Introduction
Bacterial effectors are proteins secreted by
pathogenic bacteria into their host cells, using a type III, IV, or
VI secretion system (T3/4/6SS) (1). These effectors alter the cellular
processes to promote intracellular bacterial replication,
intercellular spread and evasion of the host immune system
(2). To attain full catalytic
activity, effectors must be localized in their specific subcellular
compartments, such as the nucleus, mitochondria, endoplasmic
reticulum (ER), or plasma membrane (PM) (3). However, the mechanisms of
translocation of effectors to their target locations are still not
well understood.
Bacterial pathogens including enterohemorrhagic
Escherichia coli (EHEC) and enteropathogenic Escherichia
coli (EPEC) carry a type III secretion system (T3SS) and a
number of associated effectors, which contribute to bacterial
virulence (4). The attaching and
effacing lesions (A/E lesions), which are characterized by
accumulation of polymerized actin beneath sites of bacterial
attachment, result from the interaction between T3SS effectors and
the host proteins (4). Actin
pedestals are formed when EHEC or EPEC inject the translocated
intimin receptor (Tir) into the host cell PM, where it serves as a
bacterial surface receptor for intimin (5). In EHEC, the intimin-Tir interaction
is promoted by the Tir cytoskeleton coupled protein (6,7),
which recruits the insulin receptor tyrosine kinase substrate p53
(IRSp53), and the actin nucleation-promoting factor Wiskott-Aldrich
syndrome protein (N-WASP) (8).
However, in EPEC, Tir is phosphorylated, and activates N-WASP with
the help of the adapter protein Nck (9). These two pathways generate similar
pathological A/E lesions (10,11).
Tir, an essential effector in A/E lesions signaling
transduction and a type III transmembrane protein with an
approximate molecular weight of 58 kDa (12), is injected into host cells and
integrated into the PM. However, the detailed mechanism of Tir
transportation and integration into the PM remains unknown. In this
study, we investigated the possible pathway(s) of Tir
transportation. We found that the EHEC Tir colocalizes with the 58K
Golgi protein of the host cells. Damage to Golgi structure by
brefeldin A (BFA) blocks the localization of Tir on host plasma
membrane. Our results suggest that Tir is translocated to plasma
membrane through the Golgi apparatus.
Materials and methods
Cell lines, bacterial strains, and
antibodies
HeLa cells were purchased from the American Type
Culture Collection (ATCC, CCL-2) and were grown in DMEM (Hyclone,
Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS)
(Hyclone) and antibiotics at 37°C in a 5% CO2
atmosphere. The EHEC O157:H7 Sakai strain was obtained from the
ATCC (BAA-460). The monoclonal antibody against EspA of EHEC (1H10)
was used as a marker for EHEC staining and was produced as
described by Yu et al (13). The recombinant carboxyl terminus of
the Tir protein, containing residues of Ala405-Ala523
(reTirC405–523), was produced for the preparation of
anti-Tir antibodies. In brief, the gene fragment encoding
reTirC405–523 was amplified from whole genome of EHEC
Sakai (ATCC BAA-460) using primer pairs PtirC-F
and PtirC-R (Table
I). The amplified gene fragment was cloned into pET22b vector
at NdeI and XhoI restriction sites. The
pET22b-reTirC405–523 was transformed into E. coli
BL21 and the recombinant protein was induced by adding of 1 mM of
isopropyl β-D-1-thiogalactopyranoside (IPTG). Cells were then
harvested and homogenized in PBS (pH 7.2). The soluble fraction was
collected and went through a Ni2+-NTA resin (Qiagen) and
the His-tagged reTirC405–523 was eluted with buffer A
containing 250 mM imidazole. Elution from Ni2+-NTA resin
was then bound to Resource Q column (GE Healthcare, Piscataway, NJ,
USA) and eluted with a gradient concentration buffer (containing
PBS and 1 M NaCl, pH 7.2). Finally purified
reTirC405–523 was concentrated and verified by
N-terminal sequencing and SDS-PAGE.
 | Table I.Primers used in this study. |
Table I.
Primers used in this study.
| Primers | Sequence | Notes |
|---|
| PtirC-F |
5′-CATATGCGTACGGTAGAGAATAAGCCTG-3′ | Amplication of
tirC405-523 |
| PtirC-R |
5′-CTCGAGCGCCAGACGCGCATAAGTGCTTTG-3′ | Amplication of
tirC405-523 |
| Ptir1 |
5′-CAATGTGAATAATTCAATTCCTCCTGCACCTCCATTACCTTCACAAACCGTGTGTAGGCTGGAGCTGC-3′ | Knockout of
tir |
| Ptir2 |
5′-CCCGTTAATCCTCCCATGTCATGGCGTAATCCACCACTTAGCGCCAGACGCATATGAATATCCTCCTT-3′ | Knockout of
tir |
| Ptir3 |
5′-ATGCCTATTGGTAATCTTGGTCATA-3′ | Detection of
tir |
| Ptir4 |
5′-TTAGACGAAACGATGGGATCCCGGCGCT-3′ | Detection of
tir |
| Ptir-F |
5′-CGGGGTACCCCGCACGTGGTGTTTACTTTTA-3′ | Amplication of
tir and its promoter |
| Ptir-R |
5′-CCCAAGCTTGGGTTAAGCGTAGTCTGGGACGTCGTATGGGTAGACGAAACGATGGG-3′ | Amplication of
tir and its promoter |
| PtirHA-R |
5′-CCCAAGCTTGGGTTAAGCATAATCTGGAACATCATATGGATAAGCGTAGTCTGGGACGTC-3′ | Amplication of
tir and HA tag (underlined sequence encodes HA tag) |
For the production of anti-Tir antibodies, purified
reTirC405–523 was formulated with complete or incomplete
Freund's Adjuvant as needed (F5881 and F5506; Sigma-Aldrich, St.
