Introduction
Osteosarcoma, which is most prevalent in childhood
and adolescence, is an aggressive, malignant neoplasm that exhibits
osteoblastic differentiation and produces malignant osteoid
(1). Although the majority of
patients are able to have limb surgery, various risk factors,
including infection, complications of surgery and local tumor
recurrence may induce the need for further surgery (2). In addition, although the 5-year
survival rates of patients that received combined treatments, such
as chemotherapy and surgery, may be as high as 70%, rates in
patients with lung metastasis remain unsatisfactory (20–40%)
(3,4). Thus, it is necessary to investigate
the pathogenesis and molecular mechanisms of osteosarcoma in
further depth to further advance treatment.
To date, various studies have investigated the
molecular mechanism of osteosarcoma, including investigation of
associated genes and pathways. Of those investigated, the
retinoblastoma (RB) gene and p53 gene are commonly
implicated in the activation of metastatic osteosarcoma (5). Furthermore, it has been demonstrated
that the integrity of the RB pathway has an important role
in tumor behavior, clinical progression and outcome in patients
with osteosarcoma (6). Notably,
restoration of RB also corrected the activity of the p53
pathway in an aggressive osteosarcoma (7,8). A
previous study demonstrated that the methylation of heterozygous
RB and the RB promoter have been exhibited in several
patients with osteosarcoma (9).
Previously, CpG island methylation, which is an epigenetic form of
gene regulation that disturbs the function of tumor suppressor
genes or oncogenes, has also been demonstrated to contribute to
carcinogenesis (10). Skarn et
al (11) demonstrated that
methylation of CpG islands had an important role in the regulation
of microRNA expression in osteosarcoma. However, the understanding
of the epigenetic alterations implicated in osteosarcoma is
currently limited. Therefore, the role of CpG methylation in the
pathogenesis and progression of osteosarcoma remains to be defined
in full.
A previous study made progress in the investigation
of the molecular mechanisms of osteosarcoma dependent on
methylation. Kresse et al (12) demonstrated the association between
the copy number of DNA, mRNA expression and DNA methylation in
osteosarcoma. Specifically, the present study focused on the
effects of CpG island methylation in transcriptional regulation
that contributes to the tumorigenesis of osteosarcoma. Based on the
DNA methylation and gene expression profiles in osteosarcoma,
differentially expressed genes (DEGs) with CpG methylation were
identified, and the transcriptional regulatory relationship was
analyzed by building a regulatory network. Functional annotation
was also performed to investigate the biological role of abnormally
expressed genes.
Materials and methods
Data source
Gene expression data of GSE36001 (ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE36001) and
DNA methylation profiles of GSE36004 (ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE36004) were
downloaded from the Gene Expression Omnibus (GEO) based on the
platform of Illumina human-6 v.2.0 expression BeadChip (Illumina,
Inc., San Diego, CA, USA) and Illumina HumanMethylation27 BeadChip
(Illumina, Inc.) (12). In each of
these datasets, 19 osteosarcoma cell lines were included, while two
normal osteoblast cell lines (OB1 and OB2, two primary osteoblast
cultures isolated from human calvaria of different donors) were
purchased from ScienCell Research Laboratories (California, USA)
(12) and four normal bone samples
were included as controls. The following osteosarcoma cell lines
were included: 143B; HAL; HOS; ΩIOR/OS9; IOR/OS10; IOR/OS14;
IOR/OS15; IOR/OS18; IOR/MOS; IOR/SARG; KPD; MG-63; MHM; MNNG/HOS;
OHS; OSA; Saos-2; U-2 OS; and ZK-58. Details of the origin of each
cell line or bone sample involved in the datasets can be obtained
via the web links provided for each dataset.
Data preprocessing
Raw data of all probes were normalized by the median
method (13). Following
normalization, the probe name was converted into a gene symbol
based on the annotation information. If more than one probe mapped
to one gene, an aggregate function in R (14) was performed to calculate the mean
expression value for this gene. Probes with missing values were
imputed with the nearest neighbor averaging method (15) of imputation (impute) package
(version 1.0; https://bioconductor.org/packages/release/bioc/html/impute.html)
in R (16). The DNA methylation
data obtained from GEO was preprocessed with BeadStudio software
(version 3.1) from Illumina, Inc. where the methylated locus for
each sample with a missing value was removed.
