Introduction
Chinese hamster cell mutants, obtained by chemical
mutagenesis of wild type cells from the lung or ovary, have served
as a valuable model in research on various biological processes in
eukaryotes. At present, despite the expansion of numerous novel
molecular techniques, such as gene silencing and the use of
transgenic animals, Chinese hamster cell mutants remain to serve a
significant role, particularly in the research of novel therapeutic
strategies and drug resistance mechanisms in cancer cells (1–3). It
has been established that fibroblast cell lines sensitive to
various carcinogenic agents are used as a basic tool, contributing
to the identification of numerous genes involved in various DNA
repair pathways. UVC-sensitive Chinese hamster cell lines have been
used to identify more than a half of nucleotide excision repair
(NER) genes, including excision repair cross-complementation group
1 (Ercc1), Ercc2 [Xeroderma pigmentosum
complementary group D (XPD) in humans], Ercc3
(XPB), Ercc4 [(XPF/Fanconi anemia (FA)
complementation group Q (FANCQ)], Ercc5 (XPG)
and Ercc6 (Cockayne syndrome B protein, CSB)
(4,5). In addition, genes involved in other
DNA repair pathways, e.g. base excision repair (X-ray repair
complementing defective repair in Chinese hamster cells 1,
Xrcc1), homologous recombination (Xrcc2 and
Xrcc3) and non-homologous end joining [Xrcc4,
Xrcc5/KU80, Xrcc7/protein kinase,
DNA-activated, catalytic polypeptide (PRKDC)] were
identified during the studies of the ionizing radiation-sensitive
mutants (6). The cellular
phenotype determined by mutations in Ercc1 and
Rad51c/Fanco repair genes was described before
identification of patients with these lesions (7–9). Due
to phenotypic similarity between Chinese hamster cell mutants
sensitive to various DNA-damaging agents and the cells of patients
whose diseases are associated with abnormal DNA repair (including
ataxia-telangiectasia, Xeroderma pigmentosum or FA), rodent
mutants remain as a useful model for studying mechanisms of DNA
repair (10,11).
While the majority of repair mechanisms are
relatively well known, ICLs removal remains to be fully understood.
These highly toxic lesions are introduced to DNA by crosslinking
agents, including derivatives of nitrogen mustard, platinum
compounds e.g. cisplatin (CDDP), mitomycin C (MMC) and psoralens,
which are commonly used in the treatment of various malignancies
(12). Removal of ICLs is a
complicated process involving proteins from the majority of the
known DNA repair pathways. The process can be simplified to three
main steps: i) Cell cycle arrest induced by the presence of ICLs
within DNA and recruitment of DNA repair proteins, depending
primarily on the FA pathway and ataxia telangiectasia and
Rad3-related protein (ATR), ii) excision of ICLs from DNA with the
participation of NER- and FA-associated proteins, which leads to
formation of DNA double strand breaks (DSBs), and iii) repair of
DSBs by homologous recombination (12).
Defective ICLs repair has been observed in several
human hereditary diseases associated with genetic instability,
predominantly in FA, however additionally in Xeroderma
pigmentosum, cerebro-oculo-facio-skeletal syndrome (COFS),
Roberts syndrome, Cornelia de Lange syndrome and Warsaw breakage
syndrome (13–16). In addition, heterozygous mutations
of FA genes, namely breast cancer 2, early onset (BRCA2;
also referred to as FANCD1), BRCA1 interacting protein
C-terminal helicase 1 (BRIP1, also termed FANCJ),
partner and localizer of BRCA2 (PALB2, also termed
FANCN) and RAD51C/FANCO, may predispose to
cancer, in particular to breast and/or ovarian malignancies
(17). Despite major progress in
research on ICLs repair, the molecular backgrounds of FA and
Cornelia de Lange syndrome remain unclear in 5 and 35% of patients
with these conditions, respectively (http://fanconi.org) (18). This in turn implies that certain
genes involved in ICLs removal remain to be identified.
A total of 8 complementation groups of Chinese
hamster cell mutants sensitive to DNA crosslinking agents, carrying
mutations of Ercc1, Ercc4, Xrcc2, Xrcc3, Brca2, Rad51C,
Fanca and Fancg genes, have been described at present
(4,6–8,19–22).
The current study demonstrated a novel Chinese hamster mutant,
CL-V8B, with an unknown genetic background and different phenotype
than that identified in previously described cell lines
hypersensitive to DNA crosslinking agents. CL-V8B cells share
numerous features of HR mutants, which points to the likely role of
mutated genes in this DNA repair process.
Materials and methods
Cell lines and culture conditions
Cell lines and hybrids used in the present study
were provided by the Department of Toxicogenetics, Leiden
University Medical Centre, The Netherlands. Wild type and
chemically (N-ethyl-N-nitrosourea, ENU) mutated fibroblasts of
Chinese hamster were routinely cultured in 94-mm culture dishes
(Greiner Bio-One International GmbH, Kremsmünster, Austria) in
Ham's F10 medium (Sigma-Aldrich; Merck Millipore, Darmstadt,
Germany) supplemented with 10% fetal calf serum (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) and antibiotics
(penicillin 1 U/ml, streptomycin 0.1 mg/ml; Sigma-Aldrich; Merck
Millipore). The cells were maintained at 37°C in a 5%
CO2 atmosphere and relative humidity of 95%. The cells
for subcultures were washed in phosphate-buffered saline (PBS;
Sigma-Aldrich; Merck Millipore) and detached with 0.25% trypsin
containing 1 mM EDTA (Sigma-Aldrich; Merck Millipore). The cells
were stored at −85°C in ampules (Nunc; Thermo Fisher Scientific,
Inc.) containing 1×106 cells in Ham's F10 medium with the addition
of 6% dimethyl sulfoxide (DMSO; Sigma-Aldrich; Merck Millipore).
Prior to the experiment, the cells were taken from ampules, seeded
for 2 days, then subcultured and grown for another 2 days. The
study was approved by the Ethics Committee of the Collegium Medicum
in Bydgoszcz, Poland.
Clonogenic survival assay
To establish the sensitivity of CL-V8B to bleomycin
(BLM; Dagra Pharma B.U., Diemen, Netherlands), camptothecin (CPT;
Sigma-Aldrich; Merck Millipore), CDDP (Sigma-Aldrich; Merck
Millipore), hydroxyurea (HU; Sigma-Aldrich; Merck Millipore),
hydrogen peroxide (H2O2; Merck Millipore),
PARP inhibitor KU0058948 (donated by Dr Graeme C. M. Smith; KuDOS
Pharmaceuticals Limited; Astra Zeneca), MMC (Kyowa Hakko Co., Ltd.,
Tokyo, Japan) and ultraviolet radiation (UVC light of 254 nm;
Philips Lamp, 6 W), cell cultures in the exponential growth phase
were trypsinized and then 300 cells were plated onto culture dishes
in duplicate (untreated controls in triplicate). The cells were
treated continuously with BLM, CPT, CDDP, HU, KU0058948 and MMC. In
assays with CPT and KU0058948, these two reagents were dissolved in
DMSO, the concentration of the latter in all plates, including the
controls, was maintained at 0.5%. Prior to
H2O2 treatment and UVC exposure, the cells
were allowed to attach for 4 h and then were incubated with
H2O2 for 1 h or were irradiated. The dose of
radiation was determined with the VLX 254 radiometer (Vilber
Lourmat Deutschland GmbH, Eberhardzell, Germany). Subsequently, the
cells were washed with PBS, and fresh growth medium was added. The
cells were grown for 8–10 days, and then the dishes were rinsed
with 0.9% NaCl (Sigma-Aldrich; Merck Millipore), dried, stained
with 0.25% methylene blue (Sigma-Aldrich; Merck Millipore) and
dried again prior to counting the visible colonies. Parental cells
used as the controls were treated in the same manner as the mutant
cells. Survival curves were prepared with GraphPad Prism 5.0
software (GraphPad Software, Inc., La Jolla, CA, USA). Each
survival curve illustrated a percentage of surviving cells as a
function of the analyzed compound concentration or UVC dose.
Cellular sensitivity was expressed as the D10 value,
i.e. the concentration of a chemical agent or the irradiation dose
necessary to reduce the number of surviving cells to 10%.
Genetic complementation analysis by
cell fusion
Cell fusion was achieved with polyethylene glycol
(PEG; Sigma-Aldrich; Merck Millipore), as previously described
(23). Briefly, a total of 2×106
cells (1:1 ratio of each type) were seeded and incubated overnight
in Ham's F10 medium. Then, the cells were rinsed with the
serum-deprived medium and exposed for 1 min to 2 ml fusion medium
containing 47% PEG and 10% DMSO. Subsequently, the cells were
washed 3 times with the growth medium and incubated for 24 h to
form hybrids. Then, the cells were trypsinized and plated (2
replicates from each cross) into hypoxanthine-aminopterin-thymidine
(HAT) medium containing 1 mM ouabain (Sigma-Aldrich; Merck
Millipore). The populations of hybrid clones (>100) were then
collected from each cross and subjected to genetic complementation
analysis (correction of MMC sensitivity) with a clonogenic survival
assay.
Analysis of chromosomal aberrations
and sister chromatid exchanges
Chromosomal aberrations (CAs) and frequencies of
sister chromatid exchanges (SCEs) were determined in exponentially
growing cells. The cells for CAs determination were treated with
various concentrations of MMC for 2 h (V79B: 25, 45 and 70 ng/ml;
CL-V8B: 1.5, 2.5 and 5 ng/ml), and the cells for SCEs number
determination were irradiated with UVC at a dose rate of 2.5, 5 and
10 J/m2 or exposed to MMC (V79B: 15, 30 ng/ml; Cl-V8B: 1, 1.5
ng/ml), also for 2 h. Subsequently, 10 µM 5-bromo-2′-deoxyuridine
(BrdU; Sigma-Aldrich; Merck Millipore) was added to both the CAs
and SCEs cultures. A total of 16 h (CAs) or 30 h (SCEs) subsequent
to exposure of cells to MMC or UVC, colcemid (1 µg/ml;
Sigma-Aldrich; Merck Millipore) was added to the growth medium for
2 h. Following this, the cells were harvested, treated with
hypotonic solution of 0.075 M KCl (Sigma-Aldrich; Merck Millipore)
for 20 min at 37°C and fixed three times with methanol
(Sigma-Aldrich; Merck Millipore)/glacial acid (Avantor Performance
Materials Poland, Gliwice, Poland) solution (3:1 ratio). Air-dried
slides were washed with 10 µM bisbenzamide (Sigma-Aldrich; Merck
Millipore) in the dark for 20 min, incubated with McIlvaine
citrate/phosphate buffer (pH 8; Avantor Performance Materials
Poland) using a slide drying hotplate (60°C; Barnstead
Electrothermal; Thermo Fisher Scientific, Inc.) exposed to UVC
(Philips Lamp, 15 W) for 25 min and stained with 10% Giemsa
solution (NBS Biologicals, Huntingdon, UK). For CAs, 100 mitotic
cells were analyzed for each MMC dose and for each experiment
separately, and 30 cells were used for SCEs.
Immunofluorescence labelling and
microscopy
To examine centrosomes and Rad51 focus formation,
the cells were seeded onto 15-mm round coverslips (Menzel Gläser,
Braunschweig, Germany). A total of 24 h after the seeding, the
cells were washed with PBS and fixed on ice with methanol/acetone
solution (7:3 ratio; Sigma Aldrich; Merck Millipore) for 15 min for
centrosome analysis. The cells for determination of Rad51 foci were
treated with equitoxic doses of MMC (V79 and V79B: 45 ng/ml;
CL-V8B: 2 ng/ml; V-C8: 0,5 ng/ml) or BLM (0.4 µg/ml) for 1 h and
then transferred again to Ham's F10 medium. At the indicated time
points (8, 24, 48 and 56 h) after the MMC or BLM treatment, the
cells were washed and fixed in 2% formaldehyde (MP Biomedicals LLC,
Solon, OH, USA) for 15 min. Then, the cells were permeabilized with
0.1% Triton X-100 (MP Biomedicals LLC) and blocked for 30 min with
PBS (PBS+) containing 0.15% glycine and 0.5% bovine serum albumin
(Sigma-Aldrich; Merck Millipore). For centrosome analysis, all the
stages mentioned above were held on ice. Subsequently, the cells
were incubated for 1.5 h with mouse monoclonal antibodies against
γ-tubulin (dilution 1:1,000; T655T; GTU-88 clone; Sigma-Aldrich;
Merck Millipore) or rabbit polyclonal antibodies against Rad51
(2307; donated by Dr Fiona Benson; Department of Public Health,
Academic Medical Center, University of Amsterdam, Amsterdam, The
Netherlands), washed with 0.1% Triton X-100 and PBS+, and incubated
for 1 h with Alexa-488 conjugated polyclonal goat antibodies
against mouse (R37120) or rabbit (A-11070) IgG (dilution 1:1,500;
Molecular Probes Life Technologies; Thermo Fisher Scientific,
Inc.). Then, the cells were washed again with 0.1% Triton X-100 and
their nuclei were stained with 4′,6-diamino-2-phenylindole (0.15
µg/ml; Sigma-Aldrich; Merck Millipore). Subsequently, the round
coverslips were coated with AquaPoly/Mount (Polysciences, Inc.,
Warrington, PA, USA), placed on standard glass slides (Menzel
Gläser) and stored at 4°C in the dark. In each out of the three
independent experiments, a total of 100 or 300 cells were examined
under Nikon Eclipse E800 fluorescence microscope (Nikon
Corporation, Tokyo, Japan) for the presence of centrosomes or Rad51
foci, respectively. The cells with more than five Rad51 foci per
nucleus were considered as representing an active DNA repair
process.