Louis, MO, USA) and used to immunize rabbits. Then, serum from
immunized rabbits was collected and anti-reTirC405–523
IgG was purified by affinity chromatography using a protein G
column. The purified rabbit anti-Tir polyclonal antibody was
further analyzed by SDS-PAGE, western blotting and
immunofluorescence assay.
Construction of Δtir EHEC O157 mutants
and its complemented mutant
Strain Δtir EHEC O157:H7 (tir knockout) was
constructed using the λ Red recombinase method (14). Primer pairs Ptir1
and Ptir2 (Table
I) were designed to amplify the chloramphenicol resistance gene
from plasmid pKD3 (kindly provided by George F. Gao, Institute of
Microbiology, Chinese Academy of Sciences). The purified PCR
product was electroporated into E. coli O157:H7 carrying the
λ Red recombinase expression plasmid pKD46 (kindly provided by
George F. Gao, Institute of Microbiology, Chinese Academy of
Sciences). Primers located inside the chloramphenicol resistance
cassette were used to verify deletion of tir by PCR
(Ptir3 and Ptir4) (Table I). EHEC O157:H7 clones were grown
on LB medium containing 25 µg/ml of chloramphenicol to select for
chloramphenicol resistance. The pKD46 plasmid was inactivated by
growing bacteria at 42°C.
To construct complementary strains, the gene
encoding Tir plus its promoter was amplified using primer
pairs Ptir-F and Ptir-R
(Table I). The gene encoding
Tir-HA was amplified by Ptir-F and
PtirHA-R. They were cloned into pBluescript II SK
(−) vector (Agilent Technologies, Inc., CA, USA), and transformed
into Δtir EHEC O157:H7. The expression of Tir was detected
by western blotting using purified rabbit anti-Tir polyclonal
antibodies as primary antibody.
Infection of HeLa cells and BFA
treatment
HeLa cells were infected with EHEC strains as
described by Wang et al (15). with minor modifications. Briefly,
HeLa cell monolayers were grown on 20×20 mm coverslips in a 6-well
tissue culture plate. Monolayers were co-incubated with wild-type
or Δtir EHEC O157:H7 Sakai (multiplicity of infection, 100:1) for 5
h at 37°C and 5% CO2 in the infection medium (2% FBS
DMEM). Additionally, monolayers were pretreated with 2.5 µg/ml of
BFA (Beyotime Biotech, Jiangsu, China) for 30 min, prior to EHEC
infection (16). At 3 h
post-infection, cells were washed gently twice with
phosphate-buffered saline (PBS), followed by addition of an equal
volume of fresh infection medium with similar BFA
concentration.
Immunofluorescence microscopy
HeLa cells were fixed in 2.5% paraformaldehyde
(Boster, Wuhan, China) and then permeabilized with 0.1% Triton
X-100 (Sigma-Aldrich) with gentle shaking. Permeabilized cells were
incubated with rabbit polyclonal anti-Tir-antibody diluted 1:600 in
0.1% bovine serum albumin (BSA) (Boster Inc., Wuhan, China) in PBS
for 1 h, and treated with FITC-labeled goat anti-rabbit secondary
antibody (1:500) (Beyotime Biotech) for 1 h. HA-tagged Tir
constructs were visualized by staining with a mouse anti-HA
antibody (Sigma-Aldrich) followed by Alexa Fluor 633-conjugated
goat anti-mouse secondary antibody (Invitrogen Life Technologies,
Carlsbad, CA, USA). To visualize Golgi, cells were incubated with
mouse anti-58K Golgi protein monoclonal antibody (Abcam, Cambridge,
MA, USA) at a dilution of 1:500. Cells were then washed and
incubated with Alexa Fluor 633-conjugated goat anti-mouse secondary
antibody diluted 1:500 for 1 h.
Polymerized actin was visualized by incubation of
cells with FITC-labeled phalloidin (Sigma) diluted 1:200 in PBS for
40 min. The monoclonal antibody 1H10 was used as a marker for EHEC
staining. Cells were stained with 1H10 diluted at 1:500 for 1 h
followed by incubation with Alexa Fluor 633-conjugated goat
anti-mouse antibody as the secondary antibody. All steps were
performed at room temperature. Cells were then examined using a
fluorescence microscope (Zeiss Axioimager, Oberkochen, Germany).
Images were analyzed using LAS AF Lite Leica confocal imaging
software (Leica, Heidelberg, Germany).
To quantify the colocalization of Tir and 58K Golgi
protein, the colocalization index was calculated by Pearson's
correlation with the colocalization finder plugin of ImageJ (US
National Institutes of Health, Bethesda, MD, USA). The
quantification of actin polymerizations was performed as described
previously (9). The number of
actin pedestals and attached bacterial foci were counted manually
in at least 40 fields and used to calculate the percentage of
pedestal formation.
Immunoelectron microscopy
The infected HeLa cells were prepared as described
above. Subsequently, cells were fixed with 2% paraformaldehyde and
0.2% glutaraldehyde in PBS (pH 7.4) for 24 h at 4°C. Cells were
then washed twice with PBS and dehydrated with serial diluted
ethanol (30, 50, 70, 90 and 100% ethanol, 15 min each). The
specimens were embedded in LR-White acrylic resin (SPI Supplies,
West Chester, PA, USA). Polymerization of the LR-White was
performed at −20°C for 72 h, and at room temperature for 48 h,
under ultraviolet light. Primary antibody against Tir was used at a
dilution of 1:500. Gold-labeled secondary antibody with a gold
particle size of 10 nm (Sigma) was used at a dilution of 1:30.
