Introduction
Parkinson's disease (PD) is the second most common
neurodegenerative disease characterized by a selective loss of DA
neurons in the substantia nigra pars compacta (SNpc) (1). Such a loss produces the depletion of
striatal dopamine (DA) which leads to movement disorders such as
resting tremor, rigidity, bradykinesia and postural instability
(2). Despite the various
researches conducted in this field, the precise etiology of PD
still remains unclear. Some evidences indicate that the
neuroinflammation mediated by microglial activation may play an
important role in PD pathogenesis and notably in the degeneration
of the SNpc dopaminergic neurons (3,4).
Once activated, microglia includes classically activated M1
phenotype (pro-inflammatory function) and also, alternatively,
activated M2 phenotype (anti-inflammatory function) (5,6).
Microglial activation is believed to lead to neuroinflammation by
production of pro-inflammatory cytokines such as interleukin-1
(IL-1), IL-6, tumour necrosis factor-α (TNF-α), nitric oxide (NO)
and prostaglandin E2 (PGE2) (7),
which contribute to dopaminergic neuronal death in PD.
The Sonic hedgehog (SHH) diffusible protein, a
member of the hedgehog family, become active in binding Patched
receptors (Ptch). This binding relieves Ptch-mediated inhibition
exerted on Smoothened (Smo) receptor (a key transducer of the
Hedgehog, HH, signaling pathway). Subsequently, the Smo receptor
promotes the transcription factor ‘Glioma-associated oncogene’
(Gli) which translocate to the nucleus and regulates the
transcription and expression of target genes (8,9). SHH
signaling pathway is also activated in brain injuries,
neurodegenerative diseases and neurogenesis processes taking place
in adult (10–13). SHH signaling can also protect
dopaminergic cells from MPP (+) toxicity in vitro and
improve nigrostriatal pathway by restoring tyrosine hydroxylase
(TH) positivity in vivo (14). In this frame, a recent report
suggests that SHH-N overexpression can improve the motor function
in PD model, restore the nigrostriatal pathway and reduce the loss
of DA neurons in vivo (15). Finally, various other reports
indicate that PI3K/Akt pathway, related with inflammation, is
impaired in PD animal models (16,17).
In this sense, Fasudil, a vasodilatator and a potent Rho-kinase
inhibitor, is suggested for protecting dopaminergic neurons through
the inflammatory inhibition running through the activation of
PI3K/Akt signaling pathway (18).
Further evidences also suggest that SHH pathway may protects
cortical neurons and astrocytes from oxidative stress by activating
the PI3K/Akt pathway (19,20). To date, despite the various
approaches mentionned above, the precise mechanisms involving a
neuroprotective effects of the SHH signaling pathway still remain
unclear.
In the present study, by way of in vitro
(LPS-treated BV2 microglial cells) and in vivo (MPTP-induced
mouse model of Parkinson disease) approaches, we demonstrate that
the SHH signaling through the PI3K/AKt pathway is capable to:
Attenuate the inflammatory response, inhibit the microglial
activation, protect dopaminergic neurons and reduce behavioral
impairments.
Materials and methods
Materials
The following reagents were used in the present
study: Dulbecco's modified Eagle's medium (DMEM) and fetal bovine
serum (FBS), from Gibco (Grand Island, NY, USA); TH antibody, p-AKt
antibody and AKt antibody, from Abcam (Cambridge, UK); Ionized
calcium binding adaptor molecule 1 (Iba1) antibody, from Wako
(Osaka, Japan); β-actin antibody, HRP-conjugated goat polyclonal
anti-rabbit IgG antibody, Purmorphamine and Cyclopamine, from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA); LPS and MPTP, from
Sigma-Aldrich (St. Louis, MO, USA); LY294002, from Cell Signaling
Technology, Inc. (Danvers, MA, USA).
Cell culture
The murine BV2 microglial cell line was grown in
DMEM supplemented with 10% FBS (Gibco), 100 U/ml penicillin, and
100 mg/ml (Sigma, St. Louis, MO, USA). In a humidified 5%
CO2 incubator maintained at 37°C, streptomycin and the
culture medium were renewed every day. Cells were plated at 5×105
concentration and grown for 24 h prior to the experiments.
In vitro treatments
Dishes of cultured BV2 cells were randomly divided
into six groups: Including: i) Control group, ii) LPS group, iii)
PM+LPS group, iv) Cyclopamine+PM+LPS group, v) LY294002+PM+LPS
group and vi) LY294002+LPS group. For LPS group: LPS (1 µg/ml) was
employed during 24 h to obtain an inflammatory response with BV2
cells. For PM+LPS group: Purmorphamine (PM, 1.5 µmol/l) was used to
activate the SHH pathway in BV2 cells 24 h before LPS treatment;
For Cyclopamine+PM+LPS group: BV2 cells were pretreated with a
specific SHH signal inhibitor (Cyclopamine) to further explore the
role of PI3K/Akt pathway on the effects of SHH pathway; in this
respect, Cyclopamine (20 µmol/l) was administered to block the SHH
pathway (1 h before PM treatment); then, PM was used to treat BV2
cells for 24 h; then after, a LPS treatment was applied. For
LY294002+PM+LPS group: A selective inhibitor of PI3K/AKt, LY294002
(20 µmol/l) was used during 30 min in order to block PI3K/Akt
pathway before PM treatment; then PM was used to treat BV2 cells
for 24 h; this pharmacological situation was completed with a LPS
treatment. For LY294002+LPS group: LY294002 was used to treat BV2
cells without PM treatment; then after, a LPS treatment was
applied.
In vivo treatment
All animal studies were approved by the
Institutional Animal Care and Use Committee at Guangzhou Medical
University. Male C57BL/6 mice (8–10 weeks, 22-25 g) were housed
under a 12-h light/dark cycle with free access to food and water.
All animals were randomly divided into five group, including: i)
Control (n=10); ii) MPTP (n=10); iii) PM+MPTP (n=11, PM+MPTP); iv)
LY294002+PM+MPTP (n=11, LY+PM+MPTP); and v) LY294002+MPTP (n=11,
LY+MP). For the MPTP group, mice received four intraperitoneally
(i.p.) injections of MPTP (20 mg/kg) in a 2 h interval (21); For the PM+MPTP group, mice received
i.p. injection of PM (1 mg/kg) 24 h before the first MPTP
injection; For the LY+PM+MPTP group, 30 µl of LY294002 (5 mg/ml)
were administered intranasally 1 h before PM treatment; then after,
PM was administered i.p. and, 24 h later, MPTP was given. For the
LY294002+MPTP group, LY294002 was administered intranasally and
MPTP was injected four times.