DNA fiber assay
Exponentially growing cells were labeled with
5-iodo-2′-deoxyuridine (50 µM; IdU; Sigma-Aldrich; Merck Millipore)
for 20 min, and then exposed to hydroxyurea (4 mM) for 1.5 h.
Subsequently, the cells were washed 3 times with PBS and a fresh
growth medium containing 5-chloro 2′-deoxyuridine (50 µM; CldU;
Sigma-Aldrich; Merck Millipore) was added for 20 min. To examine
unperturbed replication, the HU exposure stage was omitted in the
controls. DNA fibers were prepared as described previously, with
certain modifications (24).
Subsequent to labeling, the cells were trypsinized, centrifuged at
room temperature for 10 min at 175.5 × g and resuspended in
ice-cold PBS to obtain a concentration of 0.75×106 cells per ml.
Subsequently, 2 µl suspension was dropped on a glass slide, left
for 2 min and mixed with 8 µl lysis buffer (50 mM EDTA, 0.5% sodium
lauryl sulfate, 200 mM Tris-HCl; Sigma-Aldrich; Merck Millipore).
Then, the slides were carefully tilted to a 45° angle to allow the
DNA fibers to spread. Subsequently, the slides were air-dried,
fixed with methanol/glacial acid solution (3:1 ratio) for 10 min,
washed with dH2O and left overnight at 4°C. Next day,
the slides were treated with 2.5 M HCl (Merck Millipore) for 90
min, washed in PBS and blocked for 1 h with 5% BSA in PBS
containing 0.1% Triton X-100. Then, the slides were incubated with
a mixture of primary α-BrdU mouse (dilution 1:25; 347580; BD
Biosciences, San Jose, CA, USA) and rat antibodies (dilution 1:400;
ab6326; Abcam, Cambridge, MA, USA) for 2 h, washed and incubated
with secondary antibodies: Alexa-488 conjugated goat antibodies
against mouse (1:200; R37120) and Alexa-555 conjugated goat
antibodies against rat (1:350; A-21434) from Molecular Probes Life
Technologies for 1 h. DNA fibers were examined under the Nikon
Eclipse E800 fluorescence microscope. A minimum of 150–200
individual fibers were analyzed for each experiment (repeated three
times) using ImageJ software, version 1.49 (National Institutes of
Health, Bethesda, MD, USA). The frequencies of various classes of
replication structures were expressed as the percentages of all
counted structures. Fork velocity was determined on the basis of
the conversion factor (1 µm=2.59 kb) established by Jackson and
Pombo (24).
Cell cycle analysis by flow
cytometry
Cells from the exponential growth phase were exposed
to MMC (50 and 250 ng/ml) for 8, 24 and 48 h. Then, the cells were
trypsinized, centrifuged, washed with PBS and centrifuged again.
Subsequently, approximately 1–2×106 cells were stained with a
solution of propidium iodide (50 µg/ml; Sigma-Aldrich; Merck
Millipore) and 0.03% Nonidet P-40 (Sigma-Aldrich; Merck Millipore)
for 15 min at room temperature in the dark, and incubated with
RNAse A (100 µg/ml, 88 Kunitz U/ml; Sigma-Aldrich; Merck Millipore)
under the same conditions. The number of cells from
sub-G1, G0/G1, S and
G2/M phases was determined on the basis of their DNA
contents, using FACS Canto II flow cytometer (BD Biosciences). The
results were analyzed with FlowJo software, version 7.5.5 (Tree
Star LLC, Ashland, OR, USA). Each experiment was repeated
independently 3 times.
Statistical analysis
Statistical analysis was conducted with Statistica
package (version 12; StatSoft Polska Sp. z o.o., Kraków, Poland).
The descriptors of cell cycle distribution (percentages of cells
from various cell cycle phases) at three time points (8, 24 and 48
h) were subjected to univariate one-way analysis of variance with
repeated measures (dose 0, 50 and 250 ng/ml), followed by post hoc
analysis with the least significant difference test. The results
are presented as the means of biological replicates with their
standard errors unless otherwise specified. All graphs and figures
were prepared with GraphPad Prism 5.0 software (GraphPad Software
Inc., La Jolla, CA, USA).
Results
Cross-sensitivity of CL-V8B to various
genotoxic agents
The CL-V8B cell line originates from ENU mutagenized
Chinese hamster wild type fibroblasts derived from lungs (V79B). It
was isolated at the Department of Toxicogenetics, Leiden University
Medical Centre, The Netherlands, on the basis of its
hypersensitivity to MMC, using the replica plating method as
described previously (25).
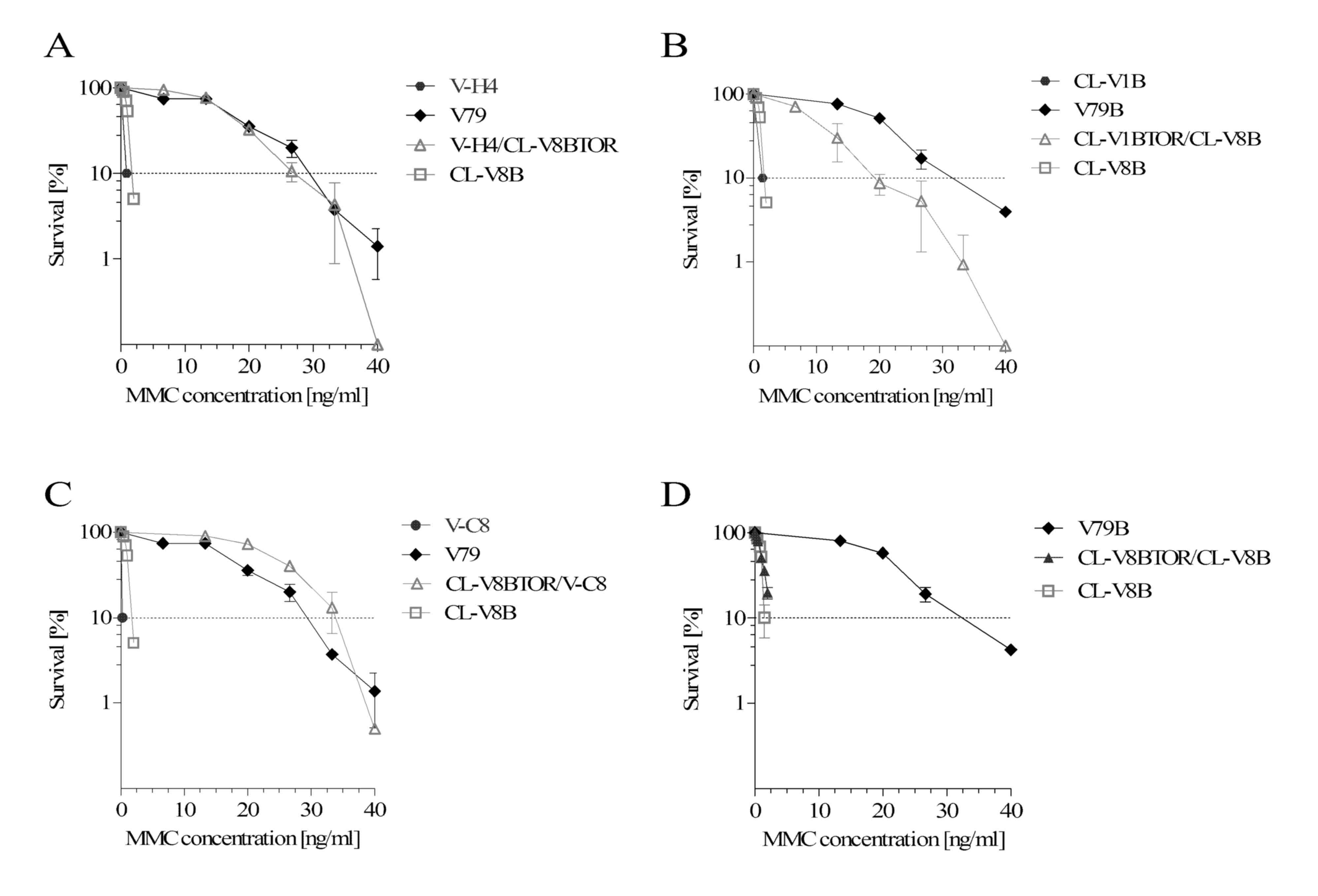
The results of clonogenic survival assays (Fig. 1A and B) imply that CL-V8B mutant
were significantly more sensitive to crosslinking agents, MMC
(~17-fold) and CDDP (~7.4-fold) than the parental V79B cell line.
MMC is an antibiotic and a powerful bifunctional alkylating agent,
forming six covalent DNA adducts (26). The covalent link between two DNA
strands (ICLs) interferes with fundamental cellular processes, such
as replication and transcription, and is responsible for MMC
cytotoxicity. Compared with MMC, cisplatin forms fewer ICLs and a
large percentage of intrastrand crosslinks (27). The fact that the CL-V8B mutant is
hypersensitive to crosslinking agents, suggests strongly that this
cell line has defective ICLs repair. To test whether CL-V8B cells
were also sensitive to other genotoxic agents, a series of
clonogenic survival assays were conducted.
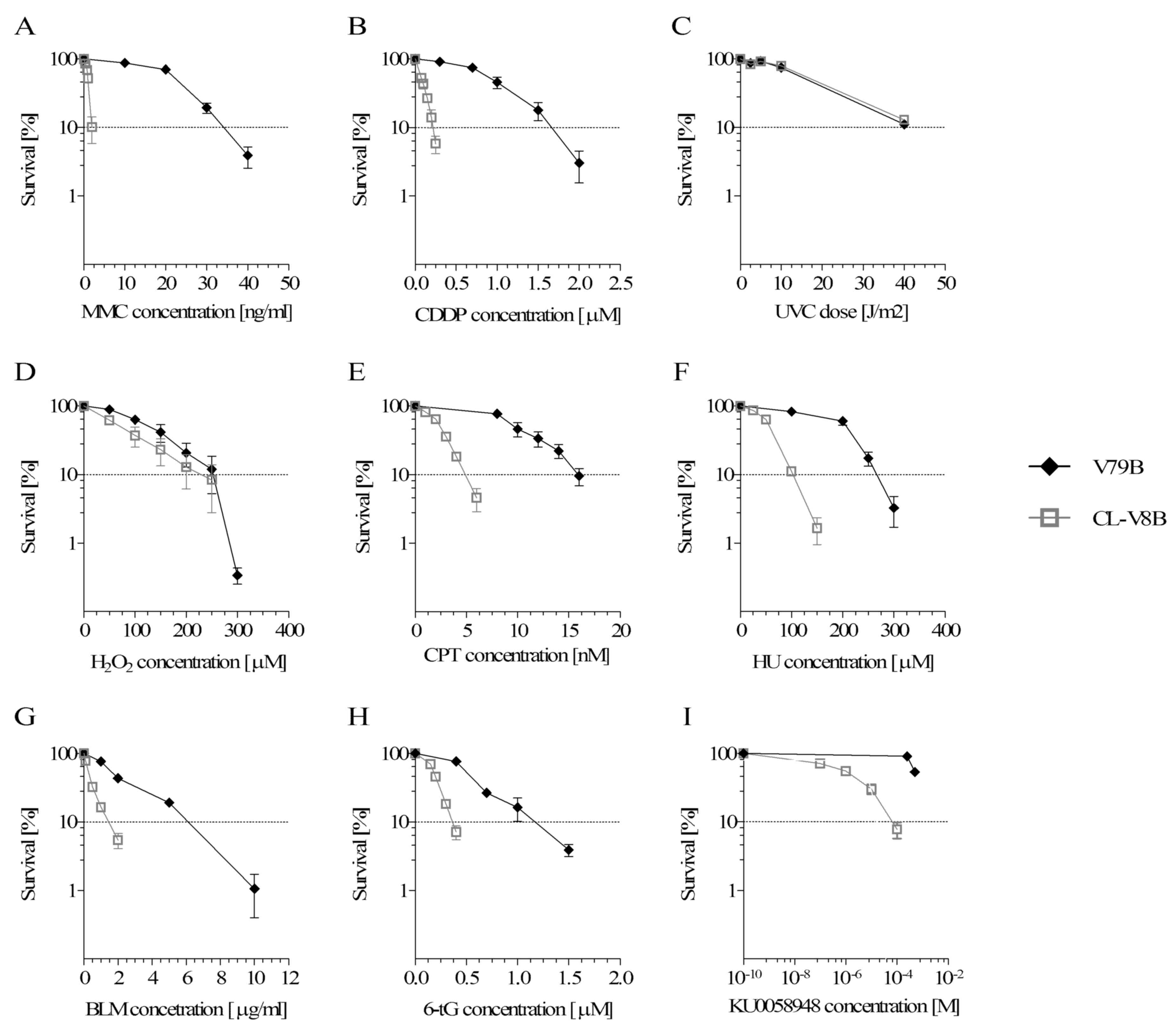 | Figure 1.Clonogenic survival of CL-V8B and the
parental cell line V79B following exposure to (A) MMC, (B) CDDP,
(C) UVC radiation, (D) hydrogen peroxide, (E) CPT, (F) HU, (G) BLM,
(H) 6-tG, (I) poly (adenosine diphosphate-ribose) polymerase
inhibitor KU0058948. Data are presented as the mean values from at
least three independent experiments. Error bars represent standard
error. MMC, mitomycin C; CDDP, cisplatin; CPT, camptothecin; HU,
hydroxyurea; BLM, bleomycin; 6-tG, 6-tioguanine. |
It was demonstrated that CL-V8B was not sensitive to
ultraviolet radiation and oxidative DNA stress caused by
H2O2 (Fig. 1C
and D). However, CL-V8B displayed a moderate-degree sensitivity
to agents that interfere with replication forks, e.g. topoisomerase
I inhibitor CPT (~3.2-fold) and HU (~2.5-fold; Fig. 1E and F, respectively). The
distinctive features of the mutant included its ~4-fold greater
sensitivity to BLM (Fig. 1G), a
radiomimetic compound associated with formation of single strand
breaks (SSBs) and DSBs within DNA, in addition to hypersensitivity
to 6-thioguanine (~3.5-fold; Fig.