Sections were observed using a TECNAL-10 transmission electron
microscope (Philips Medical Systems B.V., Eindhoven, The
Netherlands).
Cellular fractionation and western
blotting
Fractionation of HeLa cells was accomplished
according to the procedure described previously (17). Briefly, after 5 h of infection,
HeLa cells were washed with ice-cold PBS and resuspended in a
homogenization buffer (3 mM imidazole, 250 mM sucrose, 0.5 mM EDTA,
pH 7.4) supplemented with protease inhibitor cocktail (Roche
Diagnostics GmbH, Mannheim, Germany). Cells were mechanically
disrupted using a grinder. The homogenate was centrifuged at 3,000
× g for 15 min at 4°C to pellet adherent bacteria, the host nuclei
and cytoskeleton. The supernatant was then subjected to high-speed
ultracentrifugation (41,000 × g) for 20 min at 4°C in a JA-21 rotor
in a Avanti® J-E centrifuge (Beckman Coulter, Inc., Brea, CA, USA)
to separate host cell membranes (pellet) from cytoplasm
(supernatant). The membrane pellet for alkaline phosphatase
treatment was solubilized in 50 mM Tris (pH 8.0)-1% Triton X-100
without phosphatase inhibitors and incubated with 2U of alkaline
phosphatase (New England Biolabs) for 1 h at 37°C (12). The samples were resuspended in
Laemmli sample buffer and boiled for 5 min.
Equal volumes of all fractions were separated using
SDS-PAGE, transferred onto PVDF membranes, and probed with
anti-Tir, anti-HA (Sigma), anti-Calnexin (Beyotime Biotech) (marker
for membrane fraction) and anti-Tubulin (Beyotime Biotech) (marker
for cytosol fraction), followed by treatment with the appropriate
horseradish peroxidase-conjugated secondary antibodies (Jackson
Immunoresearch Laboratories, West Grove, PA, USA). The protein of
interest was visualized using Supersignal® West Dura Duration
substrate reagent (Thermo Fisher Scientific, Inc., Waltham, MA,
USA).
Statistical analysis
The colocalization index and the percentage of