Identification of DEGs in osteosarcoma
cells
To identify DEGs between osteosarcoma cells and
normal controls, one way analysis of variance (ANOVA) in the
genefilter package of R was performed (15). P-values were generated using
Benjamini-Hochberg (BH) multiple testing correction method and
P<0.05 was considered to indicate a statistically significant
difference. The ratio of mean expression of normal and osteosarcoma
cell groups was used to determine whether genes were up or
downregulated.
Identification of disease-associated
methylated regions (DMR)
To identify disease-associated CpG methylated sites
in osteosarcoma cells, compared with normal control for
disease-association analysis, CpG loci with beta values that were
not significantly different between case and control were
eliminated. Similarly, all CpG loci on the X, Y and mitochondrial
chromosomes were removed. Subsequently, the CpGassoc package of R
(17) was used for
disease-association analysis, which is designed to investigate the
association between methylation at CpG loci across the genome and a
phenotype of interest. CpGassoc algorithm of R package was used to
determine the association between CpG loci and osteosarcoma. In
addition, CpGassoc can also be used to create quantile-quantile
plots, manhattan plots and scatterplots for individual CpG sites.
BH multiple testing correction was used to estimate the false
discovery rate (FDR) in disease-associated analysis. FDR≤0.05 was
chosen as the threshold.
Integration analysis of gene
expression and methylation profiles
To investigate the association between methylation
and gene expression in osteosarcoma cell lines, methylation data
was measured on ±2 kb genomic regions around the transcriptional
start sites (TSS) of each gene. The obtained genes were
differentially methylated. Integration analysis was performed to
identify genes where there was an overlap between differential
expression between control and osteosarcoma, and the presence of
methylation. In order to investigate the influence of methylation
on gene expression, the transcription factor binding sites were
searched within the UCSC database (18). Subsequently, methylated DEGs in
transcription factor binding regions in osteosarcoma cells were
screened.
Construction of transcriptional
regulatory networks