1H), an antimetabolite causing DSBs with resultant Rad51 foci
formation and repair of these damages through the HR process
(2). The cells carrying mutations
of various HR genes, e.g. BRCA1/2, RAD51,
NBS1 and ATM, are known to be highly sensitive to
poly (ADP-ribose) polymerase inhibitors (iPARP) (3,28).
Analysis of clonogenic survival of CL-V8B cells treated with a
potent PARP inhibitor KU0058948 indicated that mutant was extremely
sensitive to this agent (~430-fold, Fig. 1I). Inhibition of PARP interrupts
repair of DNA SSBs by base excision repair and leads to
accumulation of these lesions. SSBs are then transformed into DSBs
(28). Since DSBs are formed
during ICLs removal and repaired via HR, the sensitivity of
CL-V8B cells to crosslinking agents is likely linked to impairment
of HR.
Complementation of MMC
sensitivity
To establish whether CL-V8B belongs to any of
previously identified groups of Chinese hamster cell mutants
hypersensitive to crosslinking compounds, a genetic complementation
analysis was conducted. This method, based on cell fusion combined
with clonogenic survival assay, allows it to be established whether
a gene defective in one cell line is complemented by proper copy of
the same gene from another cell line used to create a hybrid. If
the phenotype of the hybrid cells is similar to the wild type, they
belong to different complementation groups. CL-V8B cells were fused
with all four previously identified Chinese hamster cell lines with
established HR defects, in addition to seven other cell mutants
sensitive to crosslinking agents (Table I). Representative survival curves
for CL-V8B hybrids with cells from various complementation groups
and a control hybrid CL-V8BTOR/CL-V8B, all exposed to MMC, are
presented on Fig. 2. All analyzed
hybrids, apart from the control hybrid created exclusively of
CL-V8B cells, exhibited similar sensitivity to MMC as the parental
V79B line; this implies that the ICLs repair defect present in
CL-V8B was corrected due to fusion with other cells. This points to
CL-V8B as a representative of a new previously uncharacterized
complementation group of Chinese hamster cell mutants being
sensitive to crosslinking agents.
 | Table I.Complementation of MMC sensitivity in
hybrids of CL-V8B with other Chinese hamster cell mutants
representing different complementation groups. |
Table I.
Complementation of MMC sensitivity in
hybrids of CL-V8B with other Chinese hamster cell mutants
representing different complementation groups.
| Mutant | Defected
gene/pathway | Complementation of
MMC sensitivity after fusion with CL-V8B cells |
|---|
| irs1 |
Xrcc2/HR | a,b |
| irs1SF |
Xrcc3/HR | a,b |
| CL-V4B |
Rad51C/HR | a,b |
| V-C8 |
Xrcc11/Brca2/Fancd1/HR and FA | a |
| UV40 |
Xrcc9/Fang/FA | a,b |
| V-H4 |
Fanca/FA | a |
| CL-V1B, CL-V5B,
CL-V101B |
Fanccd/FA | a,b |
| UV20 |
Ercc1/NER | a,b |
| UV41 |
Ercc4/NER | a,b |
| CL-V8B | ? | c |
Kinetics of Rad51 foci formation
To determine the status of homologous recombination
in CL-V8B, Rad51 foci formation was analyzed subsequent to exposure
of the cells to MMC and radiomimetic BLM. Mutations in the majority
of HR genes were previously demonstrated to impair Rad51 foci
formation (8,20–22,29,30).
Monomers of Rad51 recombinase are recruited to stalled replication
forks above the DSBs and create nucleoprotein filaments, which are
essential for strand invasion during DSBs repair. Presence of the
DNA damage-induced Rad51 foci in the nucleus is postulated to be a
marker of DSBs repair by homologous recombination (29).
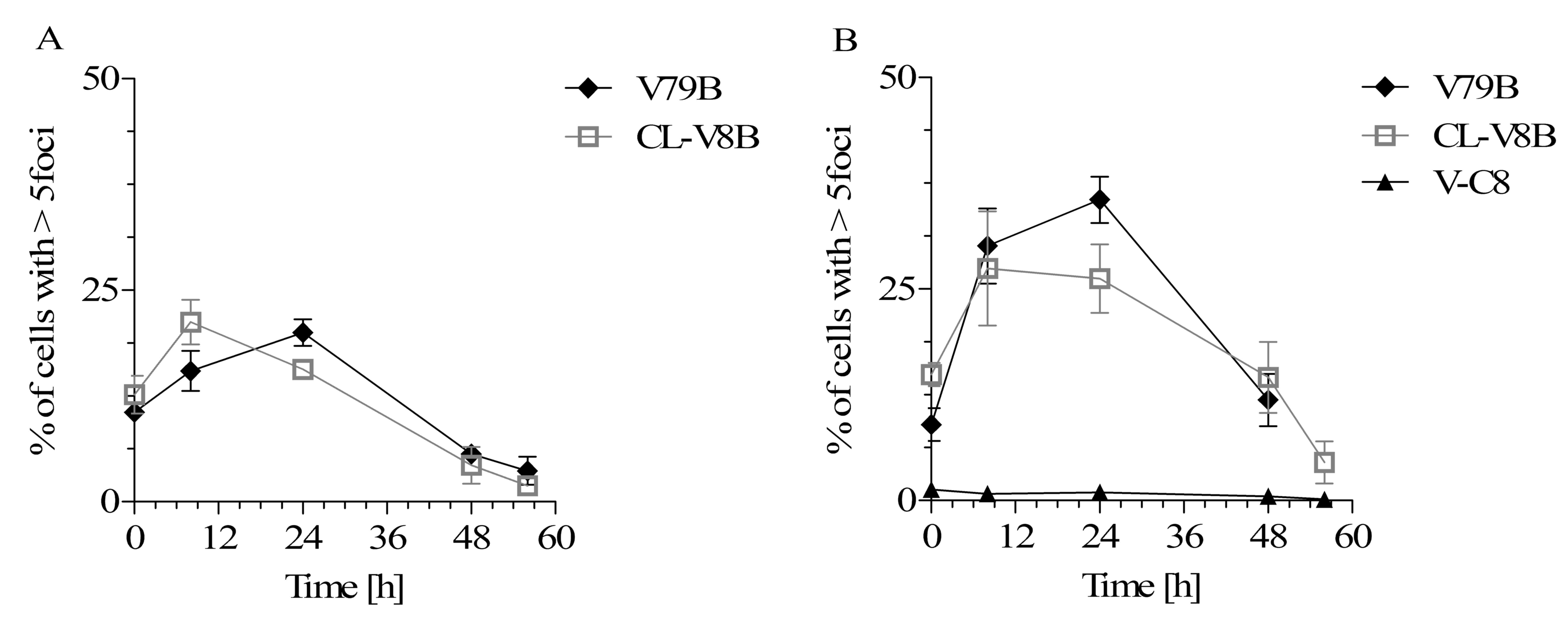
Although the results of clonogenic survival analysis
suggested the presence of HR disturbances in CL-V8B, notably,
induction of Rad51 foci was observed in these mutant cells
subsequent to their exposure to either BLM or MMC (Fig. 3). In contrast, Rad51 foci were not
identified in the nuclei of control HR mutant V-C8 cells with
defective Brca2/Fancd1 genes (Fig. 3B), which is consistent with
previously published data (29).
The CL-V8B and parental V79B cell lines differed marginally
regarding their Rad51 foci formation kinetics following treatment
with BLM. Although the initial percentage of Rad51 foci-positive
cells in both cell lines was similar, the fraction of the positive
CL-V8B cells reached its maximum (21.2%) after 8 h and then started
to decrease, whereas the percentage of the positive V79B cells
continued to increase up to 24 h (20.0%, Fig. 3A). Following 48 and 60 h, the
proportions of Rad51 foci-positive cells decreased below their
baseline levels and were again similar for both cell lines. In
contrast, exposure to MMC did not alter significantly the kinetics
of Rad51 foci formation in the mutant cell line (Fig. 3B). Maximum percentage of CL-V8B
cells containing Rad51 foci was reached after 8 h (27.4%) and
remained mostly stable up to 24 h (26.2%). In turn, maximum
fraction of the positive V79B cells was observed no earlier than
after 24 h (35.6%). These data imply that while the formation of
Rad51 foci in CL-V8B cells is generally normal, the kinetics of
this process at 8 and 24 h post-exposure is marginally different
than in the parental cells.
Spontaneous and mutagen-induced sister
chromatid exchanges
SCEs are a cytological manifestation of DNA brakeage
and rejoining. They represent the exchange of homologous DNA
fragments between sister chromatids during physiological processes,
including meiosis and replication. SCEs are additionally markers of
HR function, since the number of sister chromatid exchanges in
cells with defective HR was demonstrated to be lower than in normal
cells (8,21,30–32).