pedestal formation were analyzed using ANOVA method from three
independent experiments. P<0.05 was considered to indicate a
statistically significant difference.
Results
Tir localizes to host Golgi
apparatus
Tir plays an important role in the transportation
and modification of proteins in eukaryotic cells (18). In order to observe the subcellular
localization of Tir, we generated the rabbit
anti-reTirC405–523 antibody, against residues
Ala405-Ala523. The purified rabbit anti-reTirC405–523
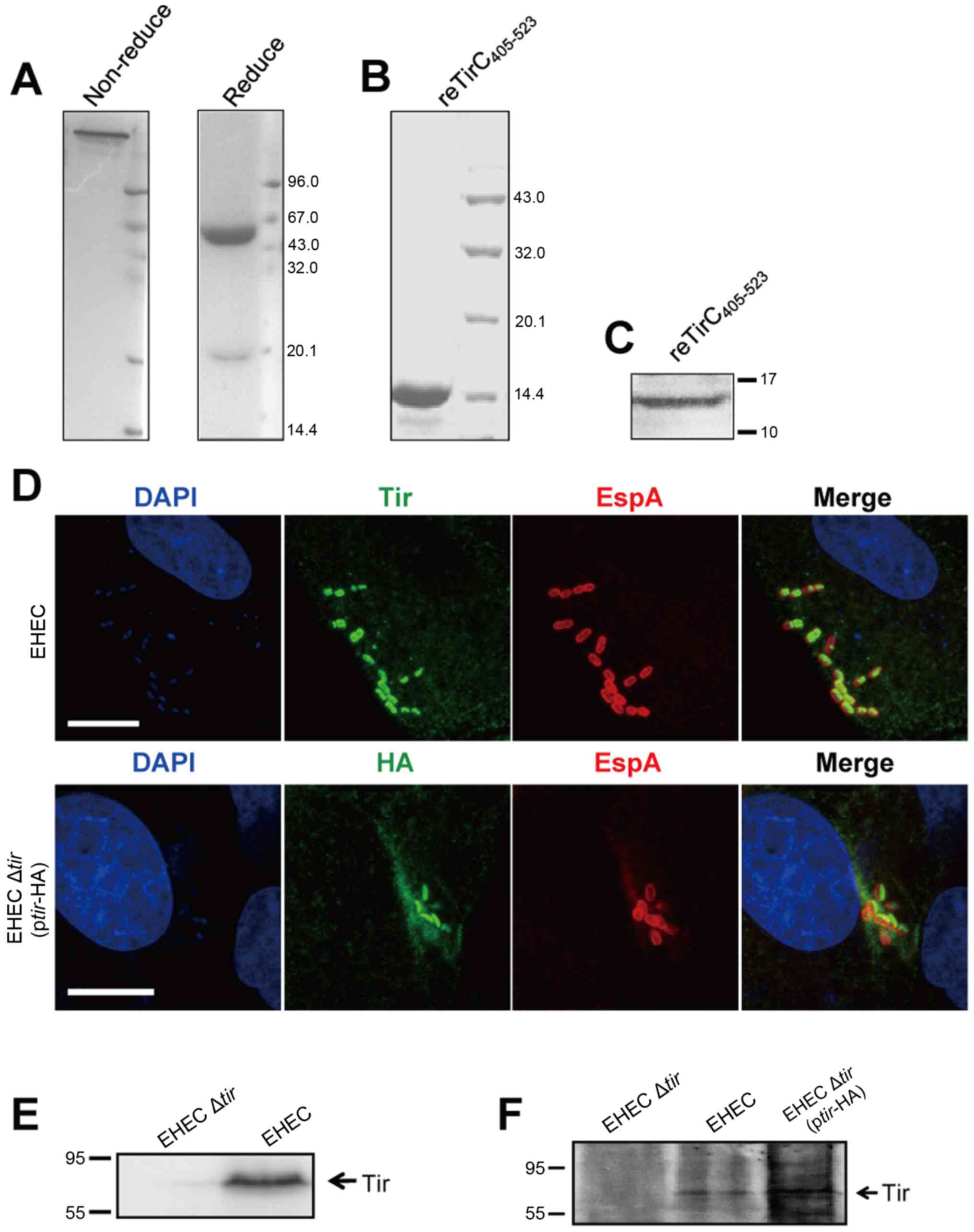
antibody was analyzed by SDS-PAGE (Fig. 1A and B). To confirm the specific
binding of anti-reTirC405–523 antibody to Tir, we
performed the western blotting and immunofluorescence assay, and
the effective immune responses were observed (Fig. 1C and D). To better understand the
function of the EHEC Tir, we first examined the localization of Tir
in the host cells using immunofluorescence microscopy. As shown in
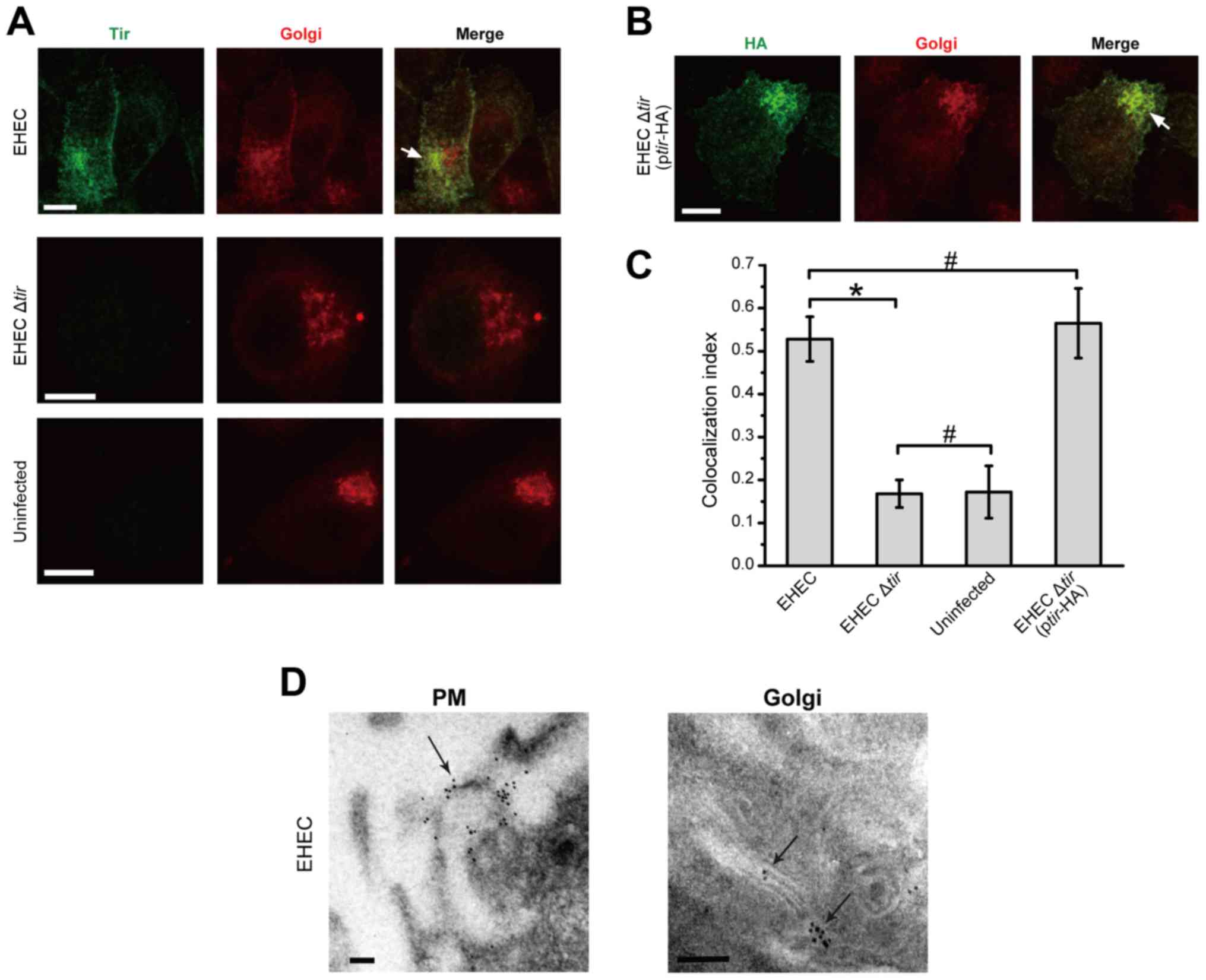
Fig. 2A, when HeLa cells were
infected with EHEC, Tir was clearly localized inside the host cells
surrounding the nuclei. Co-staining with the anti-58K Golgi protein
indicated that the two proteins were co-localized extensively.
In addition, we constructed the Δtir EHEC
deletion and its complemented mutants using the λ Red recombinase
method (14), and the deletion was
confirmed by western blotting (Fig. 1E
and F). No apparent Tir staining was observed in HeLa cells
that were not infected or infected with EHEC Δtir (Fig. 2A). When EHEC Δtir was
transformed with the Tir-containing plasmid pBluscript-tir, Tir was
detected in HeLa cells similar to the indigenous Tir. The HA-tagged
Tir was colocalized with the 58K Golgi protein (Fig. 2B). To further confirm the
colocalization of Tir and 58K Golgi protein, the colocalization
index was calculated using the Pearson's correlation coefficient.