Behavioral tests
Traction behavior (TR) test was described previously
(22). The stainless steel bar
(diameter: 1.5 mm, 25 cm long) was fixed 30 cm over ground. Each
animal was hanged by its forelimbs and left on the bar. The time
lapse before the fall down was checked for each animal. For the
score of TR, the following standards were employed: 0–4 sec=0; 5–9
sec=1; 10–14 sec=2; 15–19 sec=3; 20–24 sec=4; 25–29=5; over 30=6.
Each animal was submitted three times to the test with an interval
of 1 min between each trial. The general mean of the trials was
taken.
Quantitative PCR (qPCR)
For qPCR analysis, total RNA was isolated from BV2
cells and from the brain substantia nigra by TRIzol reagent (Takara
Bio, Inc., Otsu, Japan). Total RNA (1 µg) was reverse transcribed
into cDNA using PrimerScript™ RT reagent kit (Takara Bio, Inc.).
The mRNA expression levels were measured to use SYBR® Premix Ex
Tag™ and primers. The sequences for qPCR promers were as follows:
IL-1β forward, 5′-TGCCACCTTTTGACAGTGATG-3′; reverse, 5′-GGAAG for
each GTCCACGGGAAAGAC-3′; TNF-α forward, 5′-ATGGCCTCCCTCTCATCAGT-3′;
reverse, 5′-ATAGCAAATCGGCTGACGGT-3′; Nurr1 forward,
5′-AAGACCTTCTCCCAAGCACG-3′; reverse, 5′-GAACTGGACACTTCAACCAGC-3′;
TGF-β1 forward, 5′-GGTCCTTGCCCTCTACAACC- 3′; reverse,
5′-CCACGTAGTAGACGATGGGC-3′; β-actin forward,
5′-GTTACAGGAAGTCCCTCACCC-3′; reverse, 5′-CAGAAGCAATGCTGTCACCTT-3′.
Cycling condition included one cycle at 95°C for 5 sec, 30 cycles
at 95°C for 30 sec and 60°C for 1 min, followed by one cycle 95°C
for 15 sec, 60°C for 1 min and 95°C for 15 sec. β-actin was used as
an internal control and the transcript levels are expressed as
2−∆∆Cq values.
Western blotting
BV2 cells and substantia nigra were lysed in ice
cold lysis buffer (1X PBS, 1% Nonidet P-40, 05% sodium deoxycholate
and 0.1% SDS; RIPA) containing phosphatase inhibitor protease
inhibitor. Lysates (50 µg protein) from each sample were resolved
on SDS 10% polyacrylamide gel (10% PAGE) and electrotransferred to
PVDF membrane. The membrane was incubated in 5% non-fat dry milk to
block non-specific antibody binding site and then incubated
overnight at 4°C with rabbit anti-p-AKt (1:1,000), rabbit anti-AKt
(1:1,000), rabbit anti-TH (1:200), rabbit anti-Iba1 (1 µg/ml),
rabbit anti-β-actin (1:1,000). After washing in TBST (0.01 M TBS
and 0.1% Tween-20), membranes were incubated with horseradish
peroxidase-conjugated secondary antibodies (goat anti-rabbit
immunoglobulin G [IgG], 1:1,000) for 2 h. This step was followed by
a washing in TBST and protein visualized. In this respect,
chemiluminescence (ECL) reagents were employed.
Immunohistochemistry
Mice were anesthetized and perfused transcardially
with phosphate-buffered saline (PBS) followed by a 4% fresh
paraformaldehyde (PFA) solution (pH 7.4). Brains were removed and
kept in 4% PFA overnigth at 4°C. Then after, they were inserted in
a 30% sucrose solution for 48 h at 4°C. Afterwards, brains were
cutted (slices of 30 m depth) with a freezing microtome (Leica,
Germany) and incubated during 30 min in 3%
H2O2 solution. Then, slices were rinsed three
times and during 5 min in PBST (0.01 M PBS and 0.3% Triton-X 100)
and incubated with a 5% normal goat serum at room temperature for 1
h. Brain section were then incubated with polyclonal rabbit anti-TH
(1:750; cat. no. ab112; Abcam), Ibal (0.5 µg/ml; Wako) overnight at
4°C. The next day, the sections were washed with PBST three times
for 5 min and incubated with corresponding secondary antibodies at
room temperature for 2 h. They were washed again in PBS and
subsequently incubated with streptavidin-peroxidase for 30 min.
After a new wash in PBS (3×5 min.), they were incubated with a DAB
chromogenic substrate (Fuzhou Maixin Biotech Co., Ltd. (Fuzhou,
China) for 5 min and then after washed again in distilled water.
Afterwards, brain sections were mounted on gelatin-coated slides,
air-dried, dehydrated, and covered with a glass plate. Brain
sections were examined using a light-field microscope. Finally,
TH+ cells were identified with the Image-Pro Plus
software and visually counted.
Statistical analysis
The Statistical Package for the Social Sciences
(SPSS version 13.0) was used for the statistical analyses. All data
were expressed as mean ± standard deviation (SD) for three
independent experiments, at least. The analysis of variance (ANOVA)
was performed for all tests, followed by post hoc Fisher's LSD
multiple comparison test. P<0.05 was considered to indicate a
statistically significant difference.
Results
SHH signaling pathway activates the
PI3K/Akt signaling pathway
Data obtained indicate that LPS-induced inflammatory
response in BV2 microglial cells produces a significant reduction
in p-AKt protein content when compared with the saline group of
animals. This effect, however, does not reach significance vs.
total AKt proteins. Activation of SHH pathway by PM enhances the
expression of p-AKt protein, which can be reversed by the SHH
pathway inhibitor cyclopamine. No change are observed in total AKt
proteins after PM and cyclopamine treatments (Fig. 1A, D and G). Western blot analysis
shows that treatment of BV2 cells with LY294002 suppresses the SHH
signaling mediated by the AKt activation and accelerates the LPS
mediated AKt signalling suppression (Fig. 1B, E and H).
