Introduction
Alcohol is a widely-consumed hepatic toxicant, and
excessive alcohol consumption may lead to acute and chronic fatty
liver diseases, including steatosis, steatohepatitis, liver
fibrosis and liver cirrhosis. Although the pathogenesis of
alcoholic liver disease (ALD) remains to be fully elucidated,
oxidative stress is considered to be one of the principal factors
responsible for alcoholic liver damage (1). Previous studies have demonstrated
that ethanol-induced oxidative stress may lead to the production of
cytokines and chemokines, including tumor necrosis factor (TNF)-α,
transforming growth factor (TGF)-β1 and reactive oxygen species
(ROS), which are hypothesized to serve a role in the development of
ALD (1,2).
Aldose reductase (AR; additionally termed AKR1B or
EC1.1.1.21) catalyzes the reduction of glucose to sorbitol with the
aid of the co-factor reduced nicotinamide adenine dinucleotide
phosphate, in the polyol pathway (3). The role of AR in the development of
complications in diabetes is well-established (4). Additionally, hepatic AR expression
has been observed to be induced in a number of liver disease
conditions, including alcoholic liver disease, non-alcoholic fatty
liver disease, hepatitis, cirrhosis and hepatocellular carcinoma
(5–8). It was previously reported that
overexpression of AR induced the production of ROS and TNF-α in
liver cells, and that administration of the AR inhibitor
zopolrestat attenuated methionine-choline-deficient diet-induced
hepatosteatosis and liver inflammation (8,9).
However, the role of AR in the development of ALD remains unknown.
Therefore, the aim of the present study was to investigate the
effect of AR inhibition on ethanol-induced hepatic steatosis and
injury in mice and AML12 mouse liver cells, and to investigate the
mechanism through which AR affects the development of ALD.
Materials and methods
Animal experiments
Male Kunming mice (age, 6 weeks; weight, 22–25 g;
total n=40) were obtained from the Shanghai SLAC Laboratory Animal
Co., Ltd. (Shanghai, China) and 5 mice/cage were maintained under a
12-h light/dark schedule, with a temperature range of 20 to 24°C.
Experiments were conducted according to protocols and guidelines
approved by the Longyan University Institutional Animal Care and
Use Committee (Longyan, China). Liquid diets were based on the
modified Lieber-DeCarli formulation and provided 1 kcal/ml
(prepared by Trophic Animal Feed High-tech Co., Ltd., Nantong,
China). The ethanol diet consisted of 18% of the total energy as
protein, 35% as fat, 19% as carbohydrate and 28% as ethanol. In the
control diet, ethanol was replaced isocalorically with
carbohydrate. Following a 1-week period of adaptation to the
environment with free access to the liquid diet, mice were randomly
divided into four experimental groups (n=10 mice/group): Control
diet-fed mice; control diet-fed mice + zopolrestat (zopol); ethanol
diet-fed mice and ethanol diet-fed mice + zopol. Mice were
administered 50 mg/kg/day zopol as a single daily intraperitoneal
injection for 4 weeks. Equivalent volumes of saline were
administered to the control groups of mice. On the final day, mice
were sacrificed under anesthesia. Tissues were snap-frozen in
liquid nitrogen immediately following resection and stored at −80°C
until further analysis.
Histological examination
Liver tissues were fixed in 10% formalin for 24 h at
room temperature and were routinely embedded in paraffin. Briefly,
tissues were rinsed in tap water for 5 min at room temperature
prior to dehydration with a series of alcohol (70, 80 and 90%,
respectively, for 5 min each, followed by 3 incubations with 100%
alcohol for 5 min each at room temperature). The tissues were then
treated twice with xylene for 5 min each at room temperature, prior
to paraffin embedding. Sections (5-µm) were deparaffinized in
xylene twice for 5 min each and treated with 100% alcohol twice for
3 min, then 95, 70 and 50% alcohol, respectively, for 3 min each at
room temperature. Sections were stained with hematoxylin and eosin
(H&E) for histological analysis. Hepatic steatosis was graded
as previously described (10),
according to the percentage of lipid-laden hepatocytes: 0, 0%; 1,
0–25%; 2, 26–50%; 3, 51–75% and 4, 76–100%.
Cell culture
AML12 mouse hepatocyte cells were obtained from the
American Type Culture Collection (Manassas, VA, USA) and cultured
in Dulbecco's modified Eagle's medium/Ham's F12 containing 100 U/ml
penicillin, 100 µg/ml streptomycin and 10% fetal bovine serum
(Hyclone; GE Healthcare Life Sciences, Logan, UT, USA), in a 37°C
incubator with 5% CO2.