The colocalization index of EHEC-infected cells was significantly
higher than that of EHEC Δtir infected or uninfected cells
(P<0.05, Fig. 2C). The
colocalization index of EHEC Δtir (+pBluscript-tir) was
similar to that of the wild type EHEC (P>0.05; Fig. 2C). However, no colocalization was
observed with antibody against Calreticulin (Abcam), a marker for
endoplasmic reticulum (data not shown). These results suggest that
Tir accumulates in the Golgi complex after the EHEC infection.
The subcellular location of Tir was further analyzed
by immunoelectron microscopy. Consistent with immunofluorescence
microscopy, our result showed that gold particles were localized on
the cell membrane of EHEC-infected HeLa cells, especially at the
site of bacterial invasion (Fig.
2D, left). Gold particles were also observed on Golgi structure
(Fig. 2D, right). No gold particle
was detected in uninfected cells, or cells infected with EHEC
Δtir (data not shown). Taken together, these results
indicate that the EHEC Tir was translocated to the plasma membrane
and Golgi body of the host cells.
BFA blocks the formation of actin
pedestals
During infection, EHEC injects effector proteins,
including Tir, into the host cell to activate the actin
cytoskeleton and promote formation of actin pedestals. To further
confirm the critical role of Golgi-localized Tir in infection, the
formation of actin pedestals was analyzed using BFA, a fungal
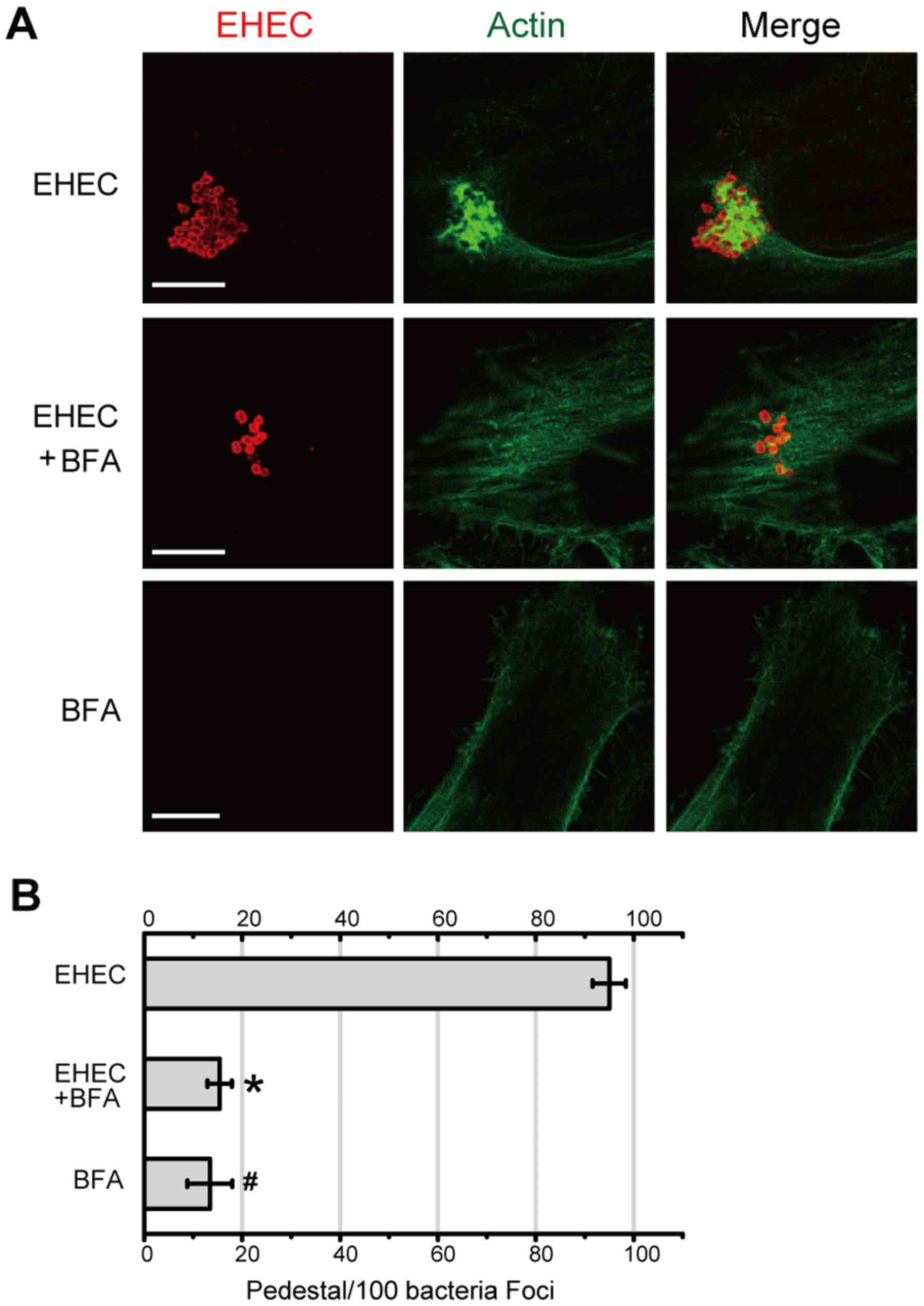
metabolite that disrupts Golgi structure (16). As shown in Fig. 3A, actin pedestals were clearly
observed when host cells were infected with EHEC (upper panel).