Based on the transcription factor and target gene
information provided by the UCSC database, transcriptional
regulatory networks were constructed and further visualized by
Cytoscape software (version 2.8.0; www.cytoscape.org) (19). In the network, the node degree was
calculated by igraph package in R (20).
Functional annotation of target genes
of transcription factors
To understand the biological roles of target genes
of the transcription factors blocked by DNA methylation, gene
ontology (GO) function and pathway enrichment analysis was
performed by DAVID (database for annotation, visualization, and
integrated discovery) online tool (21). P<0.05 was considered to indicate
a statistically significant difference.
Results
Identification of DEGs in osteosarcoma
cells
Following preprocessing of methylation profile data,
a total of 20,006 methylation sites were identified from the 25
samples Similarly, a total of 24,214 genes were identified from 25
samples following preprocessing of expression profile data. By
applying ANOVA, a total of 6,419 DEGs were identified in
osteosarcoma cells compared with normal control, including 3,236
upregulated and 3,183 downregulated genes.
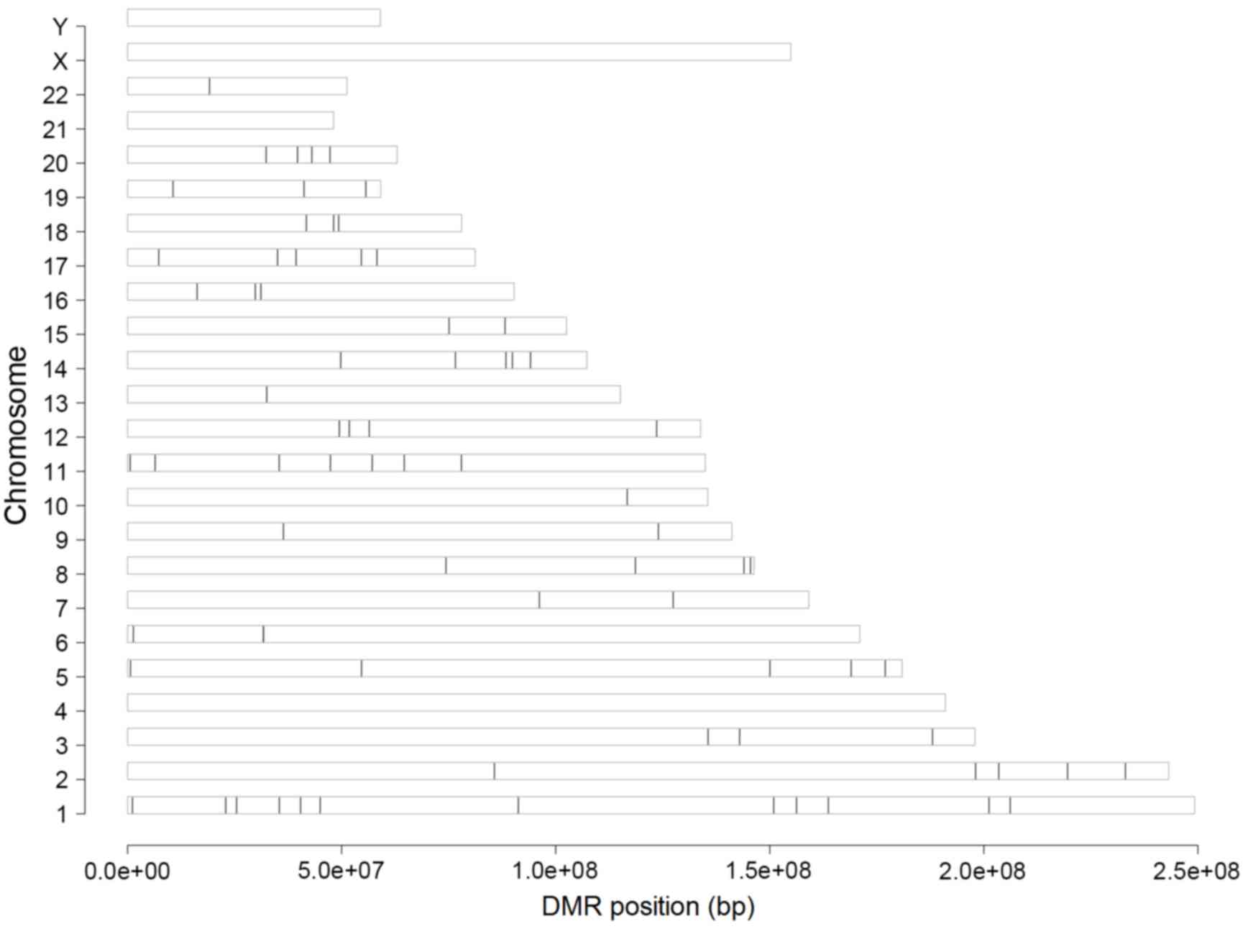
Identification of DMRs
To identify differentially methylated genes, the
present study performed three types of analyses. Initially
preprocessing of raw methylation data was performed and 20,006
methylated sites were identified in the 25 samples included in the
dataset. Subsequently, methylated sites that were located on the X,
Y and mitochondrial chromosomes and imputation analysis was
performed; as a result, 5,921 sites were eliminated from further
analysis. Finally, following disease-associated analysis, 13,750
differentially methylated genes located in different chromosome
regions, with an FDR<0.05, were identified in osteosarcoma cells
(Fig. 1).
Integration analysis of DEGs and
differentially methylated genes
Integration analysis indicated that 3,625 genes were
differentially expressed in osteosarcoma and control, and also
methylated around their TSS. Based on the UCSC database for
transcription factors, a total of 75 methylated sites were located
in the transcription factor binding regions, which may affect 83
transcription factors and 75 downstream target genes.