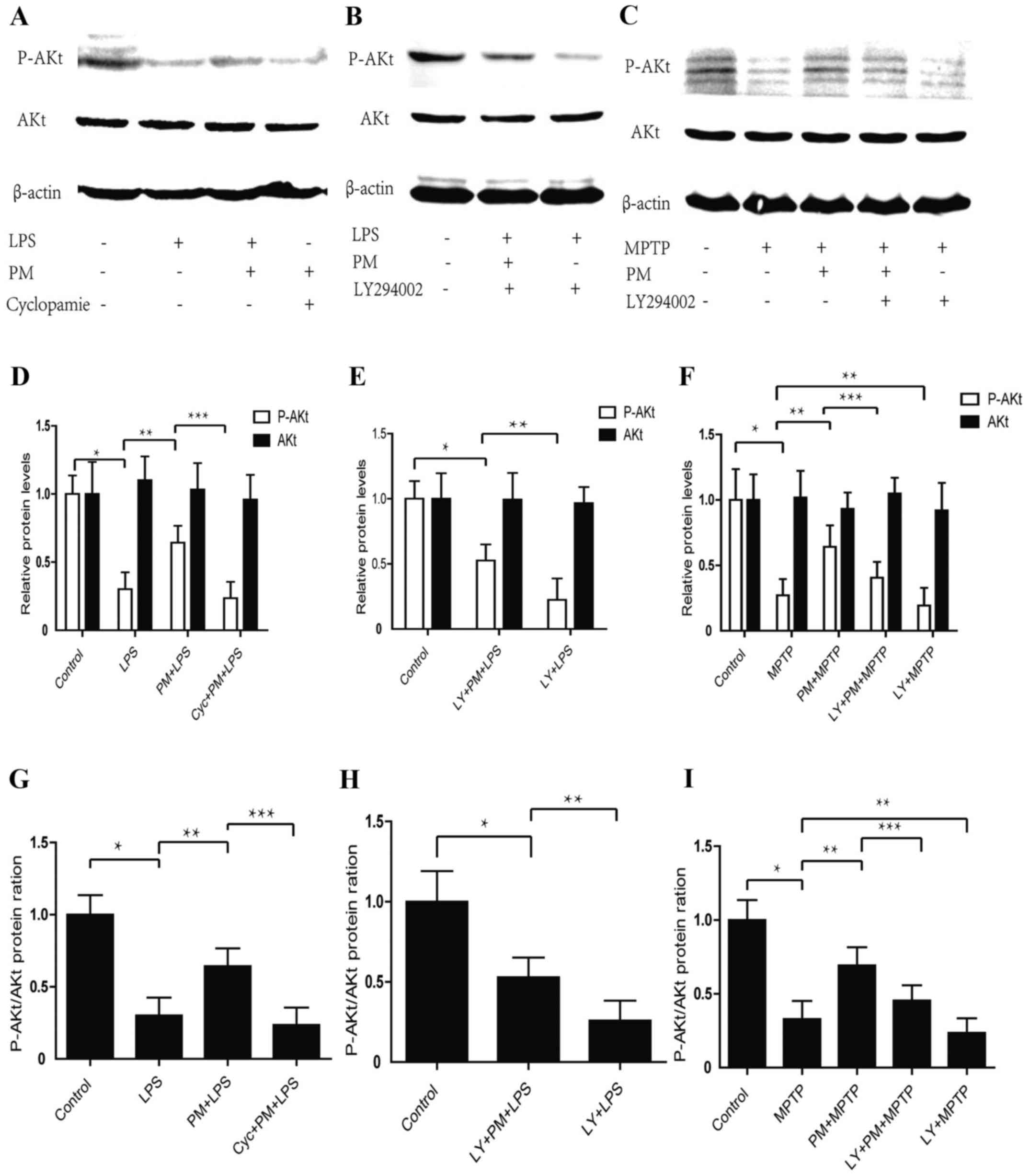 | Figure 1.Purmorphamine (PM) and LY294002
respectively activates and inhibits Sonic hedgehog (SHH) signaling
by way of PI3K/AKt pathway either in vitro or in
vivo. (A) BV2 cells successive treatments were as it follows:
At first, Cyclopamine (20 µmol/l) was given; 1 h later PM (1.5
µmol) was added; after a 24 h time lapse LPS (1 µg/ml) was applied;
24 h after this last treatment AKt phosphorylation was checked by
Western blot and β-actin was used as an internal control. (B) BV2
cells successive treatment were as it follows: At first, LY294002
(20 µmol/l); 0.5 h later a BV2 plot received PM (1.5 µmol) while a
second plot did not received this substance; LPS (1 µg/ml) was
finally added to both plots for a 24 h time lapse. After LPS
treatment AKt phosphorylation was assessed by Western blot. (C)
C57/BL6 mouse received the following successive treatments: At
first, LY294002 (5 mg/ml, 30 µl) in nasal cavity, or saline 0.9%
i.p., were given; 1 h later, PM (1 mg/kg) was administered;
finally, 24 h later, four injections of MPTP (20 mg/kg, i.p.) were
achieved within a practical time lapse of 2 h. Mice were then
sacrificed and the entire SNpc collected to achieve AKt
phosphorylation by Western blot. (D-F) Relative levels of AKt
protein and P-AKt in vivo and in vitro. (G-I) Protein
ratio of P-AKt/AKt in vivo and in vitro. *P<0.01
compared with control group; **P<0.01 compared with LPS or MPTP
group or LY+PM+LPS; ***P<0.01 compared with PM+LPS or PM+MPTP
group; #P<0.01 compared with LY+PM+LPS group. PM,
Purmorphamine; LY, LY294002; LPS, lipopolysaccharide; i.p.,
intraperitoneally injections; SNpc, substantia nigra pars
compacta. |
We also checked whether the SHH pathway regulated
the expression of p-AKt protein in the area of SNpc in mice model
of PD. As shown in Fig. 1C, F and
I, MPTP causes a significant decrease in the level of p-AKt,
activation of SHH signaling and a significant increase in the level
of p-AKt protein when compared with MPTP-treated group of animals.
To analyze how are combined PI3K/Akt and SHH signalings, we
employed an intranasal administration of LY294002 to inhibit
PI3K/Akt component. Western blot analysis shows that intranasal
administration of LY294002 abrogated the PM-induced restoration of
p-AKt and accelerates MPTP mediated P-AKt inhibition. These results
indicate that inflammatory response may inhibit the PI3K/Akt
signaling while the SHH pathway may activate the PI3K/Akt signaling
in vitro and in vivo.