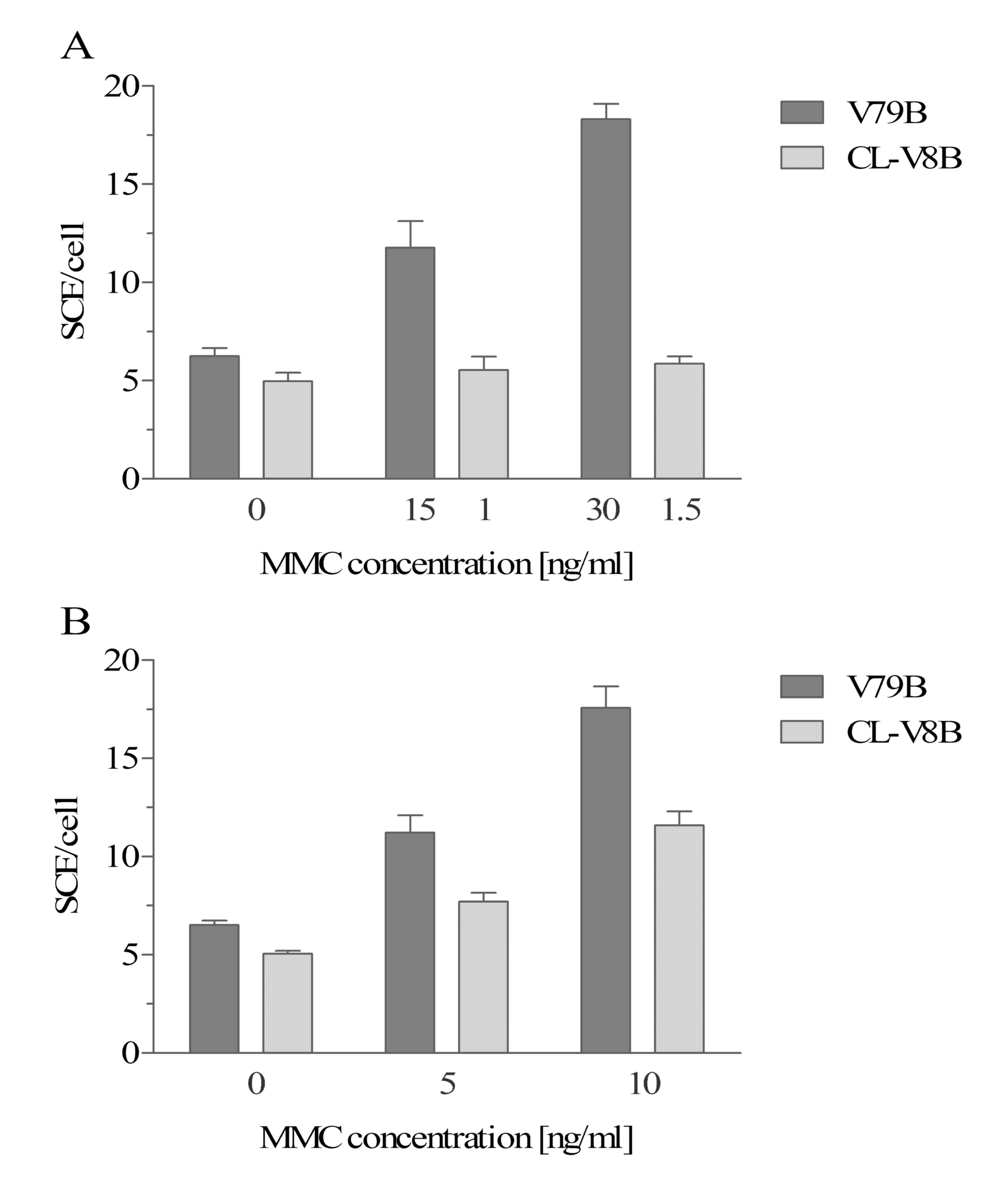
To evaluate the efficiency of homologous
recombination, the number of SCEs in CL-V8B cells and parental
cells exposed to MMC was evaluated. The number of spontaneous SCEs
in CL-V8B cells was observed to be marginally lower than in the
parental V79B cells (4.96±0.44 and 6.25±0.4 SCEs per cell,
respectively; Fig. 4A). Exposure
to MMC, regardless of its concentration, resulted in only a small
increase in the number of SCEs in CL-V8B cells (to 5.53±0.69 and
5.87±0.36 at 1 and 1.5 ng/ml, respectively); in contrast, the
number of SCEs in the wild type cells exposed to MMC was markedly
higher and increased in a dose-dependent manner (to 11.77±1.35 and
18.31±0.77 at 15 and 30 ng/ml, respectively). In order to confirm
that the variance in the SCEs number did not reflect the differing
susceptibility of CL-V8B and V79B cells to MMC, this parameter was
determined after exposure to UVC, a potent inductor of SCEs.
Irradiation with 5 and 10 J/m2 resulted in a marginal
increase in the number of SCEs in CL-V8B cells (from 7.7±0.45 to
11.62±0.58), which was markedly lower than in the parental cells
(an increase from 11.26±0.8 to 17.46±1.27; Fig. 4B). This clearly demonstrates that
CL-V8B display lower number of MMC- and UVC-induced SCEs, which
indicates a likely impairment of HR process within the mutant cell
line.
Centrosome analysis
Centrosomes constitute an organization center of
microtubules within the cell. Mutations of HR genes are often
associated with abnormalities of centrosomes, and frequently
manifest as numeric aberrations, disrupting the cellular role of
these structures and leading to genomic instability (21,33).
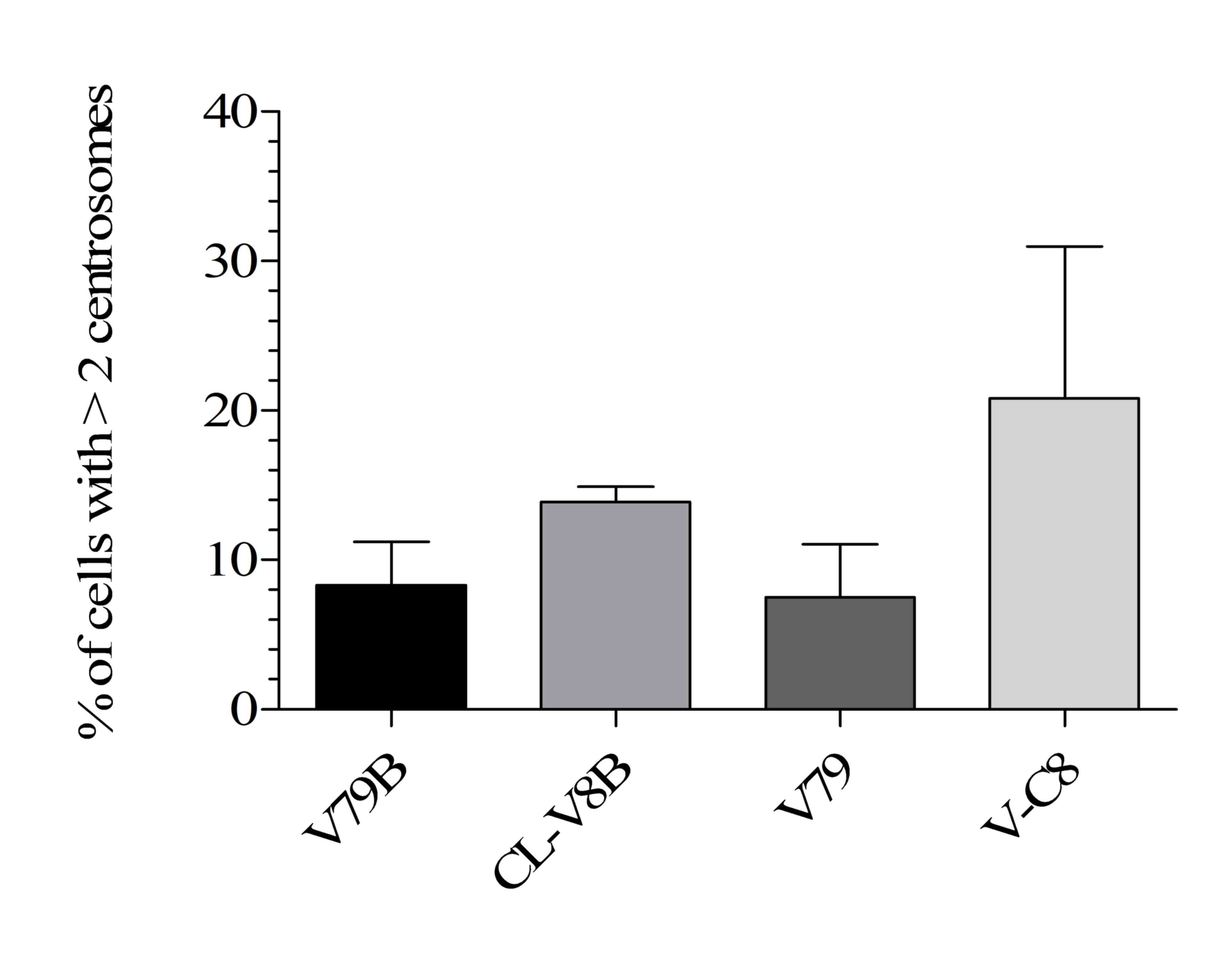
In order to establish whether the defect present in CL-V8B cells
affects the number of centrosomes, these organelles were subjected
to an immunofluorescent analysis. V-C8 cells with a mutation of the
Brca2/Fancd1 HR gene and elevated numbers of centrosomes were used
as a positive control (21).
Immunofluorescent staining indicated that the proportion of CL-V8B
cells with an abnormal number of centrosomes was twice as high as
in the parental V79B line (21.3 vs. 10.6%; Fig. 5). Notably, the percentage of the
defective cells was similar to that of the V-C8 line. In addition,
small labelled structures were identified within CL-V8B cells;
these structures may represent numerical or structural
abnormalities of centrosomes, e.g. their fragmentation, which may
result in the presence of acentriolar centrosomal fragments
(acentriolar bodies) acting as additional chromosomes (34). These observations suggest that the
defect present in the CL-V8B line affects the number of centrosomes
in these cells.
Chromosomal aberrations
Alterations in CL-V8B leading to numerical
abnormalities of centrosomes may predispose these cells to genomic
instability. Cells with compromised DNA repair often exhibit
increased numbers of spontaneous and mutagen-induced CAs (8,19,21,22).
It was demonstrated that CL-V8B cells presented with a 1.65-fold
greater number of spontaneous aberrations than the V79B cells
(25.33±7.64 and 15.33±0.58, respectively) and approximately
two-fold greater number of CAs following MMC treatment, regardless
of the concentration of this agent (Table II). The key type of spontaneous
and MMC-induced CAs in the two cell lines were chromatid gaps,
however also chromosomal gaps and chromatid breaks were observed
with a considerable frequency (Table
III). All types of CAs were more prevalent in mutants than in
wild type cells, which was particularly evident in the case of
chromatid gaps and dicentric chromosomes (Table III). Compared with wild type
cells, mutant cells displayed spontaneous and induced CAs following
exposure to markedly lower concentrations of MMC. This phenomenon
reflected hypersensitivity of CL-V8B to this compound, suggesting
that the molecular alteration present in the mutant affects its
genomic stability.
| Table II.Total number of spontaneous and
MMC-induced chromosomal aberrations. |
Table II.
Total number of spontaneous and
MMC-induced chromosomal aberrations.
| Cell line | MMC concentration
(ng/ml) | CAs ± SD |
|---|
| V79B | 0 | 15.33±0.58 |
|
| 45 | 17.33±5.77 |
|
| 70 | 23.67±4.62 |
| CL-V8B | 0 | 25.33±7.64 |
|
| 2.5 | 32.00±5.29 |
|
| 5 | 45.33±1.15 |
| Table III.Various types of spontaneous and
MMC-induced CAs. |
Table III.
Various types of spontaneous and
MMC-induced CAs.
|
| Spontaneous | MMC-induced |
|---|
|
|
|
|
|---|
| Type of structural
CAs | V79B | CL-V8B | V79B
45a | CL-V8B
2.5a | V79B
70a | CL-V8B
5a |
|---|
| ctg | 9.00±1.00 | 12.33±3.06 | 6.00±2.00 | 15.33±3.06 | 11.67±3.51 | 23.67±3.06 |
| csg | 3.00±3.00 | 4.33±2.89 | 4.00±2.65 | 4.33±1.15 | 6.33±2.52 | 7.67±3.79 |
| ctb | 1.33±0.58 | 3.67±1.53 | 3.00±1.00 | 4.00±1.73 | 2.33±2.52 | 5.67±1.53 |
| dic | 0.67±0.58 | 3.33±3.51 | 1.00±1.73 | 6.00±1.73 | 1.0±1.00 | 5.0±1.73 |
| af | 0.67±0.58 | 1.00±1.00 | 1.67±1.53 | 1.33±0.58 | 1.33±1.15 | 1.33±1.53 |
| ci | 0.33±0.58 | 0.67±0.58 | 0.33±0.58 | 0.67±1.15 | 0.33±0.58 | 1.67±1.15 |
| ring | 0.00±0.00 | 0.00±0.00 | 0.00±0.00 | 0.33±0.58 | 0.00±0.00 | 0.33±0.58 |
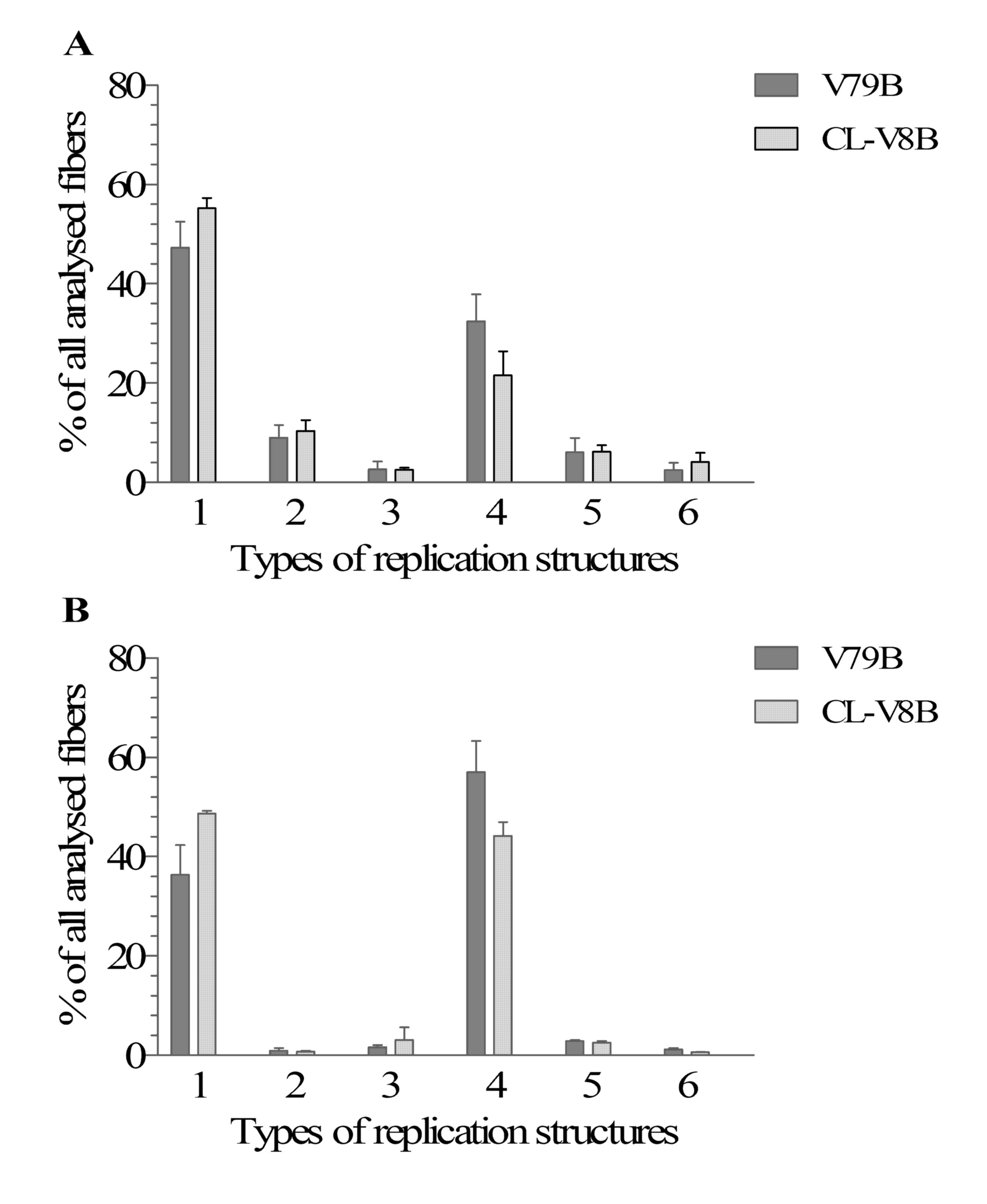
Replication analysis by DNA fiber
assay
Previous studies suggest that proteins involved in
ICLs repair, such as FA and HR, participate in additional processes
essential for maintenance of genome stability, such as DNA
replication. During this process, DNA repair proteins perform
various functions being crucial for stability of replication forks
(35–37). The DNA fiber assay allows the
establishment of the replication fork velocities and enables
various replication structures to be visualized, such as
replication fork elongation, initiation of new origins, fork
stalling and termination of replication. The replication structures
of CL-V8B cells and parental cells were analyzed without their
exposure to replication stress and following treatment with
hydroxyurea. HU blocks the replication fork due to reduction of the
deoxyribonucleotide triphosphate pool, resulting from inhibition of
ribonucleotide reductase activity necessary for synthesis of these
compounds (38).