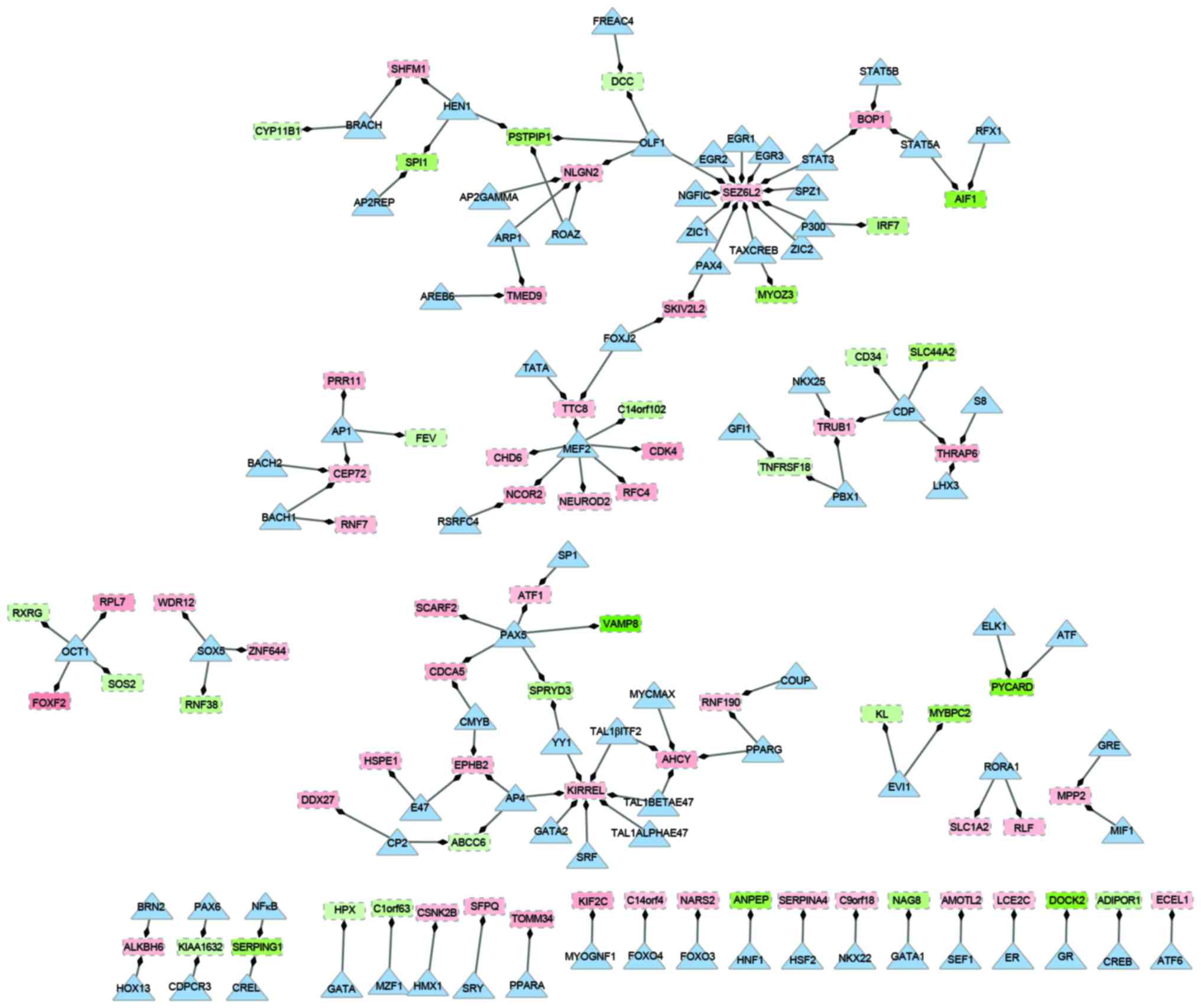
Construction of transcription
regulatory network
Based on different types of regulation, a
transcription regulatory network was constructed, which included 83
transcription factors and 75 downstream target genes (Fig. 2). In the network, there were 158
nodes and 129 edges. Of the genes in the network, the overexpressed
gene seizure related 6 homolog like 2 (SEZ6L2) had the
highest degree. It was observed that SEZ6L2 was regulated by
12 transcription factors, including signal transducer and activator
of transcription 3 (STAT3) and early growth response 1–3
(EGR1-3). Additionally, the transcription factor myocyte enhancer
factor 2 (MEF2), which had a higher degree compared with
other transcription factors, regulated seven downstream targets,
including the upregulated replication factor C (activator 1) 4,
cyclin-dependent kinase 4 (CDK4) and chromodomain helicase
DNA binding protein 6 genes, and downregulated the chromosome 14
open reading frame 102 gene. Similarly, the upregulated kin of IRRE
like gene (KIRREL) was regulated by seven different
transcription factors. Transcription factor paired box 5
(PAX5) was indicated to be involved in the regulation of
three up and two downregulated genes in the network. Upregulated
genes, centrosomal protein 72 (CEP72), block of
proliferation 1 (BOP1) and TruB pseudouridine synthase
family member 1, were regulated by a different set of three
transcription factors: STAT3, MEF2 and adaptor-related protein
complex 1 (AP1).
Functional annotation of target genes
of transcription factors
GO functional enrichment analysis was performed for
46 upregulated and 29 downregulated genes. The top five enriched
categories are listed in Tables I
and II. The upregulated genes
were predominantly enriched in the GO terms of cell cycle,
non-coding RNA metabolic process and the maturation of large
subunit ribosomal RNA (rRNA) from tricistronic rRNA transcript. In
addition, downregulated genes were predominantly involved in the
inflammatory response, regulation of the humoral immune response
and response to wounding.
 | Table I.GO function and pathway enrichment
analysis of upregulated genes in the transcriptional regulatory
network. |
Table I.