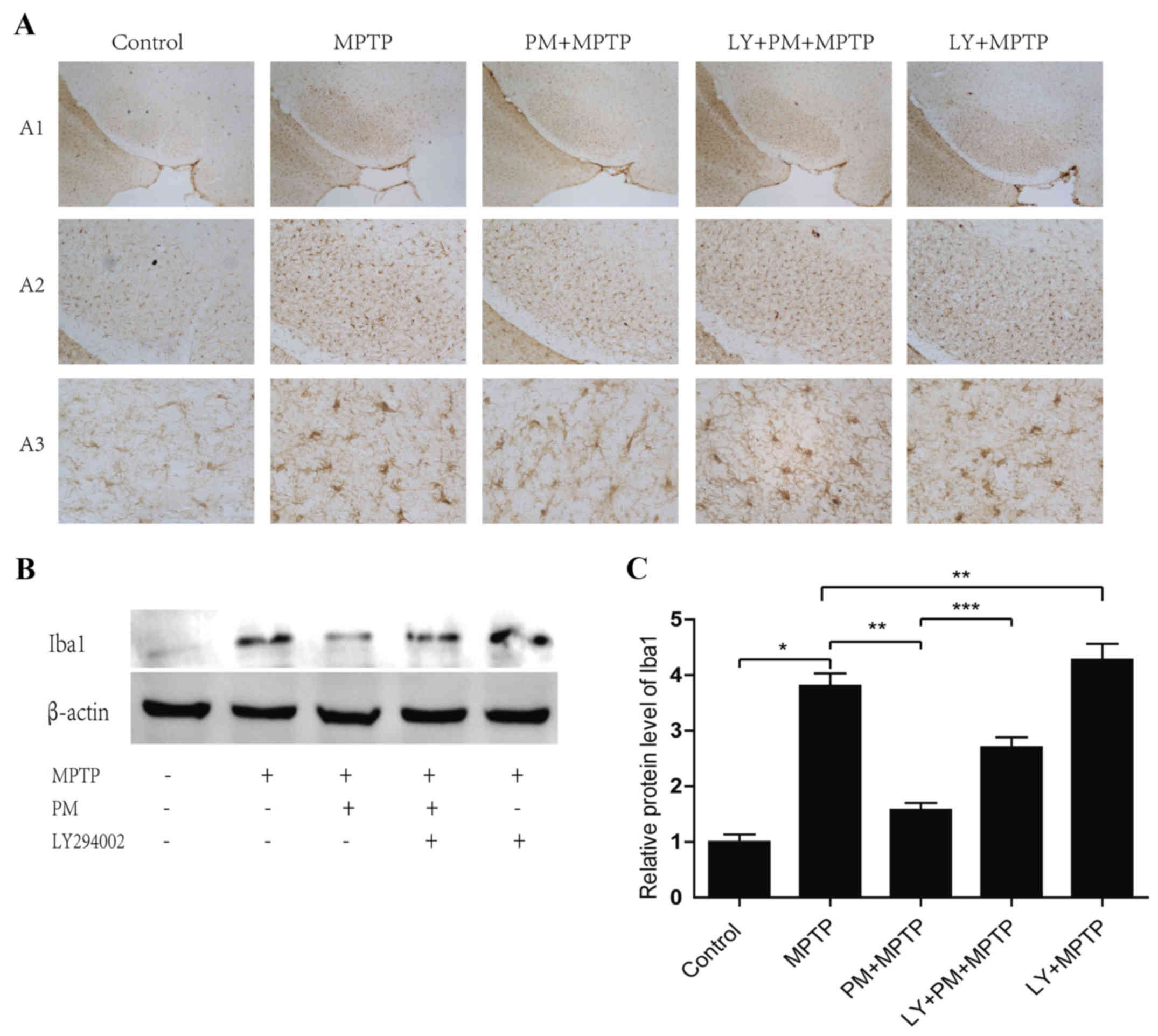
SHH signaling pathway inhibits
microglial activation through the PI3K/Akt signaling pathway
As shown in Fig.
2A, a large number of Iba1-positive microglial cells
characterized by amoeboid shape with larger cell bodies and shorter
processes are observed in MPTP group compared with saline group. In
Mice treated with PM and MPTP, small microglial cell with reduced
cell bodies, ramifications and thin processes are also observed. By
contrast, numerous activated microglial cells are accompanied by
enlarged cell bodies and thicker processes in LY+PM+MPTP group.
Morphology of Iba1-positive microglial cells in LY+PM+MPTP group
are similar to MPTP and LY+MPTP groups.
Western blot reveals that LY294002 blocked the
effect of PM treatment on microglial cells: The Iba1 protein is
significantly increased in the MPTP group. This effect, however, is
also observed when the PM group is inhibited by LY294002. LY294002
increases the expression of Iba1 protein compared with MPTP group
alone (Fig. 2B and C). These
results indicate that SHH-PI3K/Akt relayed signaling inhibit
activation of the microglia.
