effective way to
overcome the rapid degradation and related modifications.
The inhibition of STAT3 activation by SOCS3 was
presented in other human and animal studies in different
inflammatory settings. In transgenic mice that overcame the
inhibitory effect of SOCS3, dextran sulfate sodium induced stronger
STAT3 activation and more severe colitis than in their wild-type
mice, suggesting that SOCS3 serves a negative regulatory role in
intestinal inflammation by downregulating STAT3 activity (16). In another study, periarticular
injection of the SOCS3 adenovirus into the ankle joints of mice
with arthritis drastically suppressed STAT3 activation and reduced
the severity of arthritis and joint swelling compared with control
groups (17). In addition, the
current study observed a suppressive effect of SOCS3 on the STAT3
activation when lentivirally transferring SOCS3 genes into lung
CD4+ T cells isolated from mice with chronic PA
infection. The effect of SOCS3 was observed at 72 h following
transinfection, and became more obvious at 96 h. These results
supported the idea that SOCS3 functioned as a negative regulator of
STAT3 activation in the chronic PA lung infection, and maybe
beneficial to control the airway inflammation.
Th17 cell is a proinflammatory subset of effector T
cells that have been implicated in the pathogenesis of many
autoimmune diseases. Activated STAT3 regulates Th17 cell
differentiation by participating in the transcriptional activation
of several Th17-regulatory genes, including those encoding IL-23R
and RORγt (18,19). Their production of the cytokine
IL-17A is known to induce epithelial cells to express a wide range
of inflammatory mediators such as CXCL1 and CXCL5, which results in
the local recruitment of neutrophils. In the present study,
SOCS3-overexpressing lung CD4+ T cells were observed to
have lower mRNA and protein levels of RORγt than control cells, and
produced significantly decreased IL-17A in vitro. The
incubation of epithelial cells with these cells induced
significantly lower level of CXCL1 and CXCL5 secretion compared
with controls, and recruited significantly decreased number of
migrating neutrophils as indicated by chemotaxis analysis. These
results indicated that SOCS3 gene transfer maybe a potential
therapy for suppressing Th17-mediated neutrophilic airway
inflammation.
Lung CD4+ T cells were stimulated with
IL-23 in vitro to investigate the effect of SOCS3
overexpression on Th17 responses. IL-23 is a heterodimeric cytokine
recognized as an inducer of IL-17-producing cells in the pool of
activated memory T cells (20,21)
and is important to maintain the Th17 phenotype (22). Activation of STAT3 appears to be
the major signaling pathway of IL-23, and STAT3 binding sites were
identified in the IL-23R (23).
Deficiency of IL-23 was presented to be associated with lower
induction of IL-17A at sites of airway inflammation observed in
mucoid PA infection (24). The
current results reported that the overexpression of SOCS3 in lung
CD4+ T cells suppressed the IL-23-induced Th17 response, suggesting
that SOCS3 served a suppressive role in regulating the IL-23/STAT3
signaling pathway involved in the Th17-related airway
inflammation.
Although IL-17A is believed to be primarily produced
by activated CD4+ T cells, other cells, such as activated γδ T
cells, neutrophils and mast cells are also able to produce IL-17A
(25). In the current study, the
authors focused on the IL-17A expression produced by lung CD4+ T
cells because CD4+ T cells have been suggested to be a key feature
in lungs with PA infection. Both healthy individuals and patients
with cystic fibrosis had robust antigen-specific memory CD4+ T cell
responses to PA that not only contained a Th17 component, but also
Th1 and Th22 cells (26).
Inducible proliferation of Th17 with memory cell characteristics is
seen in the lung draining lymph nodes of patients with cystic
fibrosis (27). However, as the
immune responses involved in chronic PA lung infection are
complicated, the roles of IL-17A produced by other cells also need
to be investigated in future studies.