GO function and pathway enrichment
analysis of upregulated genes in the transcriptional regulatory
network.
| Term |
|
|
|---|
|
|
|
|---|
| ID | Name | Count | P-value |
|---|
|
REACTOME_PATHWAY |
|
|
|
|
REACT_152 | Cell cycle,
mitotic | 4 | 0.028263722 |
| GOTERM_BP_FAT |
|
|
|
|
GO:0034660 | ncRNA metabolic
process Maturation of LSU-rRNA | 5 | 0.002014149 |
|
GO:0000463 | from tricistronic
rRNA transcript (small subunit-rRNA, | 2 | 0.007080142 |
|
| 5.8S rRNA and
LSU-rRNA) |
|
|
|
GO:0000470 | Maturation of
LSU-rRNA | 2 | 0.007080142 |
|
GO:0006396 | RNA processing | 6 | 0.008649471 |
|
GO:0034470 | ncRNA
processing | 4 | 0.009603024 |
| GOTERM_CC_FAT |
|
|
|
|
GO:0070013 | Intracellular
organelle lumen | 12 | 0.002136088 |
|
GO:0043233 | Organelle
lumen | 12 | 0.002571635 |
|
GO:0031974 | Membrane-enclosed
lumen | 12 | 0.00301231 |
|
GO:0005654 | Nucleoplasm | 8 | 0.004426136 |
|
GO:0031981 | Nuclear lumen | 10 | 0.006006103 |
| GOTERM_MF_FAT |
|
|
|
|
GO:0005524 | ATP binding | 9 | 0.019812363 |
|
GO:0032559 | Adenyl
ribonucleotide binding | 9 | 0.021338813 |
|
GO:0008026 | ATP-dependent
helicase activity | 3 | 0.022750064 |
|
GO:0070035 | Purine nucleotide
triphosphate-dependent helicase activity | 3 | 0.022750064 |
|
GO:0042623 | ATPase activity,
coupled | 4 | 0.026532158 |
| Table II.Functional annotation of
downregulated genes in the transcriptional regulatory network. |
Table II.
Functional annotation of
downregulated genes in the transcriptional regulatory network.
| Term |
|
|
|---|
|
|
|
|---|
| ID | Name | Count | P-value |
|---|
| GOTERM_BP_FAT |
|
|
|
|
GO:0006954 | Inflammatory
response | 4 | 0.01325742 |
|
GO:0002920 | Regulation of
humoral immune response | 2 | 0.018477257 |
|
GO:0009611 | Response to
wounding | 4 | 0.047076136 |
| GOTERM_MF_FAT |
|
|
|
|
GO:0005496 | Steroid
binding | 3 | 0.004729209 |
Discussion
Osteosarcoma is the most common histological form of
primary bone cancer. To analyze the effects of genome-wide changes
in gene expression and DNA methylation in osteosarcoma cell lines,
the present study identified 75 significantly methylated genes,
which included 46 genes that were upregulated (including
SEZ6L2, KIRREL, CEP72, BOP1 and
CDK4) and 29 genes that were downregulated in osteosarcoma
cell lines compared with the normal controls. These genes were
regulated by 83 transcription factors, including MEF2 and PAX5.
SEZ6L2 encodes a seizure-associated protein
with an N-terminal signal peptide that is located on the cell
surface. In the present study, this gene was demonstrated to be
regulated by various transcription factors, including STAT3,
EGR1 and PAX4. It has been previously demonstrated
that STAT3 upregulates vascular endothelial growth factor
expression and contributes to tumor angiogenesis (22), which means it is widely regarded as
a promising target for cancer treatment. In addition, the
STAT3 inhibitor, CDDO-Me, inhibited the development of
osteosarcoma cell lines and also induced apoptosis (23). Furthermore, it was demonstrated
that microRNA (miR)-125b downregulated the expression of
STAT3, which suppressed the migration and proliferation of
osteosarcoma cells (24). In
addition, a previous study demonstrated that, by upregulating
EGR1, chemotherapy downregulated the activity of urokinase
and prevented osteosarcoma cell invasion (25). Additionally, another member of EGR
family, EGR2, was suppressed by miR-20a, which promoted the
cell cycle and proliferation of human osteosarcoma cells (26). Based on these previous results,
SEZ6L2 may modulate the cell cycle and metastasis of
osteosarcoma through regulation by STAT3, EGR1 and
PAX4.
