Introduction
Chronic kidney failure, the endpoint of chronic
kidney disease (CKD), is defined as a glomerular filtration rate
persistently below 15 ml/min per 1.73 m2. Renal
replacement therapy (RRT) is achieved by hemodialysis,
hemodiafiltration, peritoneal dialysis or kidney transplantation,
and can be lifesaving for patients with CKD. However, the mortality
rate for patients on RRT remains high, and <25% of patients with
chronic kidney failure undergo RRT in developing countries
(1).
Renal fibrosis is a complicated process
characterized by fibroblast proliferation and the accumulation of
extracellular matrix (ECM). Collectively, these changes lead to
renal tubule fibrosis, glomerular sclerosis, renal artery stenosis
and chronic inflammatory cell infiltration (2). It is currently believed that renal
fibrosis develops in response to ECM accumulation due to
epithelial-mesenchymal transition (EMT) (3), transforming growth factor (TGF)-β
signaling (4), oxidative stress
(5) and proteinuria (6). Endoplasmic reticulum stress (ERS) is
a physiological or pathological condition that can be caused by
glucose deprivation, hypoxia or virus infection. Recent studies
have shown that ERS plays a vital role in the development of CKD
(7,8). Furthermore, inhibition of ERS can
alleviate renal fibrosis progression (9–11).
Although a rich body of knowledge regarding the relationship
between ERS and CKD has been accumulated to date, how ERS
contributes to renal fibrosis has not been fully elucidated. In the
present review, the authors summarize the latest understanding of
the role of ERS during the onset of renal fibrosis, and propose
that targeted inhibition of ERS may become a promising therapeutic
strategy for renal fibrosis.
The cause and role of ERS
The ER performs a variety of cellular function,
including the regulation of protein biosynthesis, folding
trafficking and modification (8).
The ER is implicated in several cellular processes via three major
types of signaling: 1) Protein kinase RNA-like endoplasmic
reticulum kinase (PERK) signaling, 2) inositol-requiring enzyme 1
(IRE1) signaling, and 3) activating transcription factor (ATF) 6
signaling (12). The PERK pathway
is activated by PERK autophosphorylation, PERK then phosphorylates
eukaryotic initiation factor-2α (eIF2α). Phosphorylated eIF2α
promotes the expression of apoptotic proteins such as
CCAAT-enhancer-binding protein homologous protein (CHOP).
Similarly, the IRE1 pathway is activated by autophosphorylation of
IRE1, which subsequently induces unconventional splicing of
X-box-binding protein-1 (XBP1) mRNA (13). In contrast, ATF6 signaling is
initiated by ATF6 cleavage by Site 1 protease and Site 2 protease
in the Golgi apparatus, and the cleaved subunit (p50ATF6) functions
as a transcription factor (14).
ER function can change in response to environmental stimuli, such
as ischemia, glucose deprivation, or oxidative stress, as well as
to genetic mutations, which can result in abnormal protein folding
(15). Accumulation of misfolded
proteins in the ER lumen induces a range of ER dysfunctions,
collectively referred to as ERS (16). To ensure the fidelity of protein
folding and prevent the accumulation of unfolded or misfolded
proteins, cells experiencing ERS invoke a well-conserved adaptive
response known as the unfolded protein response (UPR) to ameliorate
cell damage and restore homeostasis (17).
Under prolonged or severe ERS, a variety of
molecular chaperones accumulate in the ER lumen, such as
glucose-regulated protein 78 (GRP78), GRP94 and calreticulin, which
prevent the accumulation of unfolded proteins and facilitate
protein folding (18). Moreover,
if these adaptive responses fail to alleviate ERS, apoptotic
pathways are activated to eliminate damaged cells. CHOP is thought
to be a critical mediator of ERS-induced apoptosis (19).
ERS has a crucial role in kidney diseases. ERS can
cause cellular damage and lead to renal fibrosis in podocytes,
renal tubular cells, glomerular endothelial cells (GEnCs) and
mesangial cells (11,20,21),
whereas inhibition of ERS can ameliorate renal fibrosis progression
(9,10). Collectively, these findings
suggested that the inhibition of molecular chaperones during UPR or
directly blocking ERS may offer new therapeutic strategies for
renal fibrosis.
Relationship between TGF-β and ERS
TGF-β is the primary cytokine that causes fibrosis.
Smad proteins are highly conserved transcription factors that are
central to signal transduction pathways that mediate the numerous
effects of TGF-β superfamily (22). For example, the inhibition of ERS
reduced TGF-β activity in an angiotensin II reperfusion model,
thereby alleviating myocardial hypertrophy and fibrosis (23). TGF-β induced GRP78 expression in
human and mouse lung fibroblasts, while the inhibition of GRP78
expression significantly reduced the expression of collagen and
α-smooth muscle actin (α-SMA), two biomarkers of fibrosis (24). These results indicate that ERS
serves a pro-fibrogenic role.
Prolonged or severe ERS can cause kidney cells to
undergo apoptosis, resulting in renal fibrosis, while the
inhibition of ERS can delay fibrosis development (6,25).
ERS-associated proapoptotic signals, including B-cell chronic
lymphocytic leukemia/lymphoma 2-associated protein (BAX) expression
and caspase-12 and c-Jun N-terminal kinase (JNK) phosphorylation,
are activated in the unilateral ureteral obstructed (UUO) kidney.
Prolonged ERS attenuates the expression of both unspliced and
spliced XBP-1. In addition, prolonged ERS activates IRE1α-JNK
phosphorylation and the expression of protein kinase RNA-like
endoplasmic reticulum kinase (PERK), eIF2α, ATF-4, CHOP and
cleavage activating transcription factor 6 (cATF6)-CHOP,
collectively resulting in ERS-induced apoptosis. TGF-β markedly
increases the expression of ERS-associated proteins, such as GRP78,
CHOP, ATF4, spliced XBP1 and various profibrotic factors, such as
α-SMA and connective tissue growth factor. Many of these proteins
serve as apoptotic markers in renal tubular cells (8). TGF-β also activates ERS-mediated
proapoptotic signaling via the JNK pathway, which serves an
important role in renal fibrosis and mesangial cell apoptosis
(26). Furthermore, ERS promotes
apoptosis in GEnCs by regulating TGF-β expression (21), ultimately causing renal fibrosis.