Limitations of this investigation included that, in
the part of in vitro study, lung CD4+ T cells, lung
epithelial cells and neutrophils were treated outside their normal
environment, and the in vivo exposures cannot easily be
mimicked. Although these in vitro studies provided us with
valuable insight into the influence of SOCS3 on the neutrophil
migration, subsequent in vivo studies are required to
further testify the conclusion. Since any intervention that
decreases inflammation may potentially have a detrimental effect by
promoting airway infection, in future in vivo studies,
special attention will be focused on whether the decreased amount
of pulmonary neutrophils would be adequate to cope with the PA in
the lungs. In addition, as many other immune cells, such as
CD8+ T cells, macrophages and dendritic cells, also take
part in the neutrophilic airway inflammation during chronic PA
infection, investigations regarding the interaction between
SOCS3-overexpressing CD4+ T cells and these immune cells
are needed.
In conclusion, the present results indicated that
SOCS3 was upregulated in lung CD4+ T cells in a mouse
model of chronic PA lung infection and exogenous SOCS3 suppressed
Th17-mediated neutrophil recruitment in vitro. These results
suggested that SOCS3 gene therapy could be a potential way for
immunotherapy to treat airway inflammation during chronic PA lung
infection.
Acknowledgements
The authors would like to thank Xin Zhou and Qiang
Li for their technical assistance. The present work was supported
by the National Natural Science Foundation of China (grant no.
81300005).
References
|
1
|
Rada B: Interactions between neutrophils
and pseudomonas aeruginosa in cystic fibrosis. Pathogens. 6:pii:
E10. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McAllister F, Henry A, Kreindler JL, Dubin
PJ, Ulrich L, Steele C, Finder JD, Pilewski JM, Carreno BM, Goldman
SJ, et al: Role of IL-17A, IL-17F, and the IL-17 receptor in
regulating growth-related oncogene-alpha and granulocyte
colony-stimulating factor in bronchial epithelium: Implications for
airway inflammation in cystic fibrosis. J Immunol. 175:404–412.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tiringer K, Treis A, Fucik P, Gona M,
Gruber S, Renner S, Dehlink E, Nachbaur E, Horak F, Jaksch P, et
al: A Th17- and Th2-skewed cytokine profile in cystic fibrosis
lungs represents a potential risk factor for Pseudomonas aeruginosa
infection. Am J Respir Crit Care Med. 187:621–629. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dubin PJ and Kolls JK: IL-23 mediates
inflammatory responses to mucoid Pseudomonas aeruginosa lung
infection in mice. Am J Physiol Lung Cell Mol Physiol.
292:L519–L528. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Egwuagu CE: STAT3 in CD4+ T helper cell
differentiation and inflammatory diseases. Cytokine. 47:149–156.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ivanov II, Zhou L and Littman DR:
Transcriptional regulation of Th17 cell differentiation. Semin
Immunol. 19:409–417. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Harris TJ, Grosso JF, Yen HR, Xin H,
Kortylewski M, Albesiano E, Hipkiss EL, Getnet D, Goldberg MV,
Maris CH, et al: Cutting edge: An in vivo requirement for STAT3
signaling in TH17 development and TH17-dependent autoimmunity. J
Immunol. 179:4313–4317. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yuan K, Huang C, Fox J, Gaid M, Weaver A,
Li G, Singh BB, Gao H and Wu M: Elevated inflammatory response in
caveolin-1-deficient mice with Pseudomonas aeruginosa infection is
mediated by STAT3 protein and nuclear factor kappaB (NF-kappaB). J
Biol Chem. 286:21814–21825. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yoshimura A, Naka T and Kubo M: SOCS
proteins, cytokine signalling and immune regulation. Nat Rev
Immunol. 7:454–465. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen Z, Laurence A, Kanno Y,
Pacher-Zavisin M, Zhu BM, Tato C, Yoshimura A, Hennighausen L and
O'Shea JJ: Selective regulatory function of Socs3 in the formation
of IL-17-secreting T cells. Proc Natl Acad Sci USA. 103:8137–8142.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Baker BJ, Akhtar LN and Benveniste EN:
SOCS1 and SOCS3 in the control of CNS immunity. Trends Immunol.