KIRREL, also known as NEPH1, is a
nephrin-associated member of the immunoglobulin superfamily that is
involved in cell-cell interaction and somatic cell fusion during
embryonic development (27,28).
Notably, it is thought that somatic cell fusion may be a mechanism
that contributes to cancer metastasis and chemotherapy resistance
(29,30). KIRREL has been demonstrated
to be differentially hyper-methylated in primary malignant
adrenocortical samples compared with benign samples (31). In the present study, it was
identified that KIRREL was regulated by various
transcription factors, including AP4, GATA binding protein 2
(GATA2) and serum response factor (SRF). AP4 induced the expression
of CDK2, which subsequently regulated the proliferation of
osteosarcoma cell lines (32). SRF
was previously demonstrated to be involved in the mitogen-activated
protein kinase cascade signaling pathway in human osteosarcoma
cells (33). Furthermore, it has
been demonstrated that GATA2 is required for proliferation in
various cancer cell types (34).
Thus, differential expression of KIRREL may be regulated by
methylation and transcription factors that promote tumor
development. In addition, CEP72, CDK4 and BOP1
were differentially methylated and regulated by transcription
factors, including STAT3, MEF2 and AP1. CEP72 encodes a
protein that is a member of the leucine-rich-repeat superfamily.
CEP72 has been identified as possessing a high incidence of
genomic copy number changes in the 5p15.33 region in patients with
non-small cell lung cancer (35).
As osteosarcoma has a high tendency for metastatic spread and
predominantly arises in the lungs, CEP72 may have a key role
in cancer metastasis. In addition, it was previously demonstrated
that CEP72 was regulated by AP1, which is involved in the ERK
signaling pathway (36). It is
well established that CDKs are essential drivers of cell cycle
progression and are commonly dysregulated during tumorigenesis
(37). CDK4 and other CDK
inhibitors have been identified as a class of promising anticancer
agents in cancer treatment (38).
Furthermore, regulated transcription factors STAT3 and AP1 have
been demonstrated to be involved in cancer development and
progression via promotion of the cell cycle (39,40).
The functional annotation performed in the present study was
consistent with a previous study about the enriched pathway of
CEP72 and CDK4 (41).
In conclusion, methylation of SEZ6L2,
KIRREL, CEP72 and CDK4 may have an important
role in the pathogenesis of osteosarcomas through promotion of cell
proliferation and metastasis. However, further study into the
results is required and may provide further insight into the
molecular mechanism of osteosarcoma. In addition, further
experiments, including western blot analysis and reverse
transcription-polymerase chain reaction will be performed in the
future to validate the changes in gene expression.
References
|
1
|
Luetke A, Meyers PA, Lewis I and Juergens
H: Osteosarcoma treatment-where do we stand? A state of the art
review. Cancer Treat Rev. 40:523–532. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jaffe N, Bruland OS and Bielack S:
Pediatric and adolescent osteosarcoma. Springer Science &
Business Media; 2010, View Article : Google Scholar
|
|
3
|
Buddingh EP, Anninga JK, Versteegh MI,
Taminiau AH, Egeler RM, van Rijswijk CS, Hogendoorn PC, Lankester
AC and Gelderblom H: Prognostic factors in pulmonary metastasized
high-grade osteosarcoma. Pediatr Blood Cancer. 54:216–221.
2010.PubMed/NCBI
|
|
4
|
Lewis IJ, Nooij MA, Whelan J, Sydes MR,
Grimer R, Hogendoorn PC, Memon MA, Weeden S, Uscinska BM, van
Glabbeke M, et al: Improvement in histologic response but not
survival in osteosarcoma patients treated with intensified
chemotherapy: A randomized phase III trial of the European
Osteosarcoma Intergroup. J Natl Cancer Inst. 99:112–128. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Berman SD, Calo E, Landman AS, Danielian
PS, Miller ES, West JC, Fonhoue BD, Caron A, Bronson R, Bouxsein
ML, et al: Metastatic osteosarcoma induced by inactivation of Rb
and p53 in the osteoblast lineage. Proc Natl Acad Sci USA.
105:11851–11856. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Scott MC, Sarver AL, Phan F, Gupta R,
Thayanithy V, Subramanian S and Modiano F: Abstract B19: RB
function as a central component of osteosarcoma behavior: A
comparative assessment in dogs and humans. Molecular Cancer Res.