Sox4 (SRY-related HMG box 4) is required for TGF-β-induced
apoptosis, and is widely expressed during mouse embryogenesis and
functions in the development of many tissues (27). A recent study reported that TGF-β
can induce Sox4 expression, causing pro-apoptotic responses in
pancreatic cancer cells associated with EMT-linked repression of
Kruppel-like factor 5 (28). It
remains unclear whether this mechanism of apoptosis is related to
ERS-induced renal fibrosis.
ERS may promote kidney fibroblast differentiation
and collagen formation via the upregulation of TGF-β (29,30).
Studies based on a renal fibrosis model induced by asymmetric
dimethylarginine (ADMA) demonstrated that ADMA could activate ERS
sensor proteins PERK and IRE1α, which then induce CHOP expression
and JNK phosphorylation in GenCs and mesangial cells (21,31).
These changes resulted in increased TGF-β expression, and ADMA
promoted ERS-related apoptosis by increasing TGF-β expression
(Fig. 1) (21). In contrast, TGF-β has been shown to
improve ischemia/reperfusion (I/R)-induced myocardial contractile
dysfunction in H9c2 cells via the inhibition of ERS-dependent
markers of apoptosis (GRP78, CHOP, caspase-12, and JNK) and the
modulation of Bcl2/Bax expression (32). Further studies are needed to reveal
the relationship between ERS and TGF-β in different contexts.
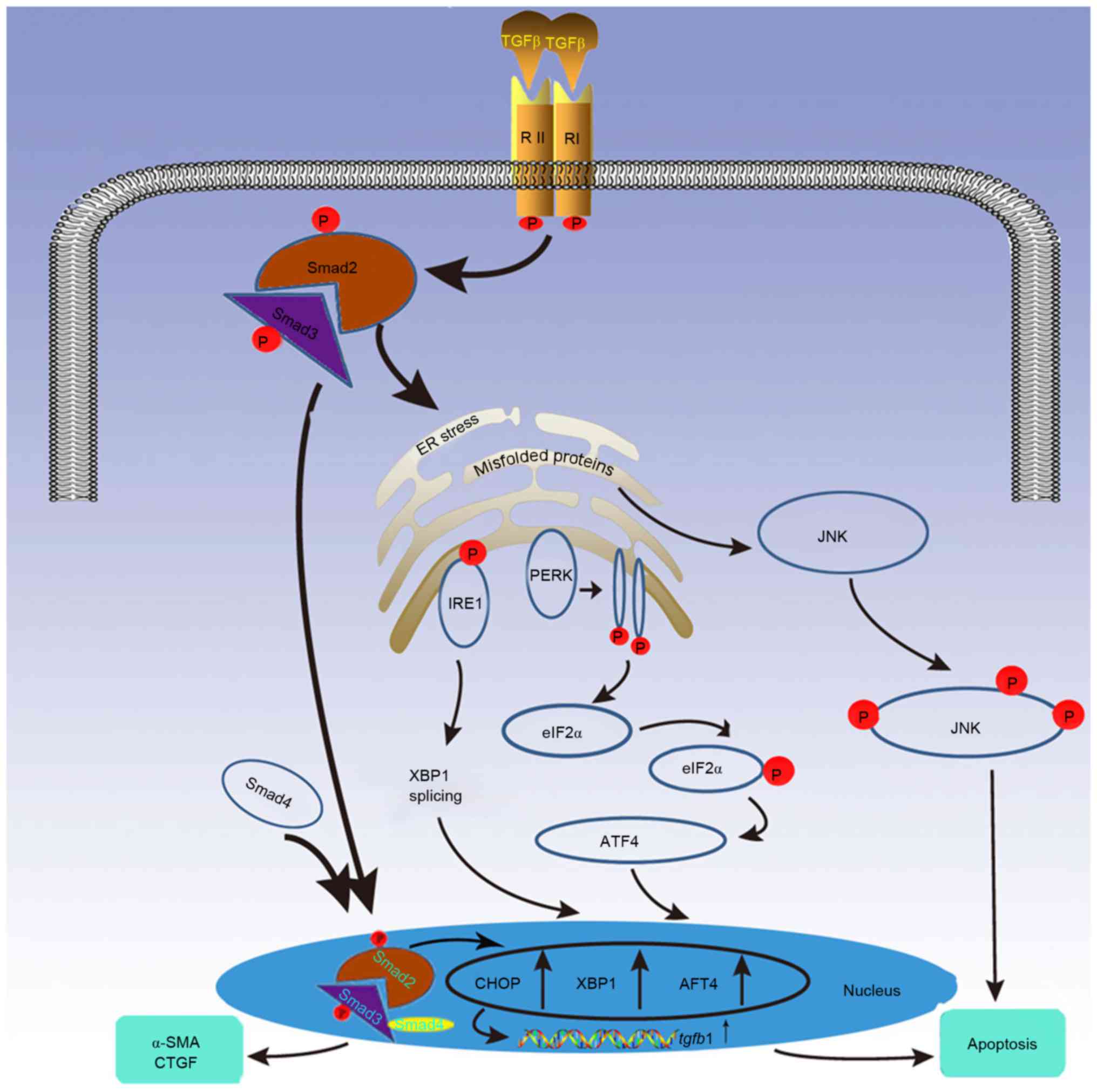 | Figure 1.TGF-β signaling induced apoptosis is
modulated by the UPR cascade. Under severe or prolonged endoplasmic
reticulum stress caused by TGF-β signaling, UPR stimulates
apoptosis through the following 3 proteins: PERK, IRE1 and ATF4.
These proteins increase the expression of transcription factors
CHOP, XBP1 and ATF4, which promote apoptosis and upregulate TGF-β1
expression, thus augmenting TGF-β signaling. Additionally, UPR
propagates apoptosis via the JNK signaling pathway, which can also
be induced by TGF-β. TGF-β, transforming growth factor-β; UPR,
unfolded protein response; PERK, protein kinase RNA-like
endoplasmic reticulum kinase; IRE1, inositol-requiring enzyme 1;
ATF, activating transcription factor; CHOP, CCAAT-enhancer-binding
protein homologous protein; XBP1, X-box-binding protein-1; JNK,
c-Jun N-terminal kinase; α-SMA, α-smooth muscle actin. |
Relationship between ERS and EMT
During EMT, renal epithelial cells are transformed
into mesenchymal cells. These mesenchymal cells serve to ameliorate
tissue damage, causing ECM accumulation and the production of
myofibroblasts, which are key effectors in ECM synthesis and
deposition (33). EMT is
characterized by a reduction in cell-cell contact, splitting of the
basement membrane, reduced expression of proteins such as
E-cadherin, nephrin, podocin and zonula occludens-1, cytoskeletal
reorganization, transition to a spindle-shaped morphology, and
increased expression of mesenchymal markers such as α-SMA,
vimentin, type I collagen and fibronectin (33). Although a large number of studies
have indicated that EMT in the kidney may lead to renal fibrosis,
the relationship remains controversial (3).