Replication fork velocity for both CL-V8B and V79B
cells approximated 1 kb per min (0.86±0.24 and 1.04±0.13,
respectively). This suggests that the alteration present in CL-V8B
does not affect the speed of replication forks. When not exposed to
any replication stress, CL-V8B and parental cells differed solely
in terms of their type 4 replication structures. Specifically,
replication fork stalling in CL-V8B cells occurred markedly less
often than in the parental cells. A reduction in the number of this
replication structure in the mutant cells correlated with an
increase in the number of replication forks being in the process of
elongation (i.e. type 1 replication structures, Fig. 6A). However, these differences were
not statistically significant. The number of other replication
structures was similar in the two cell lines. Notably, although
CL-V8B cells are marginally more sensitive to HU than the parental
V79B line (Fig. 1F), the results
subsequent to exposure to this agent were essentially the same as
without any replication-stress (Fig.
6B). This suggests that the defect present in the CL-V8B mutant
does not affect the efficiency and stabilization of replication
forks. However, the differences between CL-V8B and V79B imply that
the former may show defects in certain mechanisms responsible for
the replication stress signaling.
Cell cycle analysis
Presence of DNA damage within the cell triggers
complex damage response pathways, resulting in activation of cell
cycle checkpoints, blockade of cell cycle progression, recruitment
of repair proteins to the damaged DNA site and induction of
apoptosis. The most common type of cell response observed
subsequent to exposure to MMC is S-phase arrest,
G2/M-phase delay and increase in the number of apoptotic
cells (39,40). Taking into account available data
on cell cycle disturbances in HR-deficient cells (41,42)
and activation of apoptosis resulting from unrepaired DSBs
(43), in addition to the
observations indicating the presence of impaired homologous
recombination in CL-V8B cells, the effects of MMC on cell cycle
progression were analyzed in these mutant cells and their parental
V79B line. Cell cycle progression was analyzed 8, 24 and 48 h
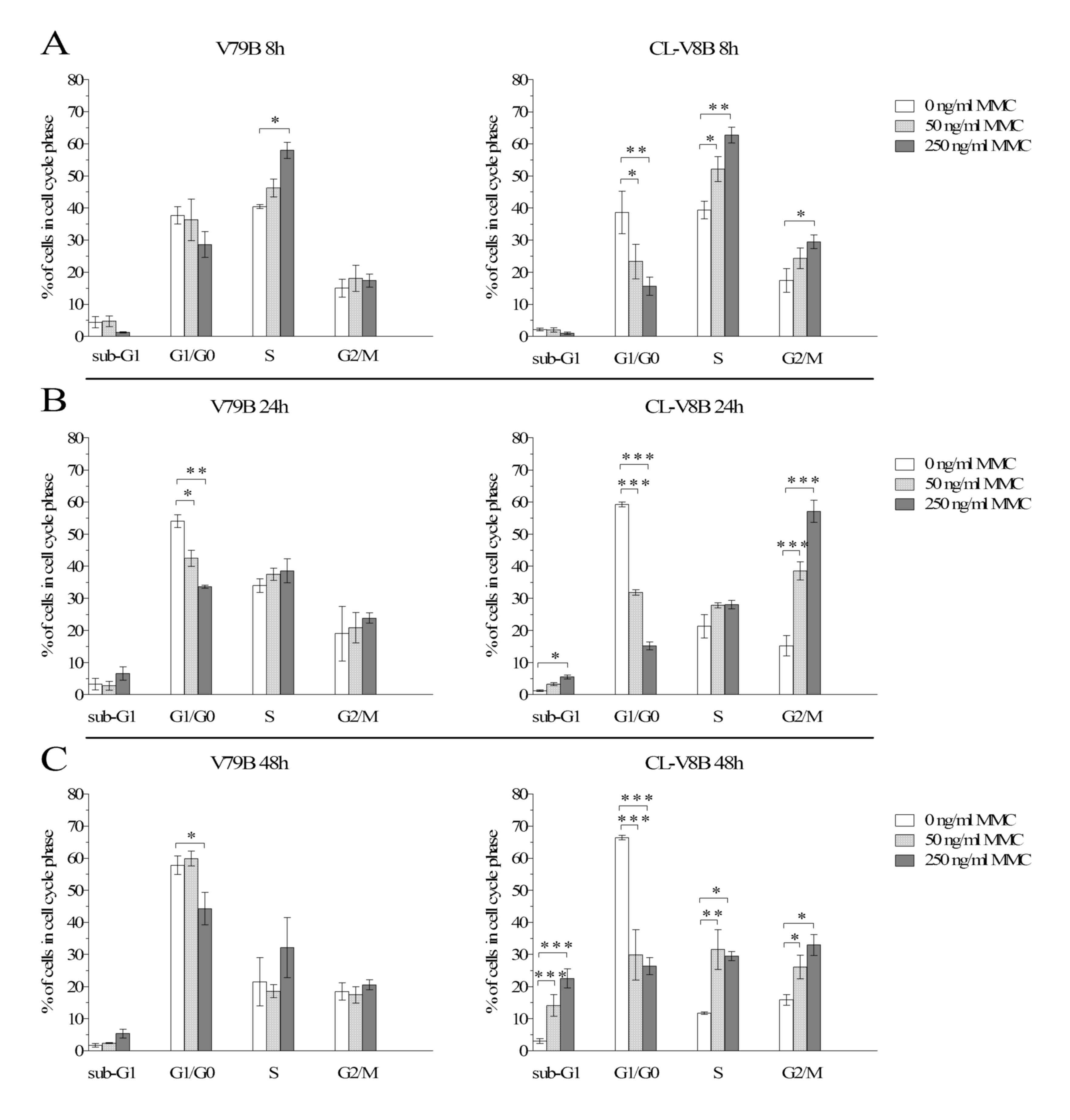
subsequent to treatment with 50 and 250 ng/ml MMC (Fig. 7 and Tables IV and V).
| Table IV.Cell cycle distribution in the
parental V79B cell line following MMC treatment. |
Table IV.
Cell cycle distribution in the
parental V79B cell line following MMC treatment.
|
| Time | 8 h | 24 h | 48 h |
|---|
|
|
|
|
|
|
|---|
| Cell line | Cell phase\MMC
(ng/ml) | 0 | 50 | P-value | 250 | P-value | 0 | 50 | P-value | 250 | P-value | 0 | 50 | P-value | 250 | P-value |
|---|
| V79B |
Sub-G1 |
4.37±1.75a |
4.70±1.65 | 0.8767 |
1.22±0.17 | 0.1412 |
3.29±1.82 |
2.75±1.36 | 0.800 |
6.64±2.04 | 0.1176 |
1.75±0.60 |
2.42±0.11 | 0,7504 |
5.35±1.37 | 0,0933 |
|
|
G1/G0 | 37.70±2.67 | 36.28±6.42 | 0.7955 | 28.61±4.04 | 0.1047 | 54.09±1.96 | 42.44±2.51 | 4.0E-2 | 33.61±0.47 | 6.8E-4 | 57.78±2.81 | 59.93±2.28 | 0.6961 | 44.29±5.01 | 1.8E-2 |
|
| S | 40.41±0.63 | 46.23±2.85 | 0.2943 | 57.98±2.48 | 2.7E-3 | 33.98±2.13 | 37.50±1.88 | 0.5238 | 38.57±3.66 | 0.4061 | 21.48±7.48 | 18.59±2.07 | 0.6000 | 32.15±9.38 | 0.0588 |
|
|
G2/M | 15.03±2.76 | 18.10±4.03 | 0.5447 | 17.38±2.04 | 0.6426 | 19.01±8.49 | 20.85±4.72 | 0.7161 | 23.85±1.65 | 0.3422 | 18.46±2.70 | 17.40±2.60 | 0.8334 | 20.56±1.55 | 0.6782 |
| Table V.Cell cycle distribution in the mutant
CL-V8B cell line following MMC treatment. |
Table V.
Cell cycle distribution in the mutant
CL-V8B cell line following MMC treatment.
|
| Time | 8 h | 24 h | 48 h |
|---|
|
|
|
|
|
|
|---|
| Cell line | Cell phase\MMC
(ng/ml) | 0 | 50 | P-value | 250 | P-value | 0 | 50 | P-value | 250 | P-value | 0 | 50 | P-value | 250 | P-value |
|---|
| CL-V8B |
Sub-G1 |
2.14±0.45 |
2.00±0.67 | 0.9482 |
0.96±0.39 | 0.5771 |
1.18±0.25 |
3.26±0.47 | 0.3257 |
5.56±0.60 | 4.3E-2 |
3.06±0.73 | 14.08±3.32 | 7.1E-6 | 22.56±2.98 | 5.1E-11 |
|
|
G1/G0 | 38.61±6.61 | 23.34±5.42 | 8.5E-3 | 15.63±2.81 | 1.8E-4 | 59.24±0.80 | 31.80±0.86 | 1.8E-5 | 15.15±1.20 | 3.2E-9 | 66.39±0.69 | 29.90±7.89 | 1.4E-7 | 26.38±2.61 | 2.4E-8 |
|
| S | 39.36±2.70 | 52.14±3.87 | 2.5E-2 | 62.72±2.46 | 1.3E-4 | 21.29±3.66 | 27.84±0.82 | 0.2383 | 28.10±1.32 | 0.2210 | 11.69±0.39 | 31.56±6.19 | 8.6E-4 | 29.50±1.43 | 2.4E-3 |
|
|
G2/M | 17.43±3.68 | 24.30±3.20 | 0.1798 | 29.48±2.20 | 2.1E-2 | 15.22±3.18 | 38.50±2.80 | 4.7E-5 | 57.07±3.44 | 7.4E-10 | 15.82±1.64 | 26.15±3.65 | 4.7E-2 | 32.94±3.27 | 1.6E-3 |
The alterations in the distribution of V79B cells
representing various phases of cell cycle were more evident
following their exposure to 250 ng/ml MMC. A significant decrease
in the percentage of G1/G0-phase cells was
observed 8 h post-exposure, along with an increase in the fraction
of S-phase cells (P=2.7E-3; Fig.