30:392–400. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ruwanpura SM, McLeod L, Brooks GD,
Bozinovski S, Vlahos R, Longano A, Bardin PG, Anderson GP and
Jenkins BJ: IL-6/Stat3-driven pulmonary inflammation, but not
emphysema, is dependent on interleukin-17A in mice. Respirology.
19:419–427. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cacalano NA, Sanden D and Johnston JA:
Tyrosine- phosphorylated SOCS-3 inhibits STAT activation but binds
to p120 RasGAP and activates Ras. Nat Cell Biol. 3:460–465. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sasaki A, Yasukawa H, Suzuki A, Kamizono
S, Syoda T, Kinjyo I, Sasaki M, Johnston JA and Yoshimura A:
Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus
tyrosine kinase by binding through the N-terminal kinase inhibitory
region as well as SH2 domain. Genes Cells. 4:339–351. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Suzuki A, Hanada T, Mitsuyama K, Yoshida
T, Kamizono S, Hoshino T, Kubo M, Yamashita A, Okabe M, Takeda K,
et al: CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3
activation and intestinal inflammation. J Exp Med. 193:471–481.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shouda T, Yoshida T, Hanada T, Wakioka T,
Oishi M, Miyoshi K, Komiya S, Kosai K, Hanakawa Y, Hashimoto K, et
al: Induction of the cytokine signal regulator SOCS3/CIS3 as a
therapeutic strategy for treating inflammatory arthritis. J Clin
Invest. 108:1781–1788. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang XO, Panopoulos AD, Nurieva R, Chang
SH, Wang D, Watowich SS and Dong C: STAT3 regulates
cytokine-mediated generation of inflammatory helper T cells. J Biol
Chem. 282:9358–9363. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou L, Ivanov I, Spolski R, Min R,
Shenderov K, Egawa T, Levy DE, Leonard WJ and Littman DR: IL-6
programs T(H)-17 cell differentiation by promoting sequential
engagement of the IL-21 and IL-23 pathways. Nat Immunol. 8:967–974.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Aggarwal S, Ghilardi N, Xie MH, de Sauvage
FJ and Gurney AL: Interleukin-23 promotes a distinct CD4 T cell
activation state characterized by the production of interleukin-17.
J Biol Chem. 278:1910–1914. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Langrish CL, Chen Y, Blumenschein WM,
Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA and
Cua DJ: IL-23 drives a pathogenic T cell population that induces
autoimmune inflammation. J Exp Med. 201:233–240. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Veldhoen M, Hocking RJ, Atkins CJ,
Locksley RM and Stockinger B: TGFbeta in the context of an
inflammatory cytokine milieu supports de novo differentiation of
IL-17-producing T cells. Immunity. 24:179–189. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Floss DM, Mrotzek S, Klöcker T, Schröder
J, Grötzinger J, Rose-John S and Scheller J: Identification of
canonical tyrosine-dependent and non-canonical tyrosine-independent
STAT3 activation sites in the intracellular domain of the
interleukin 23 receptor. J Biol Chem. 288:19386–19400. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dubin PJ and Kolls JK: IL-23 mediates
inflammatory responses to mucoid Pseudomonas aeruginosa lung
infection in mice. Am J Physiol Lung Cell Mol Physiol.
292:L519–L528. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Keijsers RR, Joosten I, van Erp PE, Koenen
HJ and van de Kerkhof PC: Cellular sources of IL-17 in psoriasis: A
paradigm shift? Exp Dermatol. 23:799–803. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bayes HK, Bicknell S, MacGregor G and
Evans TJ: T helper cell subsets specific for Pseudomonas aeruginosa
in healthy individuals and patients with cystic fibrosis. PLoS One.
9:e902632014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chan YR, Chen K, Duncan SR, Lathrop KL,
Latoche JD, Logar AJ, Pociask DA, Wahlberg BJ, Ray P, Ray A, et al:
Patients with cystic fibrosis have inducible IL-17+IL-22+ memory
cells in lung draining lymph nodes. J Allergy Clin Immunol.
131:1117–1129, e1-e5. 2013. View Article : Google Scholar : PubMed/NCBI
|