ERS mediates EMT via several pathways. Advanced
oxidation protein products (AOPPs) are formed in response to the
reactions between plasma albumin and chlorinated oxidants during
oxidative stress (34). AOPPs have
been reported to induce podocyte apoptosis, mesangial cell
perturbation (35), distal renal
tubular epithelial cell hypertrophy and EMT (36). Li et al (37) showed that AOPPs can promote the
progression of early diabetic renal fibrosis. This may be because
AOPPs induce ERS by increasing GRP78 and CHOP expression, which
causes human renal glomerular endothelial cells to undergo EMT as
they age or are exposed to hyperglycemic conditions (35). ERS mediates EMT by decreasing
E-cadherin expression and increasing α-SMA expression. These
AOPP-associated effects can be reversed by treating cells with
salubrinal, an inhibitor of ERS, but can be reproduced by treating
cells with thapsigargin (TG), an inducer of ERS (35).
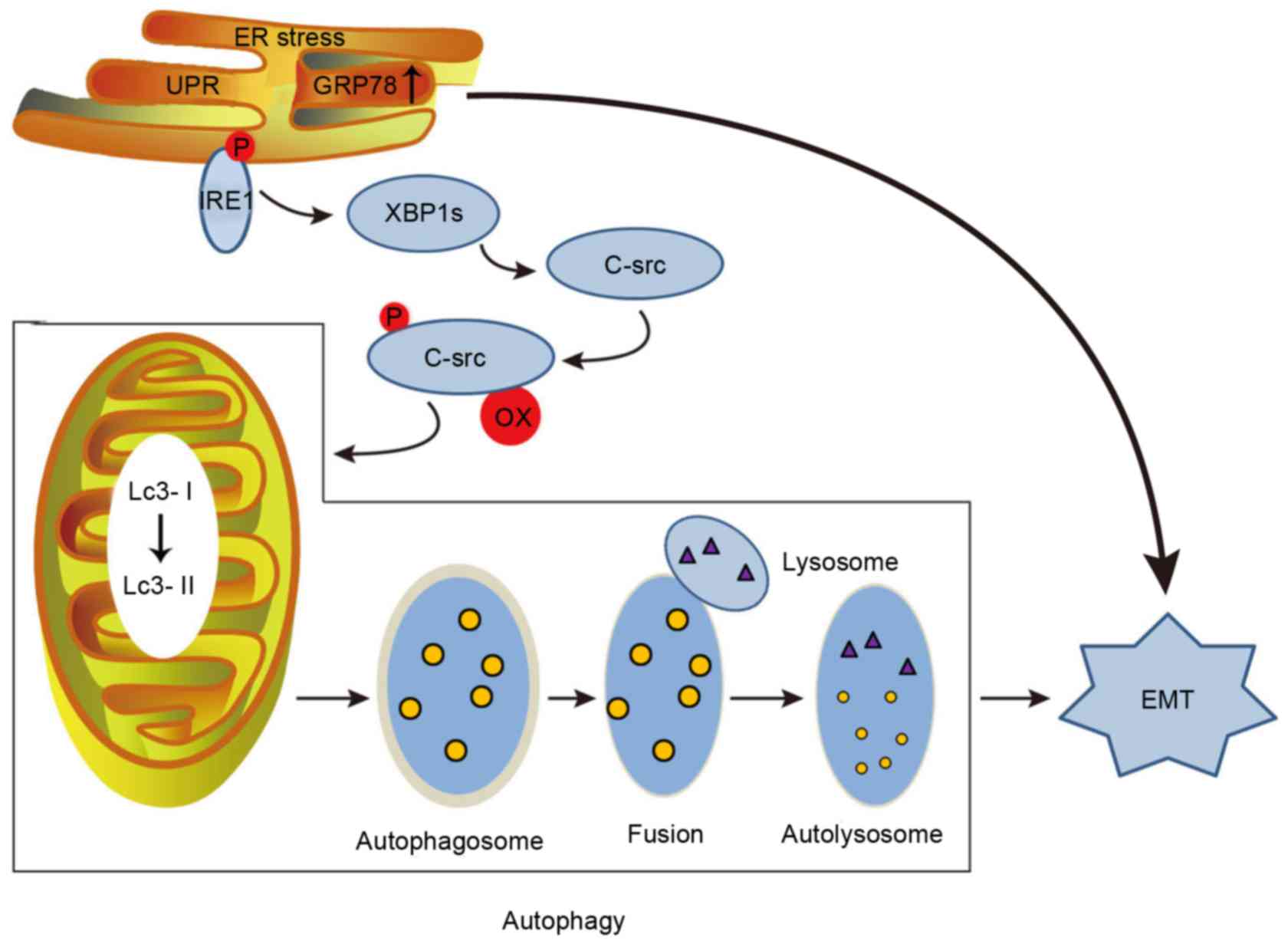
In addition, autophagy is induced during stress, and
it can alternately contribute to cell death or serve as a cellular
survival mechanism (38). ERS
resulting from tunicamycin or TG treatment can induce EMT by
causing autophagy via c-Src kinase activation in tubular epithelial
cells, ultimately leading to renal fibrosis (Fig. 2) (39). Neuronal precursor cell-expressed
developmentally downregulated protein (Nedd) 4–2, a 120 kDa highly
conserved E3 ligase present in eukaryotic cells, has been
demonstrated to regulate membrane availability of a number of ion
channels (40). Nedd4-2 has also
been reported to be involved in renal-tubular metabolism, leading
to kidney injury (41). Wang et
al (42) demonstrated that
Nedd4-2 is upregulated in response to ER stress by the spliced form
of XBP-1, which is important for inducing an appropriate autophagic
response in liver cells. Nonetheless, there is still no any report
on the relationship between ER stress and Nedd4-2 in kidney
cells.