Treatment with 2.5 µg/ml of BFA, however, revealed few actin
pedestals (Fig. 3A, middle panel).
BFA did not affect the actin polymerization per se. The
large clusters of polymerized actin were counted as pedestals and
considered as background (Fig. 3A,
lower panel and B). The number of actin pedestals in the absence or
presence of BFA was further quantified. As shown in Fig. 3B, the percentage of EHEC-induced
actin pedestals decreased significantly in the presence of BFA
(P<0.05). These results suggest that damage to the Golgi body by
BFA blocks the formation of the Tir-mediated actin pedestals.
BFA causes disassembly of the Golgi
complex and blocks Tir transportation from Golgi to plasma
membrane
To further confirm that BFA-induced damage, the
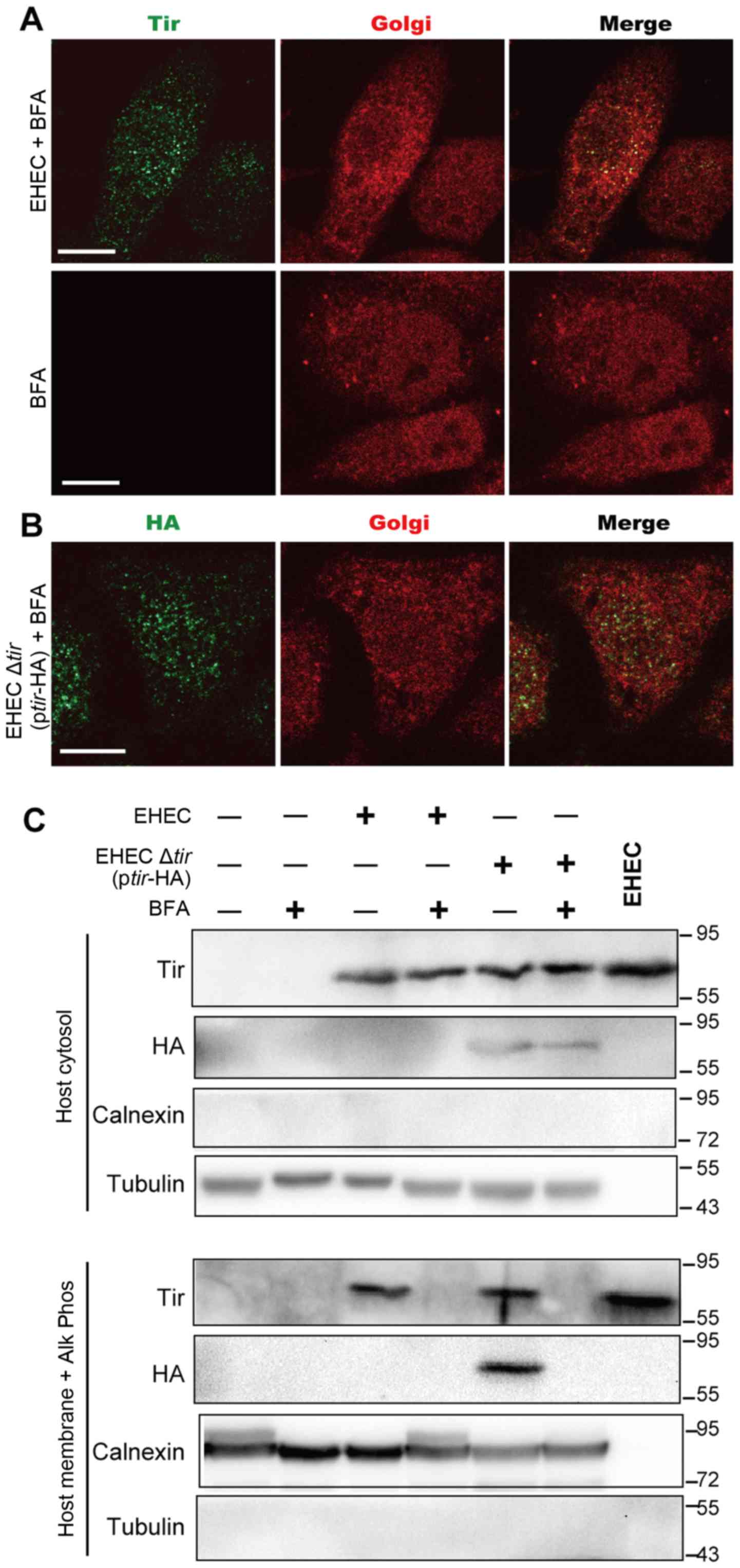
Golgi body was stained in the presence of BFA. Upon treatment with
BFA, the 58K Golgi proteins diffused throughout the cytoplasm
either in the presence or absence of EHEC infection (Fig. 4A, compare to Fig. 2A). Furthermore, Tir was diffusely
dispersed in the cytoplasm and around the nuclear envelope, in
contrast to its condensed localization in the EHEC-infected cells
(Fig. 2A and B). These results
indicate that BFA disrupts the structure of the Golgi complex and
affects the localization of Tir.
To further determine the role of Golgi body in
transporting Tir to plasma membrane, HeLa cells were infected with
wild-type EHEC or Δtir EHEC complemented with
ptir-HA. Western blotting was then used to detect Tir in
both cytosolic and membrane fractions. To ensure that both
phosphorylated and non-phosphorylated forms in membrane fraction
were detected (12,19), the membrane fraction was treated
with calf intestinal alkaline phosphatase (Alk Phos), a nonspecific
phosphatase before detection. As shown in Fig. 4C, no significant change of Tir
distribution in cytosol fraction was observed in cells treated with
or without BFA, suggesting that BFA treatment did not affect the
release of Tir into HeLa cytoplasm. Tir was also observed in the
membrane fractions when cells were infected with either EHEC or
Δtir EHEC (+ptir-HA). However, when cells were
pre-treated with BFA, Tir was not detected in the membrane
fraction. These results suggest that BFA blocks the transportation
of Tir to plasma membrane.