12:B192014. View Article : Google Scholar
|
|
7
|
Ternovoi VV, Curiel DT, Smith BF and
Siegal GP: Adenovirus-mediated p53 tumor suppressor gene therapy of
osteosarcoma. Lab Invest. 86:748–766. 2006.PubMed/NCBI
|
|
8
|
Scott MC, Sarver AL, Tomiyasu H, Cornax I,
Van Etten J, Varshney J, O'Sullivan MG, Subramanian S and Modiano
JF: Aberrant retinoblastoma (RB)-E2F transcriptional regulation
defines molecular phenotypes of osteosarcoma. J Biol Chem.
290:28070–28083. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Patiño-García A, Piñeiro ES, Díez MZ,
Iturriagagoitia LG, Klüssmann FA and Ariznabarreta LS: Genetic and
epigenetic alterations of the cell cycle regulators and tumor
suppressor genes in pediatric osteosarcomas. J Pediatr Hematol
Oncol. 25:362–367. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cui J, Wang W, Li Z, Zhang Z, Wu B and
Zeng L: Epigenetic changes in osteosarcoma. Bull Cancer.
98:E62–E68. 2011.PubMed/NCBI
|
|
11
|
Skarn M, Namlos HM, Ahmed D, Lind GE,
Meza-Zepeda LA and Myklebost O: Epigenetic regulation of miRNA
expression in osteosarcoma. Cancer Res. 72:1972012. View Article : Google Scholar
|
|
12
|
Kresse SH, Rydbeck H, Skarn M, Skårn M,
Namløs HM, Barragan-Polania AH, Cleton-Jansen AM, Serra M, Liestøl
K, Hogendoorn PC, et al: Integrative analysis reveals relationships
of genetic and epigenetic alterations in osteosarcoma. PLoS One.
7:e482622012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Irizarry RA, Hobbs B, Collin F,
Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP:
Exploration, normalization, and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang J, Xie G, Singh M, Ghanbarian AT,
Raskó T, Szvetnik A, Cai H, Besser D, Prigione A, Fuchs NV, et al:
Primate-specific endogenous retrovirus-driven transcription defines
naive-like stem cells. Nature. 516:405–409. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Altman NS: An introduction to kernel and
nearest-neighbor nonparametric regression. The American
Statistician. 46:175–185. 1992. View
Article : Google Scholar
|
|
16
|
Hastie T, Tibshirani R, Narasimhan B and
Chu G: Impute: imputation for microarray data. Bioinformatics.
17:520–525. 2001.PubMed/NCBI
|
|
17
|
Barfield RT, Kilaru V, Smith AK and
Conneely KN: CpGassoc: An R function for analysis of DNA
methylation microarray data. Bioinformatics. 28:1280–1281. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Meyer LR, Zweig AS, Hinrichs AS, Karolchik
D, Kuhn RM, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, et al:
The UCSC Genome Browser database: Extensions and updates 2013.
Nucleic Acids Res. 41:D64–D69. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kohl M, Wiese S and Warscheid B:
Cytoscape: Software for visualization and analysis of biological
networks. Data Mining in Proteomics. Springer; pp. 291–303.
2011
|
|
20
|
Csardi G and Nepusz T: The igraph software
package for complex network research. Inter J Complex Systems.
1695:2006.
|
|
21
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wei D, Le X, Zheng L, Wang L, Frey JA, Gao
AC, Peng Z, Huang S, Xiong HQ, Abbruzzese JL and Xie K: Stat3
activation regulates the expression of vascular endothelial growth
factor and human pancreatic cancer angiogenesis and metastasis.
Oncogene. 22:319–329. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ryu K, Choy E, Yang C, Susa M, Hornicek
FJ, Mankin H and Duan Z: Activation of signal transducer and
activator of transcription 3 (Stat3) pathway in osteosarcoma cells
and overexpression of phosphorylated-Stat3 correlates with poor
prognosis. J Orthop Res. 28:971–978. 2010.PubMed/NCBI
|
|
24
|
Liu LH, Li H, Li JP, Zhong H, Zhang HC,
Chen J and Xiao T: miR-125b suppresses the proliferation and
migration of osteosarcoma cells through down-regulation of STAT3.
Biochem Biophys Res Commun. 416:31–38. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Matsunoshita Y, Ijiri K, Ishidou Y, Nagano
S, Yamamoto T, Nagao H, Komiya S and Setoguchi T: Suppression of
osteosarcoma cell invasion by chemotherapy is mediated by urokinase
plasminogen activator activity via up-regulation of EGR1. PLoS One.