Finally, ERS causes EMT by increasing TGF-β
expression in renal tubular epithelial cells, and TGF-β acts as a
major regulator in inducing EMT through the PI3K/Akt pathway, the
Notch signaling pathway, the Wnt/β-catenin pathway and, perhaps,
the AOPPs (26,43). Snail, Zeb1, and TWIST1, as
transcriptional factors, have been involved in the process of EMT
(44). ERS induces EMT also via
increasing the expression of Snail (45), Zeb1 and TWIST1 in cancer cells
(44).
Relationship between ERS and oxidative
stress
Oxidative stress is usually produced by reactive
oxygen species (ROS). Oxidative stress is common in CKD and serves
an important role in renal fibrosis progression (5). ROS attack, denature and modify the
structure and function of molecules and activate redox-sensitive
transcription factors and signaling pathways, resulting in tissue
injury and dysfunction. These events increase ECM expression and
promote the development of renal fibrosis (5).
ERS promotes ROS production through multiple
pathways. Protein disulfide isomerase (PDI) is an essential enzyme
that mediates disulfide bond formation in the ER. In
chaperone-assisted disulfide bond formation between peptide chains,
two electrons are provided to a cysteine residue within the PDI
active site (46). The transfer of
electrons reduces the PDI active site and causes substrate
oxidation, contributing to ROS production in the ER (47). The NADPH oxidase protein family has
seven members, Nox 1–5 and Duox 1 and 2, among them, Nox4 is most
commonly associated with the ER (47). ERS and ROS production are
fundamental components in the acute and chronic conditions that
result from UPR signaling. Following UPR activation, peripheral
vasculature cells experience increased Nox4 level, which in turn
stimulates ROS production (48).
In addition, oxidative stress has also been indicated to initiate
and contribute to ERS (49).
Therefore, ERS and ROS have a cause-and-effect relationship with
each other.
The interaction between ERS and oxidative stress is
considered the primary driving force of renal fibrosis.
Hyperglycemia commonly leads to oxidative stress. Using a type 2
diabetic rat model, Lee et al (11) reported that ERS and oxidative
stress activated TGF-β/Smad2/3 signaling and increased α-SMA
expression, resulting in kidney cell fibrosis and apoptosis and
diabetic nephropathy (DN). Recent studies have indicated that
aldosterone/mineralocorticoid receptor (MR) is a major contributor
to CKD progression (50). For
instance, oxidant stress-mediated aldosterone/MR-induced podocyte
injury occurs in response to ERS, which then triggers both
apoptosis and autophagy to cope with the injury (50).
Relationship between ERS and
proteinuria
CKD is characterized by abnormalities in the
glomerular filtration barrier that lead to increased glomerular
permeability and abnormal filtration of macromolecules, such as
albumin. Evidence from clinical and experimental studies indicates
that albuminuria and proteinuria are not simply markers of CKD
progression, but rather are active players in the development of
CKD (6). Mechanistically, it has
been proposed that proteins released into the glomerular filtrate
have toxic effects on tubular cells and damaged tubular cells lead
to the development of interstitial fibrosis (51).
Podocyte injury is a major cause of proteinuria, and
ERS serves an essential role in podocyte damage. ERS was presented
to have a close relationship with podocyte injury in a CKD rat
model, ERS induces a series of changes such as the upregulation of
ER chaperone and ERS proteins, increased podocyte apoptosis,
accumulation of mutated and misfolded proteins in the ER, and
disruption of the glomerular filtration structure, leading to
proteinuria (Fig. 3) (52,53).
These findings suggested that UPR augmentation therapy may enhance
ER proteostasis and serve as a new therapeutic approach for renal
fibrosis.
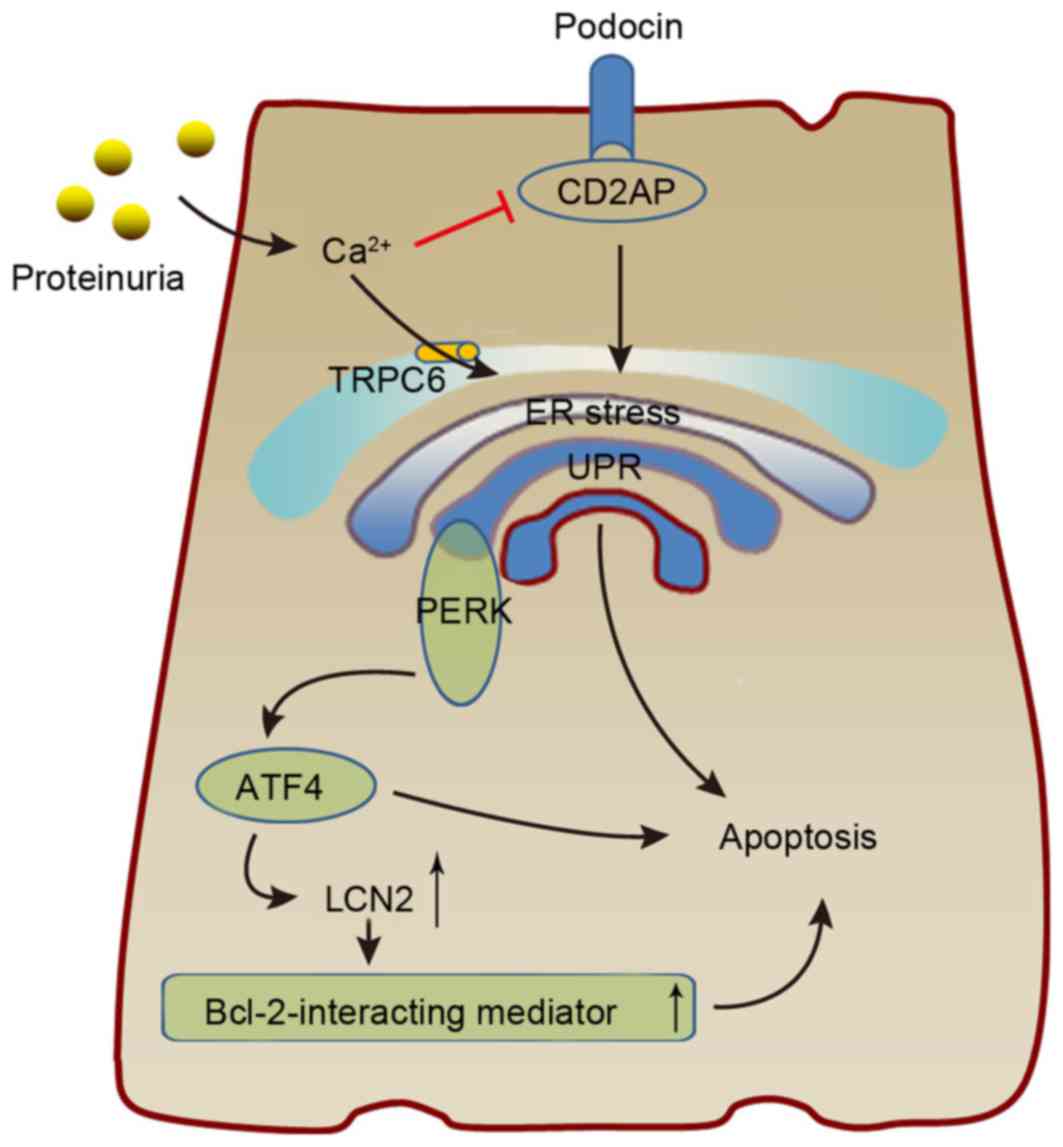 | Figure 3.The possible mechanism by which ERS
results in podocyte damage. Proteinuria causes ERS via
Ca2+, resulting in increased expression of ATF4 and
CHOP, which interact to induce LCN2 expression and cause podocyte
apoptosis. Additionally, increasing Ca2+ levels can
promote podocyte UPR by inhibiting CD2AP, which serves a critical
role in podocyte biology. ERS, endoplasmic reticulum stress; ATF,
activating transcription factor; CHOP, CCAAT-enhancer-binding
protein homologous protein; LCN2, lipocalin 2; UPR, unfolded
protein response; CD2AP, CD2-associated protein; TRPC6, transient
receptor potential C6; PERK, protein kinase RNA-like endoplasmic
reticulum kinase. |
ERS is common in the pathogenic microenvironment and
contributes to the progression of various podocyte diseases. A
proteomic study revealed a series of ERS proteins that are mainly