SHH signaling mediated
anti-inflammatory effects through the PI3K/Akt signaling
pathway
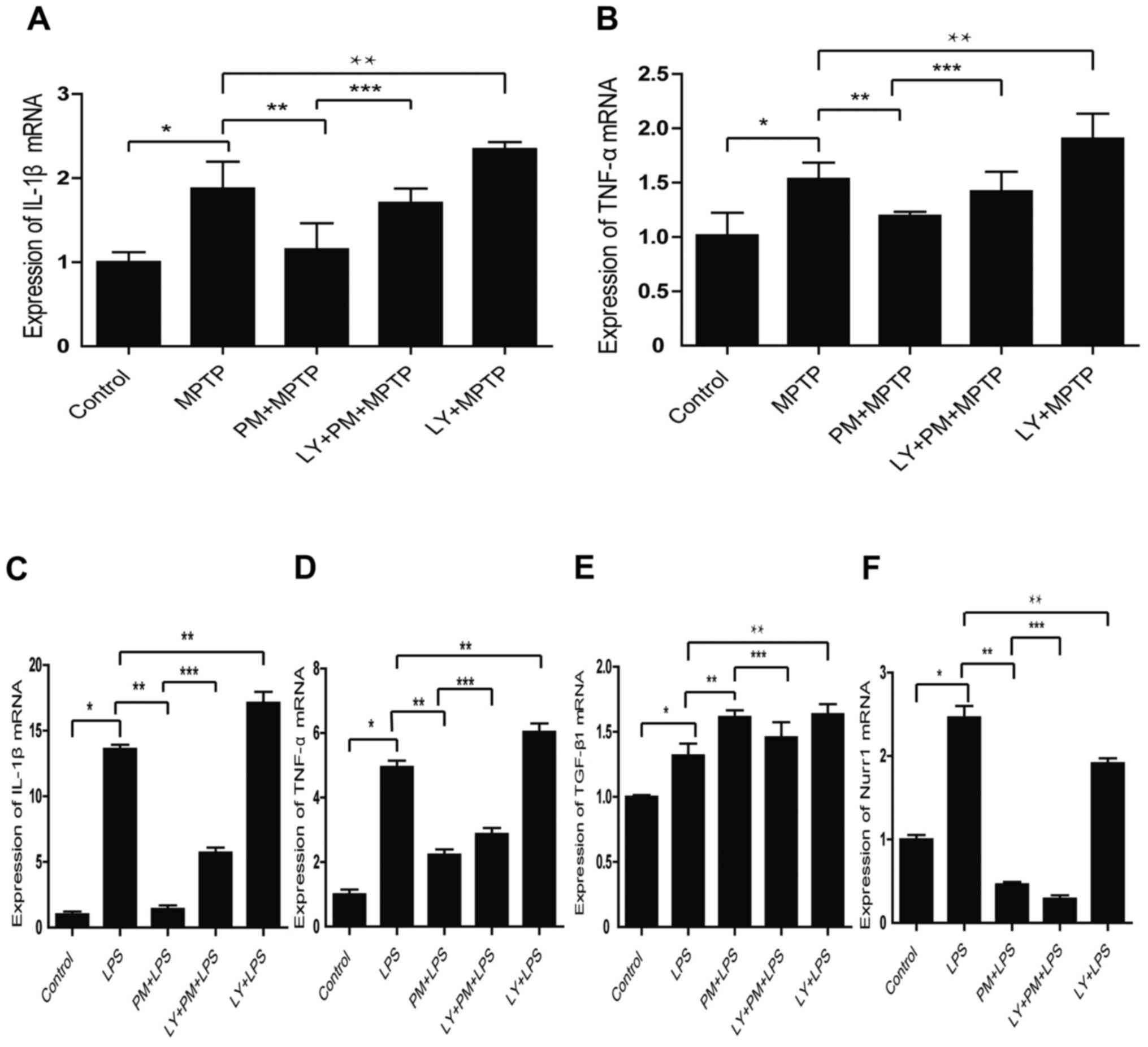
To check this aspect, SNpc tissue homogenate from
our five group of animals were performed. As shown in Fig. 3A and B PM pretreatment decreases
the expression of IL-1β and TNF-α mRNA. This aspect is reversed by
LY294002. Moreover, the inhibition of the PI3K/Akt pathway with
LY294002 in LPS treated group increases the expression of IL-1β and
TNF-α mRNA compared with LPS treated group.
We also investigated the role of PI3K/Akt signaling
in vitro. As expected, the results obtained indicate that
SHH pathway mediates the expression of pro-inflammatory factors
IL-1β, TNF-α mRNA through the PI3K/Akt signaling (Fig. 3C and D). Moreover, the inhibition
of the PI3K/Akt signaling with LY294002 reverses the increased
expression of the anti-inflammatory factor TGF-β1, induced by PM
(Fig. 3E). Finally, it appears
that SHH signaling inhibits Nurr1 independently of the PI3K/AKt
pathway (Fig. 3F).
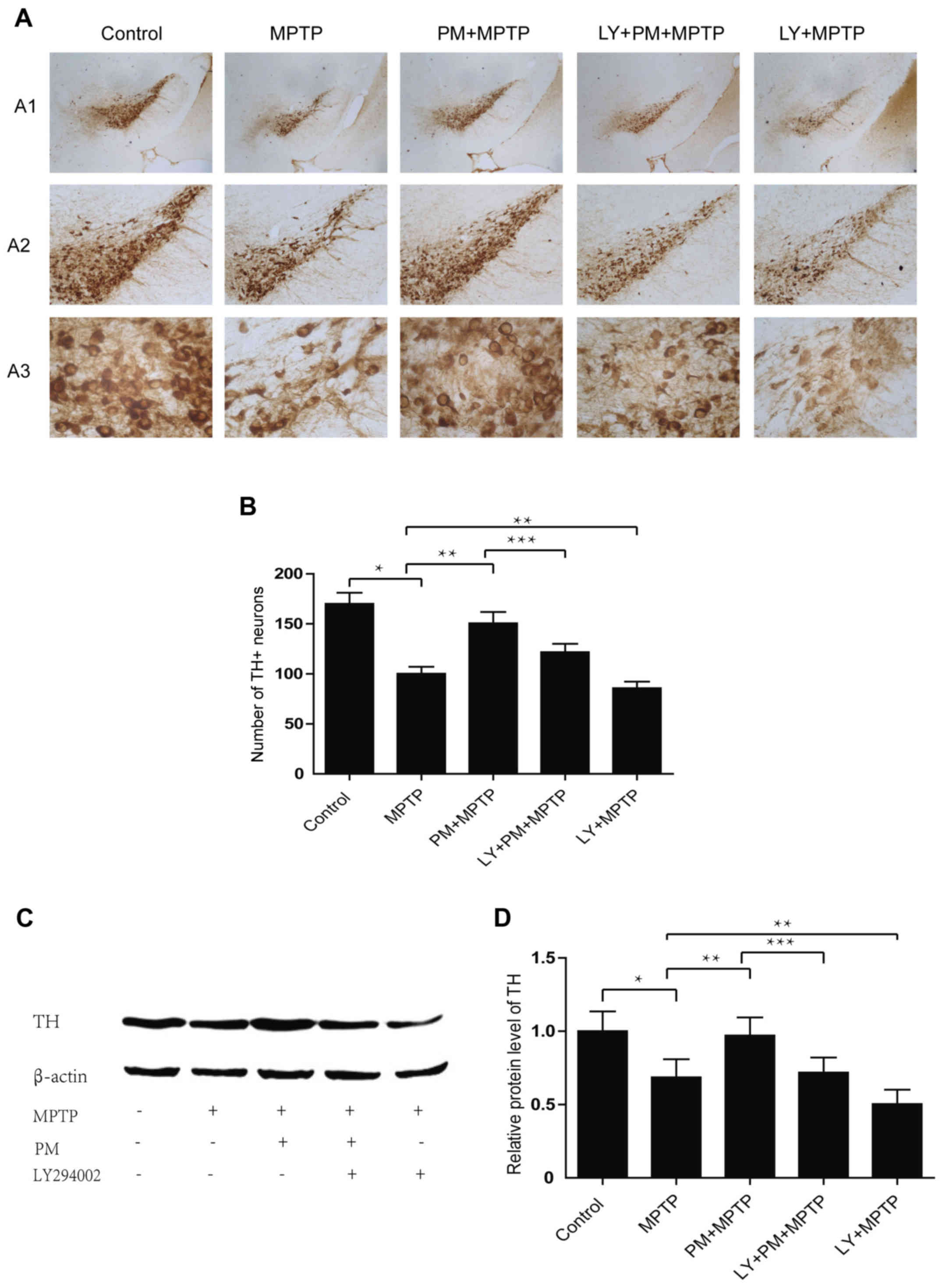
SHH signaling mediated neuroprotection
through the PI3K/Akt signaling pathway
TH immunohistochemistry reveals a significant cell
reduction in MPTP group compared with the saline one. Treatment
with PM protects dopaminergic neurons of the SNpc and enhances
their rate of survival. However, pretreatment with LY294002