6:e162342011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhuo W, Ge W, Meng G, Jia S, Zhou X and
Liu J: MicroRNA-20a promotes the proliferation and cell cycle of
human osteosarcoma cells by suppressing early growth response 2
expression. Mol Med Rep. 12:4989–4994. 2015.PubMed/NCBI
|
|
27
|
Sellin L, Huber TB, Gerke P, Quack I,
Pavenstädt H and Walz G: NEPH1 defines a novel family of podocin
interacting proteins. FASEB J. 17:115–117. 2003.PubMed/NCBI
|
|
28
|
Durcan PJ, Al-Shanti N and Stewart CE:
Identification and characterization of novel Kirrel isoform during
myogenesis. Physiol Rep. 1:e000442013. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Duelli D and Lazebnik Y: Cell fusion: A
hidden enemy? Cancer Cell. 3:445–448. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pawelek JM and Chakraborty AK: The cancer
cell-leukocyte fusion theory of metastasis. Adv Cancer Res.
101:397–444. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rechache NS, Wang Y, Stevenson HS, Killian
JK, Edelman DC, Merino M, Zhang L, Nilubol N, Stratakis CA, Meltzer
PS and Kebebew E: DNA methylation profiling identifies global
methylation differences and markers of adrenocortical tumors. J
Clin Endocrinol Metab. 97:E1004–E1013. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jackstadt R, Röh S, Neumann J, Jung P,
Hoffmann R, Horst D, Berens C, Bornkamm GW, Kirchner T, Menssen A
and Hermeking H: AP4 is a mediator of epithelial-mesenchymal
transition and metastasis in colorectal cancer. J Exp Med.
210:1331–1350. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bébien M, Salinas S, Becamel C, Richard V,
Linares L and Hipskind RA: Immediate-early gene induction by the
stresses anisomycin and arsenite in human osteosarcoma cells
involves MAPK cascade signaling to Elk-1, CREB and SRF. Oncogene.
22:1836–1847. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tsai FY and Orkin SH: Transcription factor
GATA-2 is required for proliferation/survival of early
hematopoietic cells and mast cell formation, but not for erythroid
and myeloid terminal differentiation. Blood. 89:3636–3643.
1997.PubMed/NCBI
|
|
35
|
Kang JU, Koo SH, Kwon KC, Park JW and Kim
JM: Gain at chromosomal region 5p15.33, containing TERT, is the
most frequent genetic event in early stages of non-small cell lung
cancer. Cancer Genet Cytogenet. 182:1–11. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cho HJ, Kang JH, Kwak JY, Lee TS, Lee IS,
Park NG, Nakajima H, Magae J and Chang YC: Ascofuranone suppresses
PMA-mediated matrix metalloproteinase-9 gene activation through the
Ras/Raf/MEK/ERK-and Ap1-dependent mechanisms. Carcinogenesis.
28:1104–1110. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Trovesi C, Manfrini N, Falcettoni M and
Longhese MP: Regulation of the DNA damage response by
cyclin-dependent kinases. J Mol Biol. 425:4756–4766. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Finn RS, Crown JP, Lang I, Boer K,
Bondarenko IM, Kulyk SO, Etti J, Patel R, Pinter T, Schmidt M, et
al: Results of a randomized phase 2 study of PD 0332991, a
cyclin-dependent kinase (CDK) 4/6 inhibitor, in combination with
letrozole vs. letrozole alone for first-line treatment of
ER+/HER2-advanced breast cancer (BC). Cancer Res. 72:S1–S6. 2012.
View Article : Google Scholar
|
|
39
|
Grivennikov SI and Karin M: Dangerous
liaisons: STAT3 and NF-kappaB collaboration and crosstalk in
cancer. Cytokine Growth Factor Rev. 21:11–19. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ishdorj G, Johnston JB and Gibson SB:
Cucurbitacin-I (JSI-124) activates the JNK/c-Jun signaling pathway
independent of apoptosis and cell cycle arrest in B leukemic cells.
BMC Cancer. 11:2682011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Oshimori N, Li X, Ohsugi M and Yamamoto T:
Cep72 regulates the localization of key centrosomal proteins and
proper bipolar spindle formation. EMBO J. 28:2066–2076. 2009.
View Article : Google Scholar : PubMed/NCBI
|