involved in cytoskeletal rearrangement, suggesting a potential
mechanism by which these proteins cause podocyte dysfunction
(54). In addition, abnormal
protein accumulation associated with ERS in podocytes produces
damage to the cells, which in turn leads to severe proteinuria
(55). Slit diaphragm proteins
such as CD2-associated protein (CD2AP) serve a critical role in
podocyte biology, protein permeability, cell signaling and disease
(52). The transient receptor
potential (TRP) superfamily member TRPC6 is confirmed to be the
primary Ca2+-permeable ion channel in non-excitable
cells. TRPC6, a glomerular slit diaphragm-associated channel
required for normal renal function, is expressed in the cell body,
major processes and foot processes near the slit diaphragm
(56). Downregulation of TRPC6
expression ameliorated puromycin aminonucleoside-induced podocyte
apoptosis (57). Furthermore, in
several studies of albumin overload, which mimics proteinuria,
TRPC6-mediated calcium entry into the cells and CD2AP
downregulation could induce UPR-mediated apoptosis in podocytes
(52,58). ERS also causes podocyte damage
through monocyte chemotactic protein-1, which is associated with
renal fibrosis-related chronic inflammation (59). Moreover, the activation of
rapamycin-sensitive protein kinase complex mTORC1 triggered UPR
activation in podocytes in an animal model of DN, leading to
podocyte injury (60). The carrier
protein neutrophil gelatinase-associated lipocalin (NGAL), also
known as lipocalin (LCN) 2, is associated with renal fibrosis and
has been demonstrated to promote CKD progression in mice and humans
(61). Proteinuria caused by
calcium release-induced ER stress results in LCN2 overexpression,
which in turn leads to tubular apoptosis and renal lesions.
Moreover, 4-phenylbutyric acid (PBA) was reported to delay renal
deterioration in proteinuric mice (61). This may be because albumin induces
ER stress and increases cytosolic Ca2+ concentrations in
cultured podocytes through the activation of TRPC6. In
albumin-overloaded tubular cells exhibiting UPR, ERS increases the
expression of CHOP and ATF4, and these proteins then interact with
other factors to induce LCN2 expression (Fig. 3) (58).
Potential value of ERS for the treatment of
renal fibrosis
Recently, growing evidence has suggested that
inhibiting ERS can alleviate the progression of renal fibrosis.
Downregulation of Klotho, a transmembrane protein primarily
expressed in kidney distal tubular cells, has been indicated to
cause multiple age-associated disorders (10). Aging is related to CKD development,
and Klotho, which has anti-aging properties, has been implicated in
the pathogenesis of various kidney diseases (62). Liu et al (10) reported decreased Klotho expression
and the presence of ERS in a UUO model, and Klotho administration
was able to ameliorate UUO-induced ER stress, inhibit apoptosis and
attenuate renal fibrosis. Another study reported that CHOP
deficiency not only attenuates apoptosis and oxidative stress in
experimental renal fibrosis, but also reduces local inflammation
and ameliorates UUO-induced renal fibrosis (63).
Although some drugs have shown the ability to
suppress ERS, further experimental studies are required. The
chemical chaperone sodium 4-BPA, an aromatic fatty acid analog, has
been used to treat urea cycle disorders because its metabolites
offer an alternative pathway to the urea cycle, allowing for
excretion of excess nitrogen (8).
Liu et al (8) reported that
4-PBA acts as an ER chaperone to ameliorate ERS-induced renal
tubular cell apoptosis and renal fibrosis. Valproate (VPA) is a
histone deacetylation enzyme inhibitor that increases histone
acetylation and promotes gene transcription. VPA has previously
been used as an antiepileptic and anti-tumor treatment. Recently,
Sun et al (9) demonstrated
in a rat model that VPA relieved ERS and reduced renal cell
apoptosis, thereby attenuating renal injury. Furthermore, a
pentacyclic triterpenoid compound oleanolic acid (OA) has shown
therapeutic efficacy for CKD, without apparent side effects.
Several studies have reported that OA has anti-oxidant,
microbicidal, anti-diabetic, anti-inflammatory, hypolipidemic and
anti-atherosclerotic activities (11). In mammals, N-acetylcysteine (NAC,
2-acetamido-3-sulfanylpropanoic acid) is a precursor of
intracellular cysteine and glutathione, and a previous study
reported that NAC has powerful anti-oxidant and protective effects
in β-cells in diabetic db/db mice (64). Lee et al (11) reported that OA and NAC have
potential therapeutic effects for DN based on their anti-oxidant
effects and ability to reduce ERS. Sodium citrate also has a
protective effect on chronic renal failure by affecting ERS
(65).