abolishes the neuroprotection exerted by PM in the model of PD and
also decreases the number of TH-positive neurons compared with MPTP
group (Fig. 4A and B). Results
established with the western blotting are similar to those obtained
with immunohistochemistry. They show that MPTP causes a significant
decrease in the level of TH protein. PM treatment accelerates the
restoration TH protein level, but the increase in TH protein level
in the PM group of animals is inhibited by the administration of
LY294002. This last substance when administered alone, reduces also
the expression of TH protein when compared with the MPTP group of
animals (Fig. 4C and D).
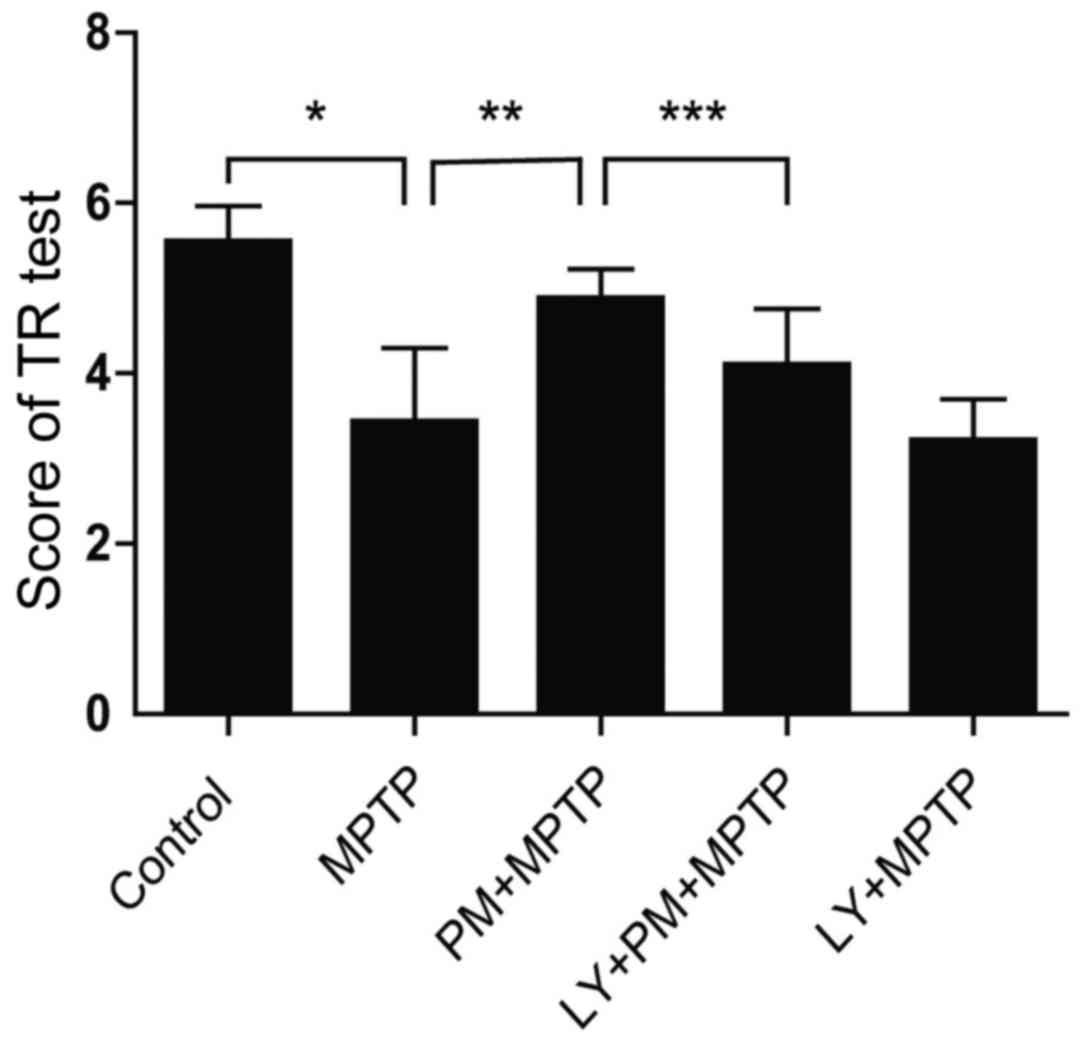
SHH signaling pathway induced
behavioral change through the PI3K/Akt signaling pathway in
MPTP-treated mice
With TR test, mice treated with MPTP spend
significantly less time on the steel compared with those treated
with saline (Fig. 5). This result
indicates an impairment of motor balance and coordination. However,
PM treatment rescued the MPTP-induced motor deficit, as reflected
by the marked increase in the time spent on the steel. This effect
can be abrogated by LY294002. Finally, TR test score was not
statistically different between MPTP and LY+MPTP groups. These data
support clearly that SHH-mediated motor function improvements are
dependent on PI3K/Akt signaling pathway.