Discussion
Bacterial effectors need to be transported to their
specific intracellular locations to participate in pathogenesis.
Two possible mechanisms of effector transport have been reported
(20–22). One is called post-translocation
modification of effectors. For example, the cysteine residue at the
C-terminus of Salmonella effector SifA is lipidated by the
protein geranylgeranyl transferase I, leading to the accumulation
of SifA at the PM (20). The
second mechanism requires dedicated structural motifs within the
effectors. Pseudomonas aeruginosa ExoS and ExoT, and YopE
from Yersinia spp. carry a shared membrane localization
domain (MLD), which mediates their initial association with the PM
(21,22). It has also been reported that the
Tir from EPEC (TirEPEC) is transported into the host
membrane via two pathways First, the TirEPEC is
translocated into the cytoplasm via a type III secretion system,
and subsequently integrated into the membrane (23). In the second pathway, extracellular
Tir directly integrates into host membranes in the presence of
limited free Ca2+ in the environment, and the
integration is independent of T3SS translocation (24). The Tir of EHEC is highly homologous
to TirEPEC and may share a similar pathway for its
translocation. In this study, we found that Tir from EHEC was
colocalized with the host Golgi structure. Tir was undetected in
plasma membrane, when the Golgi structure was disrupted by BFA
(Fig. 4C), suggesting that Tir was
first transported to cytoplasm/Golgi and then to the cell membrane
in a Golgi-dependent manner. This view is further supported by the
inhibition of the formation of actin pedestals by BFA, which
requires Tir at the plasma membrane. Further studies are needed to
understand the mechanisms mediating Tir transport into the
cytoplasm (possibly via T3SS) and then to the Golgi apparatus of
the host cells.
A possible pathway of Tir transportation to Golgi
body may involve the guanine nucleotide-exchange factors (GEFs),
which convert the GDP-bound, inactive ADP-ribosylation factor (Arf)
to GTP-bound, active Arf (Arf-GTP) (16,25).
Arf-GTP associates reversibly with the Golgi membrane to recruit
coat proteins and adaptors from the cytosol, and initiates the
formation of transport vesicles. BFA prevents the assembly of
coatomer on the membrane by inhibiting GEFs, therefore blocking the
formation of Arf-GTP (26). This
is supported by a study, in which Arf1 accumulates on the
Legionella-containing vacuoles and helps the vacuole-ER
fusion, which was abolished by BFA (27). Consistent with this report, we
observed that Tir was diffusely distributed in the cytoplasm and
around the nuclear envelope in the presence of BFA.
BFA-induced inhibition of Tir transportation is also
clear from the endosomal tubulation caused by BFA similar to that
induced in the Golgi apparatus (13). Endocytosis is an essential cellular
process involved in the transport of membrane proteins in mammalian
cells (14). It is reasonable to
speculate that endosomes may play a role in Tir transportation to
plasma membrane. The role of endosomes in Tir translocation and
Tir-induced A/E lesions needs further investigation.
In this study, we report that Tir is translocated to
the host plasma membrane in a Golgi-dependent manner. This finding
provides a novel trafficking pathway for secreted bacterial
effectors. Further studies should be focused on the functional
motifs underlying Tir trafficking and its detailed transport
mechanism within the Golgi protein delivery system.
Acknowledgements
We are grateful to Dr. Fuquan Hu for the critical
reading and editing of the manuscript. We also thank Wei Sun and
Liting Wang for technical assistance with confocal microscopy and
George F. Gao (Institute of Microbiology, Chinese Academy of
Sciences) for his kind gifts of plasmids pKD3 and pKD46. This study
was supported by the National Natural Science Foundation of China
(NSFC; no. 81271783).
Glossary
Abbreviations
Abbreviations:
|
EHEC
|
Enterohemorrhagic Escherichia
coli
|
|
EPEC
|
Enteropathogenic Escherichia
coli
|
|
Tir
|
translocated intimin receptor
|
|
Golgi
|
Golgi apparatus
|
|
PM
|
plasma membrane
|
|
MOI
|
multiplicity of infection
|
|
A/E lesions
|
attaching and effacing lesions
|
|
BFA
|
brefeldin A
|
|
T3SS
|
type III secretion system
|
References
|
1
|
Galán JE: Common themes in the design and
function of bacterial effectors. Cell Host Microbe. 5:571–579.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Troisfontaines P and Cornelis GR: Type III
secretion: More systems than you think. Physiology (Bethesda).
20:326–339. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Geissler B: Bacterial toxin
effector-membrane targeting: Outside in, then back again. Front
Cell Infect Microbiol. 2:752012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Frankel G and Phillips AD: Attaching
effacing Escherichia coli and paradigms of Tir-triggered actin
polymerization: Getting off the pedestal. Cell Microbiol.
10:549–556. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Campellone KG: Cytoskeleton-modulating
effectors of enteropathogenic and enterohaemorrhagic Escherichia
coli: Tir, EspFU and actin pedestal assembly. FEBS J.
277:2390–2402. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Campellone KG, Robbins D and Leong JM:
EspFU is a translocated EHEC effector that interacts with Tir and
N-WASP and promotes Nck-independent actin assembly. Dev Cell.