Mesencephalic astrocyte-derived neurotrophic factor
(MANF) localizes to the ER lumen and is secreted in response to ERS
in several cell types. Using a rat model, Kim et al
(66) demonstrated that MANF can
potentially serve as an urine diagnostic or prognostic biomarker
for ERS-related kidney disease to help stratify disease risk,
predict disease progression, monitor treatment response and
identify subgroups of patients who can be treated with ER stress
modulators in a highly targeted manner.
Conclusions
It is important to note that stress is a
non-specific systemic protection response and ERS acts as a
self-protecting regulatory system to promote cell survival under
different stresses that can result in various pathological
conditions. However, abnormal or excessive ERS would result in
different pathological changes. Therefore, ERS inhibitors not only
inhibit fibrosis in the kidney but also alleviate pathological role
of excessive ERS in other organs. In summary, targeted inhibition
of ERS is of great value for renal fibrosis therapy, and it is
expected that ERS inhibitors with clinical applications will become
available in the future.
Acknowledgements
The authors would like to thank Fuli Luo and Na Zhu
for their assistance in the creation of the present article. The
study was supported by a grant from the National Natural Science
Foundation of China (grant no. 81460142).
References
|
1
|
Ortiz A, Covic A, Fliser D, Fouque D,
Goldsmith D, Kanbay M, Mallamaci F, Massy ZA, Rossignol P,
Vanholder R, et al: Epidemiology, contributors to, and clinical
trials of mortality risk in chronic kidney failure. Lancet.
383:1831–1843. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ke B, Fan C, Yang L and Fang X: Matrix
Metalloproteinases-7 and Kidney Fibrosis. Front Physiol. 8:212017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Menon MC and Ross MJ:
Epithelial-to-mesenchymal transition of tubular epithelial cells in
renal fibrosis: A new twist on an old tale. Kidney Int. 89:263–266.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Meng XM, Huang XR, Xiao J, Chung AC, Qin
W, Chen HY and Lan HY: Disruption of Smad4 impairs TGF-β/Smad3 and
Smad7 transcriptional regulation during renal inflammation and
fibrosis in vivo and in vitro. Kidney Int. 81:266–279. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Habib SL and Abboud HE: Tuberin regulates
reactive oxygen species in renal proximal cells, kidney from
rodents, and kidney from patients with tuberous sclerosis complex.
Cancer Sci. 107:1092–1100. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xiao W, Fan Y, Wang N, Chuang PY, Lee K
and He JC: Knockdown of RTN1A attenuates ER stress and kidney
injury in albumin overload-induced nephropathy. Am J Physiol Renal
Physiol. 310:F409–F415. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chang JW, Kim H, Baek CH, Lee RB, Yang WS
and Lee SK: Up-regulation of SIRT1 reduces endoplasmic reticulum
stress and renal fibrosis. Nephron. 133:116–128. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu SH, Yang CC, Chan DC, Wu CT, Chen LP,
Huang JW, Hung KY and Chiang CK: Chemical chaperon 4-phenylbutyrate
protects against the endoplasmic reticulum stress-mediated renal
fibrosis in vivo and in vitro. Oncotarget. 7:22116–22127.
2016.PubMed/NCBI
|
|
9
|
Sun XY, Qin HJ, Zhang Z, Xu Y, Yang XC,
Zhao DM, Li XN and Sun LK: Valproate attenuates diabetic
nephropathy through inhibition of endoplasmic reticulum
stress-induced apoptosis. Mol Med Rep. 13:661–668. 2016.PubMed/NCBI
|
|
10
|
Liu QF, Ye JM, Deng ZY, Yu LX, Sun Q and
Li SS: Ameliorating effect of Klotho on endoplasmic reticulum
stress and renal fibrosis induced by unilateral ureteral
obstruction. Iran J Kidney Dis. 9:291–297. 2015.PubMed/NCBI
|
|
11
|
Lee ES, Kim HM, Kang JS, Lee EY, Yadav D,
Kwon MH, Kim YM, Kim HS and Chung CH: Oleanolic acid and
N-acetylcysteine ameliorate diabetic nephropathy through reduction
of oxidative stress and endoplasmic reticulum stress in a type 2
diabetic rat model. Nephrol Dial Transplant. 31:391–400. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yoshida H, Matsui T, Yamamoto A, Okada T
and Mori K: XBP1 mRNA is induced by ATF6 and spliced by IRE1 in
response to ER stress to produce a highly active transcription
factor. Cell. 107:881–891. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chiang CK, Hsu SP, Wu CT, Huang JW, Cheng
HT, Chang YW, Hung KY, Wu KD and Liu SH: Endoplasmic reticulum
stress implicated in the development of renal fibrosis. Mol Med.
17:1295–1305. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ye J, Rawson RB, Komuro R, Chen X, Davé
UP, Prywes R, Brown MS and Goldstein JL: ER stress induces cleavage
of membrane-bound ATF6 by the same proteases that process SREBPs.
Mol Cell. 6:1355–1364. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zeeshan HM, Lee GH, Kim HR and Chae HJ:
Endoplasmic reticulum stress and associated ROS. Int J Mol Sci.
17:3272016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kaufman RJ: Stress signaling from the
lumen of the endoplasmic reticulum: Coordination of gene
transcriptional and translational controls. Genes Dev.
13:1211–1233. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Inagi R: Endoplasmic reticulum stress in
the kidney as a novel mediator of kidney injury. Nephron Exp
Nephrol. 112:E1–E9. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Boot-Handford RP and Briggs MD: The
unfolded protein response and its relevance to connective tissue
diseases. Cell Tissue Res. 339:197–211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ghosh AP, Klocke BJ, Ballestas ME and Roth
KA: CHOP potentially co-operates with FOXO3a in neuronal cells to
regulate PUMA and BIM expression in response to ER stress. PLoS
One. 7:e395862012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yuan Y, Xu X, Zhao C, Zhao M, Wang H,
Zhang B, Wang N, Mao H, Zhang A and Xing C: The roles of oxidative
stress, endoplasmic reticulum stress, and autophagy in
aldosterone/mineralocorticoid receptor-induced podocyte injury. Lab
Invest. 95:1374–1386. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guo W, Ding J, Zhang A, Dai W, Liu S, Diao
Z, Wang L, Han X and Liu W: The inhibitory effect of quercetin on
asymmetric dimethylarginine-induced apoptosis is mediated by the
endoplasmic reticulum stress pathway in glomerular endothelial
cells. Int J Mol Sci. 15:484–503. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ke B, Zhang A, Wu X and Fang X: The role
of Krüppel-like factor 4 in renal fibrosis. Front Physiol.