7A, left panel). The decrease in the proportion of
G1/G0-phase cells was additionally observed
24 h post-exposure, reaching statistical significance at P=6.8E-4
(Fig. 7B). This was associated
with a marginal increase in the proportion of S- and
G2/M-phase cells, in addition to an increase in the
fraction of cells with sub-G1 DNA content, corresponding
to fragmentation of DNA at a late stage of apoptosis. 48 h
post-exposure, V79B cells indicated a significant decrease in the
proportion of G1/G0-phase cells (P=1.8E-2),
however the fraction of S-phase cells was markedly lower than at 8
and 24 h (Fig. 7C). V79B cells
were less sensitive to MMC at 50 ng/ml; exposure to this agent
resulted in only minor cell cycle distortion at all analyzed time
points, apart from a significant albeit transient
G1/G0-phase delay at 24 h (P=4.0E-2, Fig. 7B, Table IV). A total of 48 h post-exposure
to 50 ng/ml MMC, the distribution of cells representing various
phases of cell cycle was similar to that in non-treated
controls.
In the case of the CL-V8B line, severe MMC-induced
cell cycle disturbances were observed irrespective of concentration
and exposure time. At 8 h post-exposure, a dose-dependent decrease
in the proportion of G1/G0-phase cells was
observed (P=8.5E-3 and P=1.8E-4 for 50, and 250 ng/ml MMC,
respectively; Fig. 7A, right
panel; Table V) along with an
evident S-phase arrest (observed also in parental cells) and a
marginal increase in the fraction of G2/M-phase cells,
not observed in V79B. Irrespective of MMC concentration, the
percentage of S-phase cells was significantly higher than in
untreated cultures (P=2.5E-2 and P=1.3E-4 for increasing MMC
doses). Despite the dose-dependent increase in the proportion of
G2/M-phase cells, the percentage of these cells turned
out to be significantly higher than in untreated control cultures
only subsequent to exposure to 250 ng/ml MMC (P=2.1E-2).
The delay in cell cycle progression was evident 24 h
post-exposure. A significant reduction in the proportion of
G1/G0-phase cells was observed in the CL-V8B
cell line exposed to either 50 or 250 ng/ml MMC (P=1.8E-5 and
P=3.2E-9, respectively). However, in contrast to the V79B line,
mutant cells that resumed S phase were arrested at G2/M
phase (Fig. 7B). The percentage of
cells representing this phase increased with MMC dose (P=4.7E-5 for
50 ng/ml and P=7.3E-10 for 250 ng/ml). These changes were
accompanied by a dose-dependent increase in the proportion of
sub-G1 DNA cells, statistically significant for 250
ng/ml MMC (P=4.3E-2).
Similar to a previous time point, a significant
decrease in the proportion of G1/G0-phase
CL-V8B cells was also observed 48 h after the MMC treatment
(P=1.4E-7 and P=2.4E-8 for 50 and 250 ng/ml MMC, respectively).
Furthermore, the mutant exhibited S-phase delay (P=8.6E-4 and
P=2.4E-3, respectively) and a dose-dependent accumulation of
G2/M-phase cells (P=4.7E-2 and P=1.6E-3, respectively);
however, the proportion of G2/M-phase cells was lower
than at 24 h (Fig. 7C). These
alterations were accompanied by a significant increase in the
fraction of apoptotic cells (P=7.1E-6 and P=5.1E-11 for 50 and 250
ng/ml MMC, respectively), not observed in the case of the parental
V79B cell line. This implies that the MMC-induced DNA damage in
CL-V8B cells was not completely repaired, which resulted in the
cell cycle delay and a significant increase in the number of
apoptotic cells.
In addition, comparison of percentages of parental
V79B and mutant CL-V8B cells representing the same phases of cell
cycle following exposure to equal MMC concentrations at identical
time points presented with statistically significant differences.
Following 8 h, the two cell lines differed in the proportion of
G1/G0-phase cells for both MMC concentrations
(P<0.05) and in the proportion of G2/M-phase cells
for 250 ng/ml (P<0.05). At 24 h, significant differences in the
percentages of G1/G0- and
G2/M-phase cells were observed between the mutant and
parental cultures irrespective of MMC concentration (P<0.05).
After 48 h, an increase in the proportion of apoptotic cells and a
decrease in the percentage of G1/G0-phase
cells was observed in CL-V8B line exposed to either concentration
of MMC (P<0.00001 and P<0.05, respectively), which
distinguished this cell line from the parental one.
Discussion
CL-V8B represents a new complementation group among
Chinese hamster cell mutants hypersensitive to DNA crosslinking
agents. Aside from increased sensitivity to mitomycin C (~17-fold)
and cisplatin (~7.4-fold), this mutant additionally displayed
hypersensitivity to various DNA-damaging agents, which is typical
for mutants with defects of the HR and FA pathways. CL-V8B cells
demonstrated hypersensitivity to radiomimetic BLM (~4-fold), also
observed in the FA pathway mutant NM3
(Fancg−/−) and in CL-V4B cells with
mutated Rad51C gene, involved in both FA and HR pathways
(8,44). This indicates that one of these
pathways is compromised in the CL-V8B cell line. Owing their
sensitivity to HU (~2.5-fold) and 6-thioguanine (~3.5-fold), CL-V8B
seem to resemble the cells with defected DNA DSBs repair by HR,
such as V-C8 cells carrying a biallelic nonsense mutation in
Brca2/Fancd1 gene (2,21).
Another argument for defect in this repair process stems from
increased sensitivity of CL-V8B to camptothecin (~3.2-fold),
observed additionally in irs1
(Xrcc2−/−) and irs3
(Rad51C−/−), both being HR mutants
(45). Additionally, CL-V8B cells
exhibited a marked sensitivity to PARP inhibitor (~430-fold,
Fig. 1I), which is also a specific
feature of all Chinese hamster cell lines defective in HR, in
addition to human cell lines carrying mutations in BRCA1, BRCA2,
RAD51, USP11, ATM, ATR and NBS1 (3,28,46).
Chinese hamster mutants, V-C8 (Brca2/Fancd1) and
CL-V4B (Rad51C/Fanco), show clear hypersensitivity to
PARP inhibitors, up to 1,000-fold greater in the case of
Brca2-deficient cells (28,47).
Accordingly, PARP inhibition in HR mutants result in accumulation
of DSBs, which may lead to cell death (28). In contrast to HR mutants, the
sensitivity to iPARP among cells with FA pathway damage, such as
FANCD2, FANCA and FANCC, is comparable to that
observed in the case of isogenic wild type cells (3). The current study indicated that
rodent lines CL-V1B, CL-V5B and CL-V101B carrying mutations of the
Fancc gene are not sensitive to iPARP (data not shown). The
cross-sensitivity profile of CL-V8B cells suggests that the defect
inherent to this line most likely involves HR or a part of FA
pathway closely interacting with HR proteins, such as PALB2/Fancn,
Fancj and the structure-specific endonuclease subunit gene Slx4.
Mutation in the remaining FA genes encoding core and ID complexes
in FA pathway were excluded based on examination of Fancd2
monoubiquitination (data not shown). In addition, the deficiency of
NER proteins involved in ICLs repair, Ercc1 and Errc4, whose
failure results in hypersensitivity to crosslinking agents, was
excluded on the basis of complementation analysis with UV20
(Ercc1−/−) and UV41
(Ercc4−/−) mutants (Table I). Another argument for normal
function of NER in CL-V8B cells was their insensitivity to UV.
As high sensitivity of CL-V8B cells to iPARP and
DNA damaging factors, such as BLM and MMC, indicated potential
failure of homologous recombination, a series of experiments was
conducted to verify the course of this process. A key step of HR is
formation of the Rad51 nucleofilament. Induction of Rad51 foci is
impaired in cells with abnormalities of genes encoding Rad51 and
its four paralogs: Rad51C/Fanco, Rad51D,
Xrcc2 and Xrcc3, in addition to in cells with defects
of Brca2/Fancd1, PALB2/FANCN,
DSS1 and ATM genes (8,20–22,29,30,48–50).
However, abnormal nucleofilament induction is not observed in cells
with certain other mutated HR genes, including Rad52, Rad54,
Nbs1 and Usp11 (46,51,52).
Nbs1 mutation was demonstrated to be reflected by prolonged
presence of Rad51 foci in cell nucleus, and silencing of
USP11 resulted in their faster removal, which implies that
defective HR may also alter Rad51 kinetics (46,53).
Similar changes were observed in the CL-V8B mutant exposed to BLM;
these cells were characterized by earlier induction and reached
maximum number of Rad51 foci faster than the parental cells
(Fig. 3A). It is suggested that
unidentified defective proteins present in CL-V8B cells are
involved in the formation and stabilization of the Rad51
nucleofilament, however no serious disturbances of this process
were observed in the current study. It is also possible that CL-V8B
may carry a defective gene encoding a protein involved in DSBs
removal, which interacts with Rad51 nucleofilament formation,
however serves a principal role at later stages of the repair.
Sister chromatid exchanges take place during
restoration of replication after replication fork collapse due to
gaps in DNA and presence of DSBs. Mutations in homologous
recombination genes reduce the number of SCEs, as presented
previously in Chinese hamster HR mutants, V-C8 and CL-V4B (8,21).
In addition, rodent cell lines with defective Rad51D and Rad54
demonstrated a considerable decrease in the number of MMC-induced
SCEs, despite a normal number of spontaneous exchanges (30,31,54).
However, the number of SCEs in HR-defective cells appears to vary
depending on their type. Chicken DT40 with disrupted Rad54
and cells with silenced Rad51 and Rad51 paralogs,
Rad51C, Rad51D, Xrcc2 and Xrcc3, indicated
reduced frequency of both spontaneous and MMC-induced SCEs
(32). A smaller number of
spontaneous SCEs was also observed in yeast cells with mutation of
rad52, and DNA damage-associated SCEs were completely
abolished in these cells (55).
However, mutation in Nbs1 did not exert an effect on the
SCEs number (56). It was
demonstrated that the frequency of spontaneous SCEs in the CL-V8B
line was marginally reduced; however, exposure of these cells to
either MMC or UVC resulted in only small changes in the SCEs
number, which constitutes another argument for the presence of a HR
defect in these mutant cells.
Another characteristic feature of CL-V8B was the
increased proportion of cells with numeric abnormalities of
centrosomes (Fig. 5). An increase
in the number of cells with additional centrosomes was also
identified in V-C8 cells with defective Brca2/Fancd1
gene, human cells carrying mutations in NBS1 and ATR,
and mouse embryonic stem cells with deletion of the 11th exon in
Brca1 (53,57). In addition, aside from normally
labelled organelles, CL-V8B cells contained small stained
structures which may represent fragmented centrosomes. This type of
centrosome aberration, typical for cells with abnormalities of
Rad51 and its paralogs, was previously observed in other Chinese
hamster cell lines, CL-V4B
(Rad51C−/−), irs1
(Xrcc2−/−) and irs1SF
(Xrcc3−/−) (33). Fragmentation of centrosomes is also
characteristic for a Chinese hamster ovary cell line with abnormal
Rad51 protein, mouse cells with silenced Rad51D (31,58)
and human cells with haploinsufficiency of RAD51B gene
(59). These observations suggest
that the defective protein of CL-V8B mutant may serve a role in the
maintenance of correct centrosome number. Furthermore, this protein
may contribute to stabilization of these organelles, similar to
Rad51C, which was also implicated to have such a regulatory role
(33). The hypothesis of an
association between these two proteins appears to be supported by
marginal disturbances in Rad51 foci kinetics observed in CL-V8B
cells. Furthermore, abnormalities resulting from insufficient
activity of Rad51 and its paralogs are known to result in incorrect
formation of the kariokinetic spindle, which in turn leads to
chromosome instability (33). The
results of the current study are consistent with this data, as an
increased number of chromosomal aberrations were observed in CL-V8B
cells (Tables II and III). Presumably, aside from involvement
in ICLs repair, the defective protein present in this mutant is
also necessary for maintaining genomic stability. Although the
exact role of the protein impaired in CL-V8B mutant and the
mechanism of its action in centrosome stabilization remain unclear,
it is suggested that it cooperates with at least one of the HR
proteins.