7:217–228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Garmendia J, Phillips AD, Carlier MF,
Chong Y, Schüller S, Marches O, Dahan S, Oswald E, Shaw RK, Knutton
S and Frankel G: TccP is an enterohaemorrhagic Escherichia coli
O157: H7 type III effector protein that couples Tir to the
actin-cytoskeleton. Cell Microbiol. 6:1167–1183. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weiss SM, Ladwein M, Schmidt D, Ehinger J,
Lommel S, Städing K, Beutling U, Disanza A, Frank R, Jänsch L, et
al: IRSp53 links the enterohemorrhagic E. coli effectors Tir and
EspFU for actin pedestal formation. Cell Host Microbe. 5:244–258.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brady MJ, Campellone KG, Ghildiyal M and
Leong JM: Enterohaemorrhagic and enteropathogenic Escherichia coli
Tir proteins trigger a common Nck-independent actin assembly
pathway. Cell Microbiol. 9:2242–2253. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
de Groot JC, Schlüter K, Carius Y,
Quedenau C, Vingadassalom D, Faix J, Weiss SM, Reichelt J,
Standfuss-Gabisch C, Lesser CF, et al: Structural basis for complex
formation between human IRSp53 and the translocated intimin
receptor Tir of enterohemorrhagic E. coli. Structure. 19:1294–1306.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Garmendia J, Carlier MF, Egile C, Didry D
and Frankel G: Characterization of TccP-mediated N-WASP activation
during enterohaemorrhagic Escherichia coli infection. Cell
Microbiol. 8:1444–1455. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
DeVinney R, Stein M, Reinscheid D, Abe A,
Ruschkowski S and Finlay BB: Enterohemorrhagic Escherichia coli
O157: H7 produces Tir, which is translocated to the host cell
membrane but is not tyrosine phosphorylated. Infect Immun.
67:2389–2398. 1999.PubMed/NCBI
|
|
13
|
Yu S, Gu J, Wang HG, Wang QX, Luo P, Wu C,
Zhang WJ, Guo G, Tong WD, Zou QM and Mao XH: Identification of a
novel linear epitope on EspA from enterohemorrhagic E. coli using a
neutralizing and protective monoclonal antibody. Clin Immunol.
138:77–84. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Datsenko KA and Wanner BL: One-step
inactivation of chromosomal genes in Escherichia coli K-12 using
PCR products. Proc Natl Acad Sci USA. 97:6640–6645. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang H, Gu J, Yu S, Zhang W, Zhu Y, Zou Q,
Zhu F and Mao X: Characterization of enterohemorrhagic Escherichia
coli O157:H7 00B015: A Shiga toxin producing but
virulence-attenuated isolate. Can J Microbiol. 56:651–656. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fujiwara T, Oda K, Yokota S, Takatsuki A
and Ikehara Y: Brefeldin A causes disassembly of the Golgi complex
and accumulation of secretory proteins in the endoplasmic
reticulum. J Biol Chem. 263:18545–18552. 1988.PubMed/NCBI
|
|
17
|
Gruenheid S, Sekirov I, Thomas NA, Deng W,
O'Donnell P, Goode D, Li Y, Frey EA, Brown NF, Metalnikov P, et al:
Identification and characterization of NleA, a non-LEE-encoded type
III translocated virulence factor of enterohaemorrhagic Escherichia
coli O157:H7. Mol Microbiol. 51:1233–1249. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Campellone KG, Brady MJ, Alamares JG, Rowe
DC, Skehan BM, Tipper DJ and Leong JM: Enterohaemorrhagic
Escherichia coli Tir requires a C-terminal 12-residue peptide to
initiate EspF-mediated actin assembly and harbours N-terminal
sequences that influence pedestal length. Cell Microbiol.
8:1488–1503. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kenny B: The enterohaemorrhagic
Escherichia coli (serotype O157:H7) Tir molecule is not
functionally interchangeable for its enteropathogenic E. coli
(serotype O127:H6) homologue. Cell Microbiol. 3:499–510. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Reinicke AT, Hutchinson JL, Magee AI,
Mastroeni P, Trowsdale J and Kelly AP: A Salmonella typhimurium
effector protein SifA is modified by host cell prenylation and
S-acylation machinery. J Biol Chem. 280:14620–14627. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kazmierczak BI and Engel JN: Pseudomonas
aeruginosa ExoT acts in vivo as a GTPase-activating protein for
RhoA, Rac1, and Cdc42. Infect Immun. 70:2198–2205. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Black DS and Bliska JB: The RhoGAP
activity of the Yersinia pseudotuberculosis cytotoxin YopE is
required for antiphagocytic function and virulence. Mol Microbiol.
37:515–527. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kenny B, DeVinney R, Stein M, Reinscheid
DJ, Frey EA and Finlay BB: Enteropathogenic E. coli (EPEC)
transfers its receptor for intimate adherence into mammalian cells.
Cell. 91:511–520. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Michgehl S, Heusipp G, Greune L, Rüter C
and Schmidt MA: Esp-independent functional integration of the
translocated intimin receptor (Tir) of enteropathogenic Escherichia
coli (EPEC) into host cell membranes. Cell Microbiol. 8:625–633.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Klausner RD, Donaldson JG and
Lippincott-Schwartz J: Brefeldin A: Insights into the control of
membrane traffic and organelle structure. J Cell Biol.
116:1071–1080. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Donaldson JG, Finazzi D and Klausner RD:
Brefeldin A inhibits Golgi membrane-catalysed exchange of guanine
nucleotide onto ARF protein. Nature. 360:350–352. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kagan JC and Roy CR: Legionella phagosomes
intercept vesicular traffic from endoplasmic reticulum exit sites.
Nat Cell Biol. 4:945–954. 2002. View
Article : Google Scholar : PubMed/NCBI
|