6:3272015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kassan M, Galán M, Partyka M, Saifudeen Z,
Henrion D, Trebak M and Matrougui K: Endoplasmic reticulum stress
is involved in cardiac damage and vascular endothelial dysfunction
in hypertensive mice. Arterioscler Thromb Vasc Biol. 32:1652–1661.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Roberson EC, Tully JE, Guala AS, Reiss JN,
Godburn KE, Pociask DA, Alcorn JF, Riches DW, Dienz O,
Janssen-Heininger YM and Anathy V: Influenza induces endoplasmic
reticulum stress, caspase-12-dependent apoptosis, and c-Jun
N-terminal kinase-mediated transforming growth factor-β release in
lung epithelial cells. Am J Respir Cell Mol Biol. 46:573–581. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Marek I, Lichtneger T, Cordasic N, Hilgers
KF, Volkert G, Fahlbusch F, Rascher W, Hartner A and
Menendez-Castro C: Alpha8 integrin (Itga8) signalling attenuates
chronic renal interstitial fibrosis by reducing fibroblast
activation, not by interfering with regulation of cell turnover.
PLoS One. 11:e01504712016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sutariya B, Jhonsa D and Saraf MN: TGF-β:
The connecting link between nephropathy and fibrosis.
Immunopharmacol Immunotoxicol. 38:39–49. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vervoort SJ, van Boxtel R and Coffer PJ:
The role of SRY-related HMG box transcription factor 4 (SOX4) in
tumorigenesis and metastasis: Friend or foe? Oncogene.
32:3397–3409. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
David CJ, Huang YH, Chen M, Su J, Zou Y,
Bardeesy N, Iacobuzio-Donahue CA and Massagué J: TGF-β tumor
suppression through a lethal EMT. Cell. 164:1015–1030. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Baek HA, Kim DS, Park HS, Jang KY, Kang
MJ, Lee DG, Moon WS, Chae HJ and Chung MJ: Involvement of
endoplasmic reticulum stress in myofibroblastic differentiation of
lung fibroblasts. Am J Respir Cell Mol Biol. 46:731–739. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guo W, Ding J, Zhang A, Dai W, Liu S, Diao
Z, Wang L, Han X and Liu W: The inhibitory effect of quercetin on
asymmetric dimethylarginine-induced apoptosis is mediated by the
endoplasmic reticulum stress pathway in glomerular endothelial
cells. Int J Mol Sci. 15:484–503. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Park MJ, Oh KS, Nho JH, Kim GY and Kim DI:
Asymmetric dimethylarginine (ADMA) treatment induces apoptosis in
cultured rat mesangial cells via endoplasmic reticulum stress
activation. Cell Biol Int. 40:662–670. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang Y, Zong L and Wang X: TGF-β improves
myocardial function and prevents apoptosis induced by
anoxia-reoxygenation, through the reduction of endoplasmic
reticulum stress. Can J Physiol Pharmacol. 94:9–17. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Son H and Moon A: Epithelial-mesenchymal
transition and cell invasion. Toxicol Res. 26:245–252. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cao W, Hou FF and Nie J: AOPPs and the
progression of kidney disease. Kidney Int Suppl (2011). 4:102–106.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liang X, Duan N, Wang Y, Shu S, Xiang X,
Guo T, Yang L, Zhang S, Tang X and Zhang J: Advanced oxidation
protein products induce endothelial-to-mesenchymal transition in
human renal glomerular endothelial cells through induction of
endoplasmic reticulum stress. J Diabetes Complications. 30:573–579.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tang X, Rong G, Bu Y, Zhang S, Zhang M,
Zhang J and Liang X: Advanced oxidation protein products induce
hypertrophy and epithelial-to-mesenchymal transition in human
proximal tubular cells through induction of endoplasmic reticulum
stress. Cell Physiol Biochem. 35:816–828. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li HY, Hou FF, Zhang X, Chen PY, Liu SX,
Feng JX, Liu ZQ, Shan YX, Wang GB, Zhou ZM, et al: Advanced
oxidation protein products accelerate renal fibrosis in a remnant
kidney model. J Am Soc Nephrol. 18:528–538. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Luo B, Lin Y, Jiang S, Huang L, Yao H,
Zhuang Q, Zhao R, Liu H, He C and Lin Z: Endoplasmic reticulum
stress eIF2α-ATF4 pathway-mediated cyclooxygenase-2 induction
regulates cadmium-induced autophagy in kidney. Cell Death Dis.
7:e22512016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Moon SY, Kim HS, Nho KW, Jang YJ and Lee
SK: Endoplasmic reticulum stress induces epithelial-mesenchymal
transition through autophagy via activation of c-Src kinase.
Nephron Exp Nephrol. 126:127–140. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Goel P, Manning JA and Kumar S: NEDD4-2
(NEDD4L): The ubiquitin ligase for multiple membrane proteins.
Gene. 557:1–10. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Al-Qusairi L, Basquin D, Roy A, Stifanelli
M, Rajaram RD, Debonneville A, Nita I, Maillard M, Loffing J,
Subramanya AR and Staub O: Renal-tubular SGK1 deficiency causes
impaired K+ excretion via the loss of regulation of
NEDD4-2/WNK1 and ENaC. Am J Physiol Renal Physiol. 311:F330–F342.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang H, Sun RQ, Camera D, Zeng XY, Jo E,
Chan SM, Herbert TP, Molero JC and Ye JM: Endoplasmic reticulum
stress up-regulates Nedd4-2 to induce autophagy. FASEB J.
30:2549–56. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xian LW, Li TP, Wei YE, Wu SP and Ma L:
Relation of advanced oxidation protein products with VEGF and
TGF-β1 in colon cancer cells exposed to intermittent hypoxia. Nan
Fang Yi Ke Da Xue Xue Bao. 31:619–623. 2011.(In Chinese).