Since proteins of HR and FA pathways, which are
crucial for ICLs repair, are additionally involved in replication,
this process was analyzed in CL-V8B cells exposed to normal and
replication-stress conditions. CL-V8B displayed a moderate degree
sensitivity to CPT and HU (Fig. 1E and
F), i.e. to the agents that block replication forks and lead to
their collapse, respectively. Although the CL-V8B mutant was
sensitive to the replication blocking agents and appeared to carry
a defect in one of the HR genes, they did not exhibit replication
disturbances characteristic of other HR mutants. The non-stress
velocity of replication fork in CL-V8B was similar to that observed
in wild type cells, whereas cells with mutations of HR genes,
including Xrcc2, Xrcc3 and Brca2, are characterized
by reduced fork velocity. It is likely that a slower rate of fork
progression is associated with activation of specific checkpoints;
this slows down the progression of replication forks, protecting
them against damage. This effect was demonstrated to correlate with
more frequent activation of new origins, which implies that the
abovementioned HR proteins are involved both in the control of
replication fork speed and in the activation of new origins
(35). Although cells with
defective ICLs repair present with various phenotypes of
replication disturbances, none of them resemble that observed in
the CL-V8B mutant (36,37). During elongation of DNA strands,
CL-V8B cells presented with a greater number of replication forks
than the wild type cells, subsequent to treatment with HU (Fig. 6B); although this suggests the
potential impairment of mechanisms responsible for DNA damage
signaling and fork stalling in CL-V8B cells, this issue requires
further elucidation.
Commonly, exposure to MMC results in S-phase
arrest, G2/M-phase delay or increase in the proportion
of apoptotic cells (39,40). Activation of checkpoints prevents
cell cycle progression and is a prerequisite of ICLs repair and
re-entrance of cells to the cycle (41). A total of 8 h after exposure to
MMC, approximately 50–60% of cells from both CL-V8B and the
parental V79B line remained in S-phase, indicating a dose-dependent
response to cytostatic conditions (Fig. 7A). In contrast, lack of S-phase
arrest following exposure to crosslinking agents was observed in
Rad51C- (41) and
Brca2/Fancd1-deficient hamster cells (CL-V4B and
V-C8, respectively), in addition to primary fibroblasts derived
from patients with FA belonging to the complementation group
BRCA2/FANCD1 (42,60).
In addition, ATM- and NBS1-defective cells indicated
disturbances in the activation of S-phase checkpoints following
exposure to ionizing radiation (61,62).
ATM is required for the initiation of HR and DSBs repair and, as
demonstrated recently, also for completion of the HR process after
formation of the Rad51 nucleofilament (63). Furthermore, ATM was confirmed to be
involved in phosphorylation and activation of the S-phase
checkpoint following exposure to ionizing radiation (64). The S-phase checkpoint was
demonstrated to additionally be inefficient in FA lymphoblasts
(from the FANCA-, FANCB- and FANCC-deficient
lymphoblasts) that have been damaged with DNA crosslinking agents
(65). However, mutations of these
genes in CL-V8B line were excluded based on the results of genetic
complementation analysis (Table I)
and proper Fancd2 monoubiquitination (not shown). The CL-V8B mutant
indicated normal S-phase arrest and formation of Rad51 foci
following exposure to DNA-damaging agents (Figs. 3 and 7A); this implies that proteins
responsible for initiation of HR and/or Rad51 nucleofilament
formation do not contribute to damage of the mutant, which likely
occurs at later stages of HR-mediated ICLs repair.
The S-phase arrest observed following treatment of
CL-V8B with MMC was associated with a significant dose-dependent
increase in the proportion of G2/M-phase cells,
particularly evident 24 h post-exposure (Fig. 7). Accumulation of
G2/M-phase cells following exposure to crosslinking
agents is specific for both FA- and HR-deficient cells from various
species (66,67). This phenomenon was also observed in
Brca1−/−p53−/−-deficient
mouse embryonic fibroblasts, which indicated time-dependent
accumulation of S-phase cells and sustained increase in the
proportion of G2/M-phase cells following MMC treatment
(68). Spontaneous
G2/M-phase arrest was also observed in HeLa cells with
inhibited expression of other HR proteins, such as RAD51B and
RAD51C (67), and in chicken
Rad51D−/− DT40 cells. It is postulated that
the G2/M-phase arrest results from accumulation of
secondary lesions, such as single- and double-strand breaks and
gaps, during the first replication round (65), which may be associated with
impairment of DNA repair. Treatment with MMC also resulted in an
increase in the number of apoptotic CL-V8B cells, especially 48 h
post-exposure (Fig. 7B and C).
These observations are consistent with the results of previous
studies which showed that morphological consequences of the
MMC-induced apoptosis are observed 48 h post-exposure (69), and apoptosis of Brca2-deficient
V-C8 cells starts 24 h subsequent to DSBs formation (42). Induction of apoptosis in response
to DNA damage was also observed in hamster cells with defective HR
proteins, such as Xrcc2 and Rad51c, in addition to after exposure
to camptothecin. However, contrary to the results of the current
study, the apoptotic response of Xrcc2- and Rad51c-deficient cells
was similar to that observed in parental V79 or mutant cells
corrected for the HR defect (70).
Previous studies have indicated that the presence of unrepaired
DSBs may result in apoptosis (43)
and chromosomal aberrations, such as chromosome breaks (71) or chromatid gaps in the case of
unrepaired SSBs (72). Initiation
of apoptosis in CL-V8B cells likely resulted from their defects
leading to unrepaired DNA breaks; this hypothesis appears to be
supported by increased level of CAs (Table II). However, the apoptotic
response to DNA damage may also depend on the type of DNA-damaging
agent and genetic background of exposed cells. Apoptotic response
and G2/M-phase arrest were lacking in HR-deficient
hamster mutant Xrcc3−/−, however were
observed in numerous other HR mutants, in addition to in the
Xrcc3-proficient cell line (70). Research on lymphoblastoid FA cell
lines has produced inconclusive results, as treatment with MMC
either did not induce any apoptotic changes (73) or resulted in G2/M-phase
arrest and accumulation of apoptotic cells (74).
The results of the current study indicate that
CL-V8B belongs to a new complementation group of Chinese hamster
cell mutants sensitive to crosslinking agents. Phenotypic analysis
of CL-V8B suggests that a molecular defect of this mutant
contributes to defective DSBs repair. The results of clonogenic
survival tests imply that the dysfunctional protein of CL-V8B line
is directly associated with HR-related genes or cooperates closely
with this DNA repair system. This protein may additionally be
involved in the FA pathway and serve a role in DSBs removal during
ICLs repair. This hypothesis is supported by the fact that some
other proteins, e.g. BRCA2/FANCD1, Rad51C/FANCO and PALB2/FANCN,
were demonstrated to be involved in the FA and HR pathways.
Furthermore, phenotypic analysis of CL-V8B implies that molecular
disturbances present in these cells may be associated with the
final stage of DSBs formation. One argument for this hypothesis
stems from the fact that no serious alterations in Rad51
nucleofilament formation, a key step of HR process used to verify
correct functioning of this repair system, were observed. The
unique phenotype of the mutant implies that in contrast to other
cells with compromised HR, the CL-V8B line does not demonstrate
replication defects. In addition, they have normally functioning
S-phase checkpoints, which is not observed in cells with defective
HR and in certain mutants with impaired FA.
The genetic background in a small proportion of
patients with defective ICLs repair remains unclear, and the
majority of biallelic HR mutants are lethal in humans. The unique
phenotype of CL-V8B, not observed in any other known Chinese
hamster mutants sensitive to DNA crosslinking agents, suggests the
presence of a yet to be unidentified defective gene involved in ICL
repair. Subsequent to identification of this gene, CL-V8B may
constitute an interesting cellular model to study the mechanism of
DNA crosslink removal.
Acknowledgements
The present study was supported by the Polish
National Science Centre (grant no. N N401 017640). Preliminary
results were presented as a poster during the 4th EMBO Meeting
Advancing the Life Sciences (September 2012; Nice, France). The
authors would like to thank Dr D. Gackowski for assistance with
statistical analysis.
References
|
1
|
Bryant HE, Schultz N, Thomas HD, Parker
KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ and Helleday T:
Specific killing of BRCA2-deficient tumours with inhibitors of poly
(ADP-ribose) polymerase. Nature. 434:913–917. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Issaeva N, Thomas HD, Djureinovic T,
Jaspers JE, Stoimenov I, Kyle S, Pedley N, Gottipati P, Zur R,
Sleeth K, et al: 6-thioguanine selectively kills BRCA2-defective
tumors and overcomes PARP inhibitor resistance. Cancer Res.
70:6268–6276. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McCabe N, Turner NC, Lord CJ, Kluzek K,
Bialkowska A, Swift S, Giavara S, O'Connor MJ, Tutt AN, Zdzienicka
MZ, et al: Deficiency in the repair of DNA damage by homologous
recombination and sensitivity to poly (ADP-ribose) polymerase
inhibition. Cancer Res. 66:8109–8115. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thompson LH, Brookman KW, Weber CA,
Salazar EP, Reardon JT, Sancar A, Deng Z and Siciliano MJ:
Molecular cloning of the human nucleotide-excision-repair gene
ERCC4. Proc Natl Acad Sci USA. 91:6855–6859. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Weeda G, Hoeijmakers JH and Bootsma D:
Genes controlling nucleotide excision repair in eukaryotic cells.
Bioessays. 15:249–258. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Thacker J and Zdzienicka MZ: The mammalian
XRCC genes: Their roles in DNA repair and genetic stability. DNA
Repair (Amst). 2:655–672. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rolig RL, Lowery MP, Adair GM and Nairn
RS: Characterization and analysis of Chinese hamster ovary cell
ERCC1 mutant alleles. Mutagenesis. 13:357–365. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Godthelp BC, Wiegant WW, van
Duijn-Goedhart A, Schärer OD, van Buul PP, Kanaar R and Zdzienicka
MZ: Mammalian Rad51C contributes to DNA cross-link resistance,
sister chromatid cohesion and genomic stability. Nucleic Acids Res.
30:2172–2182. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vaz F, Hanenberg H, Schuster B, Barker K,
Wiek C, Erven V, Neveling K, Endt D, Kesterton I, Autore F, et al:
Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat
Genet. 42:406–409. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bracalente C, Ibañez IL, Molinari B,
Palmieri M, Kreiner A, Valda A, Davidson J and Durán H: Induction
and persistence of large γH2AX foci by high linear energy transfer
radiation in DNA-dependent protein kinase-deficient cells. Int J
Radiat Oncol Biol Phys. 87:785–794. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Maeda J, Bell JJ, Genet SC, Fujii Y, Genet
MD, Brents CA, Genik PC and Kato TA: Potentially lethal damage
repair in drug arrested G2-phase cells after radiation exposure.
Radiat Res. 182:448–457. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Deans AJ and West SC: DNA interstrand
crosslink repair and cancer. Nat Rev Cancer. 11:467–480. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jaspers NG, Raams A, Silengo MC, Wijgers
N, Niedernhofer LJ, Robinson AR, Giglia-Mari G, Hoogstraten D,
Kleijer WJ, Hoeijmakers JH and Vermeulen W: First reported patient
with human ERCC1 deficiency has cerebro-oculo-facio-skeletal
syndrome with a mild defect in nucleotide excision repair and
severe developmental failure. Am J Hum Genet. 80:457–466. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vrouwe MG, Elghalbzouri-Maghrani E,
Meijers M, Schouten P, Godthelp BC, Bhuiyan ZA, Redeker EJ, Mannens
MM, Mullenders LH, Pastink A and Darroudi F: Increased DNA damage
sensitivity of Cornelia de Lange syndrome cells: Evidence for
impaired recombinational repair. Hum Mol Genet. 16:1478–1487. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
van der LP, Oostra AB, Rooimans MA, Joenje
H and de Winter JP: Diagnostic overlap between fanconi anemia and
the cohesinopathies: Roberts syndrome and warsaw breakage syndrome.
Anemia. 2010:5652682010.PubMed/NCBI
|
|
16
|
Kim H and D'Andrea AD: Regulation of DNA
cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev.
26:1393–1408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wong MW, Nordfors C, Mossman D,
Pecenpetelovska G, Avery-Kiejda KA, Talseth-Palmer B, Bowden NA and
Scott RJ: BRIP1, PALB2, and RAD51C mutation analysis reveals their
relative importance as genetic susceptibility factors for breast
cancer. Breast Cancer Res Treat. 127:853–859. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu J and Krantz ID: Cornelia de Lange
syndrome, cohesin, and beyond. Clin Genet. 76:303–314. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zdzienicka MZ, Arwert F, Neuteboom I,
Rooimans M and Simons JW: The Chinese hamster V79 cell mutant V-H4
is phenotypically like Fanconi anemia cells. Somat Cell Mol Genet.
16:575–581. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bishop DK, Ear U, Bhattacharyya A,
Calderone C, Beckett M, Weichselbaum RR and Shinohara A: Xrcc3 is
required for assembly of Rad51 complexes in vivo. J Biol Chem.