Discussion
In the present study, we further investigated the
part played by the SHH signaling in vitro, in using BV2
microglial cells treated with LPS and, in vivo, in using the
MPTP to induced the animal model of PD. We found that SHH signaling
exerts anti-inflammatory and neuroprotective effects either in
vitro or in vivo, at least partly, through PI3K/Akt
signaling pathway. We also evidenced that SHH signaling pathway may
activate the PI3K/Akt signaling and restore the level of P-AKt
which is repressed in neuroinflammatory conditions and in animal
models of PD.
Data of literature, in keeping with our results, now
point out that neuroinflammation, triggered by microglial
activation, may play a central role in the progression of the
dopaminergic neurons degeneration in PD pathogenesis (23,24).
These data also report that the microglial activation occurs in the
early stage of PD pathogenesis and precedes the degeneration of
dopaminergic neurons (25). In
addition to these aspects, it is further stated that mesencephalic
dopaminergic neurons express SHH which is necessary to maintain
their structure and function. Different other studies indicate that
the loss of SHH signaling conduces to the dopaminergic neuron
degeneration and that the administration of exogenous SHH protein
increases the survival rate of these neurons in MPTP or 6-OHDA
treated animals (26). Our results
are thus in good agreement with previous approaches showing that
the microglial activation is linked with the progressive
degeneration of the DA neurons in PD pathogenesis (27). Microglial activation may, indeed,
exacerbate dopaminergic degeneration in releasing a large number of
pro-inflammatory cytokines such as IL-1β, IL-6, TNF-α, and NO
(28).
Beside above considerations, our results also point
out that SHH signaling negatively regulated the activation of
microglia through PI3K/Akt pathway. A study of literature
indicating that SHH signaling can alleviate inflammatory response
(29) is in agreement with our
results. Moreover, our results also indicate that the SHH signaling
is capable to downregulate the expression of pro-inflammatory
factors like IL-1β or TNF-α and to upregulate the expression of
anti-inflammatory factors like TGF-β1. Again, they are in keeping
with literature data since TGF-β1 is known as an anti-inflammatory
factor capable to regulate, in animal models of PD, the microglial
activation and reduce the neuro-inflammation through PI3K/Akt
pathway (30). It is to be
noticed, however, that SHH signaling is also capable to inhibit the
expression of Nurr1 independently of the PI3K/AKt pathway.
Finally, our data indicating that the activation of
the SHH signaling is essential to prevent, via the PI3K/AKt
signaling pathway, the MPTP-induced loss of dopaminergic neurons
and the motor deficit are in keeping with those of literature
(15,31). They further specify the nature of
the processes involved.
Apart from the data discussed above, in the full
statement of our results it remains to be further understood: How
the LY294002 inhibitor of the PI3K/Akt signaling pathway may
downregulate the expression of TGF-β1 induced by PM; how this
inhibitor can upregulate the expression of TGF-β1 induced by LPS
in vitro. Additive experiments still remain necessary to
clarify these aspects. Moreover, Nurr1, an orphan nuclear receptor,
reported as critical for the development and maintenance of
dopaminergic neurons (32), also
capable to attenuate the expression of proinflammatory molecules
released by microglial (33),
appears, according to our results, inhibited by the SHH signaling
and independent of the PI3K/AKt signaling pathway. Again, further
experiments are still necessary to clarify these last aspects. They
are part of our prospects.
In conclusion, our study demonstrates that
SHH-PI3K/Akt coupled signalings are capable to protect DA neurons,
inhibit microglia activation and improve motor performances. While
additive experiments remains, obviously, necessary to clarify the
results reported, our research appears promising for a better
understanding of the neurodegenerative pathologies.
Acknowledgments
This study was supported by grants from the
Guangdong Provincial Development of Science and Technology (no.
201301) and the Medical Scientific Research Foundation of Guangdong
Province (no. A2013239). PR Raymond CESPUGLIO contributed to the
manuscript improvement.
References
|
1
|
Mullin S and Schapira AH: Pathogenic
mechanisms of neurodegeneration in Parkinson disease. Neurol Clin.
33:1–17. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Berardelli A, Wenning GK, Antonini A, Berg
D, Bloem BR, Bonifati V, Brooks D, Burn DJ, Colosimo C, Fanciulli
A, et al: EFNS/MDS-ES/ENS [corrected] recommendations for the
diagnosis of Parkinson's disease. Eur J Neurol. 20:16–34. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Terada T, Yokokura M, Yoshikawa E,
Futatsubashi M, Kono S, Konishi T, Miyajima H, Hashizume T and
Ouchi Y: Extrastriatal spreading of microglial activation in
Parkinson's disease: A positron emission tomography study. Ann Nucl
Med. 30:579–587. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tanaka S, Ishii A, Ohtaki H, Shioda S,
Yoshida T and Numazawa S: Activation of microglia induces symptoms
of Parkinson's disease in wild-type, but not in IL-1 knockout mice.
J Neuroinflamm. 10:1432013. View Article : Google Scholar
|
|
5
|
Tang Y and Le W: Differential roles of M1
and M2 microglia in neurodegenerative diseases. Mol Neurobiol.
53:1181–1194. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Varnum MM and Ikezu T: The classification
of microglial activation phenotypes on neurodegeneration and
regeneration in Alzheimer's disease brain. Arch Immunol Ther Exp
(Warsz). 60:251–266. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim BW, Koppula S, Kumar H, Park JY, Kim
IW, More SV, Kim IS, Han SD, Kim SK, Yoon SH and Choi DK: α-Asarone
attenuates microglia-mediated neuroinflammation by inhibiting NF
kappa B activation and mitigates MPTP-induced behavioral deficits
in a mouse model of Parkinson's disease. Neuropharmacology.
97:46–57. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rubin LL and de Sauvage FJ: Targeting the
Hedgehog pathway in cancer. Nat Rev Drug Discov. 5:1026–1033. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
de Sauvage F: The HH signaling pathway in
cancer. Bull Mem Acad R Med Belg. 162:219–223. 2007.PubMed/NCBI
|
|
10
|
Banerjee SB, Rajendran R, Dias BG,
Ladiwala U, Tole S and Vaidya VA: Recruitment of the Sonic hedgehog
signaling cascade in electroconvulsive seizure-mediated regulation
of adult rat hippocampal neurogenesis. Eur J Neurosci.