PubMed/NCBI
|
|
44
|
Granados-Principal S, Liu Y, Guevara ML,
Blanco E, Choi DS, Qian W, Patel T, Rodriguez AA, Cusimano J, Weiss
HL, et al: Inhibition of iNOS as a novel effective targeted therapy
against triple-negative breast cancer. Breast Cancer Res.
17:252015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shin HS, Ryu ES, Oh ES and Kang DH:
Endoplasmic reticulum stress as a novel target to ameliorate
epithelial-to-mesenchymal transition and apoptosis of human
peritoneal mesothelial cells. Lab Invest. 95:1157–1173. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kramer B, Ferrari DM, Klappa P, Pöhlmann N
and Söling HD: Functional roles and efficiencies of the thioredoxin
boxes of calcium-binding proteins 1 and 2 in protein folding.
Biochem J. 357:83–95. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zeeshan HM, Lee GH, Kim HR and Chae HJ:
Endoplasmic reticulum stress and associated ROS. Int J Mol Sci.
17:3272016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Santos CX, Nabeebaccus AA, Shah AM,
Camargo LL, Filho SV and Lopes LR: Endoplasmic reticulum stress and
Nox-mediated reactive oxygen species signaling in the peripheral
vasculature: Potential role in hypertension. Antioxid Redox Signal.
20:121–134. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kaneto H, Matsuoka T, Nakatani Y, Kawamori
D, Miyatsuka T, Matsuhisa M and Yamasaki Y: Oxidative stress, ER
stress, and the JNK pathway in type 2 diabetes. J Mol Med (Berl).
83:429–439. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yuan Y, Xu X, Zhao C, Zhao M, Wang H,
Zhang B, Wang N, Mao H, Zhang A and Xing C: The roles of oxidative
stress, endoplasmic reticulum stress, and autophagy in
aldosterone/mineralocorticoid receptor-induced podocyte injury. Lab
Invest. 95:1374–1386. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gross ML, Hanke W, Koch A, Ziebart H,
Amann KR and Ritz E: Intraperitoneal protein injection in the
axolotl: The amphibian kidney as a novel model to study
tubulointerstitial activation. Kidney Int. 62:51–59. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
He F, Chen S, Wang H, Shao N, Tian X,
Jiang H, Liu J, Zhu Z, Meng X and Zhang C: Regulation of
CD2-associated protein influences podocyte endoplasmic reticulum
stress-mediated apoptosis induced by albumin overload. Gene.
484:18–25. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cybulsky AV, Takano T, Papillon J, Bijian
K, Guillemette J and Kennedy CR: Glomerular epithelial cell injury
associated with mutant alpha-actinin-4. Am J Physiol Renal Physiol.
297:F987–F995. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ostergaard L, Simonsen U,
Eskildsen-Helmond Y, Vorum H, Uldbjerg N, Honoré B and Mulvany MJ:
Proteomics reveals lowering oxygen alters cytoskeletal and
endoplasmatic stress proteins in human endothelial cells.
Proteomics. 9:4457–4467. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ha TS, Park HY, Seong SB and Ahn HY:
Angiotensin II induces endoplasmic reticulum stress in podocyte,
which would be further augmented by PI3-kinase inhibition. Clin
Hypertens. 21:13. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Reiser J, Polu KR, Möller CC, Kenlan P,
Altintas MM, Wei C, Faul C, Herbert S, Villegas I, Avila-Casado C,
et al: TRPC6 is a glomerular slit diaphragm-associated channel
required for normal renal function. Nat Genet. 37:739–744. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sun X, Fang Z, Zhu Z, Yang X, He F and
Zhang C: Effect of down-regulation of TRPC6 on the puromycin
aminonucleoside-induced apoptosis of mouse podocytes. J Huazhong
Univ Sci Technolog Med Sci. 29:417–422. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chen S, He FF, Wang H, Fang Z, Shao N,
Tian XJ, Liu JS, Zhu ZH, Wang YM, Wang S, et al: Calcium entry via
TRPC6 mediates albumin overload-induced endoplasmic reticulum
stress and apoptosis in podocytes. Cell Calcium. 50:523–529. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Morse E, Schroth J, You YH, Pizzo DP,
Okada S, Ramachandrarao S, Vallon V, Sharma K and Cunard R: TRB3 is
stimulated in diabetic kidneys, regulated by the ER stress marker
CHOP, and is a suppressor of podocyte MCP-1. Am J Physiol Renal
Physiol. 299:F965–F972. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Inoki K, Mori H, Wang J, Suzuki T, Hong S,
Yoshida S, Blattner SM, Ikenoue T, Rüegg MA, Hall MN, et al: mTORC1
activation in podocytes is a critical step in the development of
diabetic nephropathy in mice. J Clin Invest. 121:2181–2196. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
El Karoui K, Viau A, Dellis O, Bagattin A,
Nguyen C, Baron W, Burtin M, Broueilh M, Heidet L, Mollet G, et al:
Endoplasmic reticulum stress drives proteinuria-induced kidney
lesions via Lipocalin 2. Nat Commun. 7:103302016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Nitta K, Okada K, Yanai M and Takahashi S:
Aging and chronic kidney disease. Kidney Blood Press Res.
38:109–120. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Liu SH, Wu CT, Huang KH, Wang CC, Guan SS,
Chen LP and Chiang CK: C/EBP homologous protein (CHOP) deficiency
ameliorates renal fibrosis in unilateral ureteral obstructive
kidney disease. Oncotarget. 7:21900–21912. 2016.PubMed/NCBI
|
|
64
|
Kaneto H, Kajimoto Y, Miyagawa J, Matsuoka
T, Fujitani Y, Umayahara Y, Hanafusa T, Matsuzawa Y, Yamasaki Y and
Hori M: Beneficial effects of antioxidants in diabetes: Possible
protection of pancreatic beta-cells against glucose toxicity.
Diabetes. 48:2398–2406. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ou Y, Hou W, Li S, Zhu X, Lin Y, Han J,
Duan Z and Gui B: Sodium citrate inhibits endoplasmic reticulum
stress in rats with adenine-induced chronic renal failure. Am J
Nephrol. 42:14–21. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kim Y, Lee H, Manson SR, Lindahl M, Evans
B, Miner JH, Urano F and Chen YM: Mesencephalic astrocyte-derived
neurotrophic factor as a urine biomarker for endoplasmic reticulum
stress-related kidney diseases. J Am Soc Nephrol. 27:2974–2982.
2016. View Article : Google Scholar : PubMed/NCBI
|