273:21482–21488. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kraakman-van der Zwet M, Overkamp WJ, van
Lange RE, Essers J, van Duijn-Goedhart A, Wiggers I, Swaminathan S,
van Buul PP, Errami A, Tan RT, et al: Brca2 (XRCC11) deficiency
results in radioresistant DNA synthesis and a higher frequency of
spontaneous deletions. Mol Cell Biol. 22:669–679. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
O'Regan P, Wilson C, Townsend S and
Thacker J: XRCC2 is a nuclear RAD51-like protein required for
damage-dependent RAD51 focus formation without the need for ATP
binding. J Biol Chem. 276:22148–22153. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zdzienicka MZ, Jaspers NG, van der Schans
GP, Natarajan AT and Simons JW: Ataxia-telangiectasia-like Chinese
hamster V79 cell mutants with radioresistant DNA synthesis,
chromosomal instability, and normal DNA strand break repair. Cancer
Res. 49:1481–1485. 1989.PubMed/NCBI
|
|
24
|
Jackson DA and Pombo A: Replicon clusters
are stable units of chromosome structure: Evidence that nuclear
organization contributes to the efficient activation and
propagation of S phase in human cells. J Cell Biol. 140:1285–1295.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zdzienicka MZ and Simons JW:
Mutagen-sensitive cell lines are obtained with a high frequency in
V79 Chinese hamster cells. Mutat Res. 178:235–244. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bargonetti J, Champeil E and Tomasz M:
Differential toxicity of DNA adducts of mitomycin C. J Nucleic
Acids. 2010:pii: 6989602010. View Article : Google Scholar
|
|
27
|
Rabik CA and Dolan ME: Molecular
mechanisms of resistance and toxicity associated with platinating
agents. Cancer Treat Rev. 33:9–23. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Farmer H, McCabe N, Lord CJ, Tutt AN,
Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I,
Knights C, et al: Targeting the DNA repair defect in BRCA mutant
cells as a therapeutic strategy. Nature. 434:917–921. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Godthelp BC, Artwert F, Joenje H and
Zdzienicka MZ: Impaired DNA damage-induced nuclear Rad51 foci
formation uniquely characterizes Fanconi anemia group D1. Oncogene.
21:5002–5005. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hinz JM, Tebbs RS, Wilson PF, Nham PB,
Salazar EP, Nagasawa H, Urbin SS, Bedford JS and Thompson LH:
Repression of mutagenesis by Rad51D-mediated homologous
recombination. Nucleic Acids Res. 34:1358–1368. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Smiraldo PG, Gruver AM, Osborn JC and
Pittman DL: Extensive chromosomal instability in Rad51d-deficient
mouse cells. Cancer Res. 65:2089–2096. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sonoda E, Sasaki MS, Morrison C,
Yamaguchi-Iwai Y, Takata M and Takeda S: Sister chromatid exchanges
are mediated by homologous recombination in vertebrate cells. Mol
Cell Biol. 19:5166–5169. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lindh A Renglin, Schultz N, Saleh-Gohari N
and Helleday T: RAD51C (RAD51L2) is involved in maintaining
centrosome number in mitosis. Cytogenet Genome Res. 116:38–45.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nigg EA: Centrosome aberrations: Cause or
consequence of cancer progression? Nat Rev Cancer. 2:815–825. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Henry-Mowatt J, Jackson D, Masson JY,
Johnson PA, Clements PM, Benson FE, Thompson LH, Takeda S, West SC
and Caldecott KW: XRCC3 and Rad51 modulate replication fork
progression on damaged vertebrate chromosomes. Mol Cell.
11:1109–1117. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schwab RA, Blackford AN and Niedzwiedz W:
ATR activation and replication fork restart are defective in
FANCM-deficient cells. EMBO J. 29:806–818. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schwab RA, Nieminuszczy J, Shin-ya K and
Niedzwiedz W: FANCJ couples replication past natural fork barriers
with maintenance of chromatin structure. J Cell Biol. 201:33–48.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Koç A, Wheeler LJ, Mathews CK and Merrill
GF: Hydroxyurea arrests DNA replication by a mechanism that
preserves basal dNTP pools. J Biol Chem. 279:223–230. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mladenov E, Tsaneva I and Anachkova B:
Activation of the S phase DNA damage checkpoint by mitomycin C. J
Cell Physiol. 211:468–476. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kang SG, Chung H, Yoo YD, Lee JG, Choi YI
and Yu YS: Mechanism of growth inhibitory effect of Mitomycin-C on
cultured human retinal pigment epithelial cells: Apoptosis and cell
cycle arrest. Curr Eye Res. 22:174–181. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Somyajit K, Subramanya S and Nagaraju G:
Distinct roles of FANCO/RAD51C protein in DNA damage signaling and
repair: Implications for Fanconi anemia and breast cancer
susceptibility. J Biol Chem. 287:3366–3380. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rocca CJ, Soares DG, Bouzid H, Henriques
JA, Larsen AK and Escargueil AE: BRCA2 is needed for both repair
and cell cycle arrest in mammalian cells exposed to S23906, an
anticancer monofunctional DNA binder. Cell Cycle. 14:2080–2090.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kaina B: DNA damage-triggered apoptosis:
Critical role of DNA repair, double-strand breaks, cell
proliferation and signaling. Biochem Pharmacol. 66:1547–1554. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wilson JB, Johnson MA, Stuckert AP,
Trueman KL, May S, Bryant PE, Meyn RE, D'Andrea AD and Jones NJ:
The Chinese hamster FANCG/XRCC9 mutant NM3 fails to express the
monoubiquitinated form of the FANCD2 protein, is hypersensitive to
a range of DNA damaging agents and exhibits a normal level of
spontaneous sister chromatid exchange. Carcinogenesis.
22:1939–1946. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jones NJ, Ellard S, Waters R and Parry EM:
Cellular and chromosomal hypersensitivity to DNA crosslinking
agents and topoisomerase inhibitors in the radiosensitive Chinese
hamster irs mutants: Phenotypic similarities to ataxia
telangiectasia and Fanconi's anaemia cells. Carcinogenesis.
14:2487–2494. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wiltshire TD, Lovejoy CA, Wang T, Xia F,
O'Connor MJ and Cortez D: Sensitivity to poly (ADP-ribose)
polymerase (PARP) inhibition identifies ubiquitin-specific
peptidase 11 (USP11) as a regulator of DNA double-strand break
repair. J Biol Chem. 285:14565–14571. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Somyajit K, Mishra A, Jameei A and
Nagaraju G: Enhanced non-homologous end joining contributes toward
synthetic lethality of pathological RAD51C mutants with poly
(ADP-ribose) polymerase. Carcinogenesis. 36:13–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yuan SS, Chang HL and Lee EY: Ionizing
radiation-induced Rad51 nuclear focus formation is cell
cycle-regulated and defective in both ATM(−/−) and c-Abl(−/−)
cells. Mutat Res. 525:85–92. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gudmundsdottir K, Lord CJ, Witt E, Tutt AN
and Ashworth A: DSS1 is required for RAD51 focus formation and
genomic stability in mammalian cells. EMBO Rep. 5:989–993. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xia B, Dorsman JC, Ameziane N, de Vries Y,
Rooimans MA, Sheng Q, Pals G, Errami A, Gluckman E, Llera J, et al:
Fanconi anemia is associated with a defect in the BRCA2 partner
PALB2. Nat Genet. 39:159–161. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
51
|
Dong Z, Zhong Q and Chen PL: The Nijmegen
breakage syndrome protein is essential for Mre11 phosphorylation
upon DNA damage. J Biol Chem. 274:19513–19516. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
van Veelen LR, Essers J, van de Rakt MW,
Odijk H, Pastink A, Zdzienicka MZ, Paulusma CC and Kanaar R:
Ionizing radiation-induced foci formation of mammalian Rad51 and
Rad54 depends on the Rad51 paralogs, but not on Rad52. Mutat Res.
574:34–49. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Shimada M, Sagae R, Kobayashi J, Habu T
and Komatsu K: Inactivation of the Nijmegen breakage syndrome gene
leads to excess centrosome duplication via the ATR/BRCA1 pathway.
Cancer Res. 69:1768–1775. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Dronkert ML, Beverloo HB, Johnson RD,
Hoeijmakers JH, Jasin M and Kanaar R: Mouse RAD54 affects DNA
double-strand break repair and sister chromatid exchange. Mol Cell
Biol. 20:3147–3156. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Dong Z and Fasullo M: Multiple
recombination pathways for sister chromatid exchange in
Saccharomyces cerevisiae: Role of RAD1 and the RAD52 epistasis
group genes. Nucleic Acids Res. 31:2576–2585. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Brugmans L, Verkaik NS, Kunen M, van
Drunen E, Williams BR, Petrini JH, Kanaar R, Essers J and van Gent
DC: NBS1 cooperates with homologous recombination to counteract
chromosome breakage during replication. DNA Repair (Amst).
8:1363–1370. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Xu X, Weaver Z, Linke SP, Li C, Gotay J,
Wang XW, Harris CC, Ried T and Deng CX: Centrosome amplification
and a defective G2-M cell cycle checkpoint induce genetic
instability in BRCA1 exon 11 isoform-deficient cells. Mol Cell.
3:389–395. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bertrand P, Lambert S, Joubert C and Lopez
BS: Overexpression of mammalian Rad51 does not stimulate
tumorigenesis while a dominant-negative Rad51 affects centrosome
fragmentation, ploidy and stimulates tumorigenesis, in
p53-defective CHO cells. Oncogene. 22:7587–7592. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Date O, Katsura M, Ishida M, Yoshihara T,
Kinomura A, Sueda T and Miyagawa K: Haploinsufficiency of RAD51B
causes centrosome fragmentation and aneuploidy in human cells.
Cancer Res. 66:6018–6024. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Godthelp BC, van Buul PP, Jaspers NG,
Elghalbzouri-Maghrani E, van Duijn-Goedhart A, Arwert F, Joenje H
and Zdzienicka MZ: Cellular characterization of cells from the
Fanconi anemia complementation group, FA-D1/BRCA2. Mutat Res.
601:191–201. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Beamish H, Williams R, Chen P and Lavin
MF: Defect in multiple cell cycle checkpoints in
ataxia-telangiectasia postirradiation. J Biol Chem.
271:20486–20493. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Eggleston A: Convergence of DNA repair and
cell-cycle checkpoint control. Nat Cell Biol. 2:E952000. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Bakr A, Oing C, Köcher S, Borgmann K,
Dornreiter I, Petersen C, Dikomey E and Mansour WY: Involvement of
ATM in homologous recombination after end resection and RAD51
nucleofilament formation. Nucleic Acids Res. 43:3154–3166. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhao S, Weng YC, Yuan SS, Lin YT, Hsu HC,
Lin SC, Gerbino E, Song MH, Zdzienicka MZ, Gatti RA, et al:
Functional link between ataxia-telangiectasia and Nijmegen breakage
syndrome gene products. Nature. 405:473–477. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sala-Trepat M, Rouillard D, Escarceller M,
Laquerbe A, Moustacchi E and Papadopoulo D: Arrest of S-phase
progression is impaired in Fanconi anemia cells. Exp Cell Res.
260:208–215. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Heinrich MC, Hoatlin ME, Zigler AJ, Silvey
KV, Bakke AC, Keeble WW, Zhi Y, Reifsteck CA, Grompe M, Brown MG,
et al: DNA cross-linker-induced G2/M arrest in group C Fanconi
anemia lymphoblasts reflects normal checkpoint function. Blood.
91:275–287. 1998.PubMed/NCBI
|
|
67
|
Rodrigue A, Coulombe Y, Jacquet K, Gagné
JP, Roques C, Gobeil S, Poirier G and Masson JY: The RAD51 paralogs
ensure cellular protection against mitotic defects and aneuploidy.
J Cell Sci. 126:348–359. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Yun J, Zhong Q, Kwak JY and Lee WH:
Hypersensitivity of Brca1-deficient MEF to the DNA interstrand
crosslinking agent mitomycin C is associated with defect in
homologous recombination repair and aberrant S-phase arrest.
Oncogene. 24:4009–4016. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Pirnia F, Schneider E, Betticher DC and
Borner MM: Mitomycin C induces apoptosis and caspase-8 and −9
processing through a caspase-3 and Fas-independent pathway. Cell
Death Differ. 9:905–914. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Hinz JM, Helleday T and Meuth M: Reduced
apoptotic response to camptothecin in CHO cells deficient in XRCC3.
Carcinogenesis. 24:249–253. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Bryant PE: The signal model: A possible
explanation for the conversion of DNA double-strand breaks into
chromatid breaks. Int J Radiat Biol. 73:243–251. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Parshad R, Sanford KK and Jones GM:
Chromatid damage induced by fluorescent light during G2 phase in
normal and Gardner syndrome fibroblasts. Interpretation in terms of
deficient DNA repair. Mutat Res. 151:57–63. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Rey JP, Scott R and Müller H: Apoptosis is
not involved in the hypersensitivity of Fanconi anemia cells to
mitomycin C. Cancer Genet Cytogenet. 75:67–71. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Kruyt FA, Dijkmans LM, van den Berg TK and
Joenje H: Fanconi anemia genes act to suppress a
cross-linker-inducible p53-independent apoptosis pathway in
lymphoblastoid cell lines. Blood. 87:938–948. 1996.PubMed/NCBI
|