22:1570–1580. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sims JR, Lee SW, Topalkara K, Qiu J, Xu J,
Zhou Z and Moskowitz MA: Sonic hedgehog regulates
ischemia/hypoxia-induced neural progenitor proliferation. Stroke.
40:3618–3636. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hung HC, Hsiao YH and Gean PW: Sonic
hedgehog signaling regulates amygdalar and extinction of fear
memory. Eur Neuropsychopharmacol. 25:1723–1732. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hung HC, Hsiao YH and Gean PW: Learning
induces sonic hedgehog signaling in the amygdala which promotes
neurogenesis and long-term memory formation. Int J
Neuropsychopharmacol. 18:pyu0712015. View Article : Google Scholar :
|
|
14
|
Dass B, Iravani MM, Jackson MJ, Engber TM,
Galdes A and Jenner P: Behavioural and immunohistochemical changes
following supranigral administration of sonic hedgehog in
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated common
marmosets. Neuroscience. 114:99–109. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Y, Dong W, Guo S, Zhao S, He S,
Zhang L, Tang Y and Wang H: Lentivirus-mediated delivery of sonic
hedgehog into the striatum stimulates neuroregeneration in a rat
model of Parkinson disease. Neurol Sci. 35:1931–1940. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schabbauer G, Tencati M, Pedersen B,
Pawlinski R and Mackman N: PI3K-Akt pathway suppresses coagulation
and inflammation in endotoxemic mice. Arterioscler Thromb Vasc
Biol. 24:1963–1969. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim SN, Kim ST, Doo AR, Park JY, Moon W,
Chae Y, Yin CS, Lee H and Park HJ: Phosphatidylinositol
3-kinase/Akt signaling pathway mediates acupuncture-induced
dopaminergic neuron protection and motor function improvement in a
mouse model of Parkinson's disease. Int J Neurosci. 121:562–569.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao Y, Zhang Q, Xi J, Xiao B, Li Y and Ma
C: Neuroprotective effect of fasudil on inflammation through
PI3K/Akt and Wnt/β-catenin dependent pathways in a mice model of
Parkinson's disease. Int J Clin Exp Pathol. 8:2354–2364.
2015.PubMed/NCBI
|
|
19
|
Dai R, Xia Y, Mao L, Mei Y, Xue Y and Hu
B: Involvement of PI3K/Akt pathway in the neuroprotective effect of
Sonic hedgehog on cortical neurons underoxidative stress. J
Huazhong Univ Sci Technolog Med Sci. 32:856–860. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xia YP, Dai RL, Li YN, Mao L, Xue YM, He
QW, Huang M, Huang Y, Mei YW and Hu B: The protective effect of
sonic hedgehog is mediated by the phosphoinositide [corrected]
3-kinase/AKT/Bcl-2 pathway in cultured rat astrocytes under
oxidative stress. Neuroscience. 209:1–11. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guo Z, Xu S, Du N, Liu J, Huang Y and Han
M: Neuroprotective effects of stemazole in the MPTP induced acute
model of Parkinson's disease: Involvement of the dopamine system.
Neurosci Lett. 616:152–159. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kuribara H, Higuchi Y and Tadokoro S:
Effects of central depressants on rota-rod and traction
performances in mice. Jpn J Pharmacol. 27:117–126. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ouchi Y, Yoshikawa E, Sekine Y,
Futatsubashi M, Kanno T, Ogusu T and Torizuka T: Microglial
activation and dopamine terminal lossin early Parkinson's disease.
Ann Neurol. 57:168–75. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ouchi Y, Yagi S, Yokokura M and Sakamoto
M: Neuroinflammation in the living brain of Parkinson's disease.
Parkinsonism Relat Disord. 15 Suppl 3:S200–S204. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cappellano G, Carecchio M, Fleetwood T,
Magistrelli L, Cantello R, Dianzani U and Cormi C: Immunity and
inflammation in neurodegenerative disease. Am J Neurodegener Dis.
21:89–107. 2013.
|
|
26
|
Gonzalez-Reyes LE, Verbitsky M, Blesa J,
Jackson-Lewis V, Paredes D, Tillack K, Phani S, Kramer ER,
Przedborski S and Kottmann AH: Sonic hedgehog maintains cellular
and neurochemical homeostasis in the adult nigrostriatal circuit.
Neuron. 75:306–319. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gordon R, Singh N, Lawana V, Ghosh A,
Harischandra DS, Jin H, Hogan C, Sarkar S, Rokad D, Panicker N, et
al: Protein kinase Cd upregulation in microglia drives
neuroinflammatory responses and dopaminergic neurodegeneration in
experimental models of Parkinson's disease. Neurobiol Dis.
93:96–114. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim BW, Koppula S, Park SY, Kim YS, Park
PJ, Lim JH, Kim IS and Choi DK: Attenuation of neuroinflammatory
responses and behavioral deficits by Ligusticum officinale (Makino)
Kitag in stimulated microglia and MPTP-induced mouse model of
Parkinson's disease. J Ethnopharmacol. 164:388–397. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou X, Liu Z, Jang F, Xiang C, Li Y and
He Y: Autocrine Sonic hedgehog attenuates inflammation in
cerulein-induced acute pancreatitis in mice via upregulation of
IL-10. PLoS One. 7:e441212012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Haas SJ, Zhou X, Machado V, Wree A,
Krieglstein K and Spittau B: Expression of Tgfβ1 and inflammatory
markers in the 6-hydroxydopamine mouse model of Parkinson's
disease. Front Mol Neurosci. 9:72016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Quesada A, Lee BY and Micevych PE: PI3
kinase/Akt activation mediates estrogen and IGF-1 nigral DA
neuronal neuroprotection against a unilateral rat model of
Parkinson's disease. Dev Neurobiol. 68:632–644. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim CH, Han BS, Moon J, Kim DJ, Shin J,
Rajan S, Nguyen QT, Sohn M, Kim WG, Han M, et al: Nuclear receptor
Nurr1 agonists enhance its dual functions and improve behavioral
deficits in an animal model of Parkinson's disease. Proc Natl Acad
Sci USA. 112:8756–8761. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Maguire-Zeiss KA and Federoff HJ: Future
directions for immune modulation in neurodegenerative disorders:
Focus on Parkinson's disease. J Neural Transm (Vienna).
117:1019–1025. 2010. View Article : Google Scholar : PubMed/NCBI
|