Detection of ROS generation in AML12
cells
AML12 cells were plated on 24-well plates at a
density of ~1×105 cells/well. Following 24 h of
incubation, cells were exposed to 100 mM ethanol and co-treated
with 50 µM zopol. A total of 36 h subsequent to ethanol and zopol
treatment, ROS generation in cells was determined using ROS assay
kit (cat no S0033; Beyotime Institute of Biotechnology, Haimen,
China), according to the manufacturer's instructions.
Tissue and cell lipid peroxidation
assays
Total lipoperoxides were measured as thiobarbituric
acid reactive substances (TBARS) in liver homogenates or cell
lysates, using a lipid peroxidation malondialdehyde (MDA) assay kit
(cat no S0131; Beyotime Institute of Biotechnology), according to
the manufacturer's instructions. TBARS were quantified using MDA as
a standard and Microsoft Excel 2013 (Microsoft Corporation.
Redmond, WA, USA).
Quantitative analysis of mRNA
expression using reverse transcription-quantitative polymerase
chain reaction (RT-qPCR)
Total RNA was isolated from tissues and cells using
TRIpure reagent (Roche Applied Science, Mannheim, Germany),
according to the manufacturer's protocol. cDNA was synthesized from
mRNA using RevertAid First Strand cDNA Synthesis kits (Thermo
Fisher Scientific, Inc., Waltham, MA, USA). Expression levels of
hepatic TNF-α, TGF-β1, interleukin (IL)-6 and sterol regulatory
element binding protein (SREBP)-1c were analyzed. The specific
primers used were: Forward, 5′-CGTGCTCCTCACCCACAC-3′ and reverse,
5′-GGGTTCATACCAGGGTTTGA-3′ for TNF-α; forward,
5′-ACAACCACGGCCTTCCCTACTT-3′ and reverse,
5′-GTGTAATTAAGCCTCCGACT-3′ for IL-6; forward,
5′-CAACTTCTGTCTGGGACCCT-3′ and reverse, 5′-TAGTAGACGATGGGCAGTGG-3′
for TGF-β1; forward, 5′-ATCGGCGCGGAAGCTGTCGGGGTAGCGTC-3′ and
reverse, 5′-ACTGTCTTGGTTGTTGATGAGCTGGAGCAT-3′ for SREBP-1c and
forward, 5′-CTATTGGCAACGAGCGGTTCC-3′ and reverse,
5′-GCACTGTGTTGGCATAGAGGTC-3′ for β-actin. qPCR was performed using
the FastStart Universal SYBR-Green Master Mix (Roche Applied
Science). The reaction was performed with the following
thermocycling conditions: 95°C for 10 min, followed by 35 cycles of
95°C for 15 sec, 53–59°C for 30 sec and 72°C for 30 sec, then a
final extension of 72°C for 10 min. The 2-ΔΔCq method
(11) was used for quantification
and the data were normalized to β-actin.
Western blot analysis
Tissues and cells were homogenized in ice-cold
radioimmunoprecipitation assay buffer (Beyotime Institute of
Biotechnology). Protein concentrations of the extracts were
measured using a bicinchoninic acid protein assay kit (Beyotime
Institute of Biotechnology), according to the manufacturer's
protocol. A total of 40 µg of each protein sample was loaded and
separated using 10–12% SDS-PAGE and transferred to polyvinylidene
difluoride membranes (EMD Millipore, Billerica, MA, USA). Membranes
were blotted with 5% non-fat milk in TBS containing 0.1% Tween-20
for 1 h at room temperature. Membranes were then incubated with
anti-AR (cat no. sc-17735; 1:500; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA), anti-cytochrome P450 2E1 (cat no. ab28146;
1:1,000; CYP2E1; Abcam, Cambridge, UK), anti-phosphorylated AMPK
(cat no. 2535; 1:1,000), anti-AMPK (cat no. 2532; 1:1,000) (both
from Cell Signaling Technology), anti-GAPDH (cat no. G8795;
1:2,000) or anti-β-actin (cat no. A2228; 1:2,000) (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) at 4°C overnight, and subsequently
with horseradish peroxidase-conjugated anti-goat or anti-rabbit
immunoglobulin G (cat nos. AP307P and AB324P, respectively;
1:2,000; Sigma-Aldrich; Merck KGaA) for 2 h at room temperature.
Detection was achieved using the SuperSignal Chemiluminescent
Substrate kit (Beyotime Institute of Biotechnology).
Statistical analysis
All data were processed and analyzed using GraphPad
Prism software, version 5.0 (GraphPad Software, Inc., La Jolla, CA,
USA) and were expressed as the mean ± standard error of the mean.
One-way analysis of variance and the post hoc Newman-Keuls multiple
comparison test was used for multi-group analyses. P<0.05 was
considered to indicate a statistically significant difference.
Results
AR is induced in the livers of ethanol
diet-fed mice and in ethanol-treated mouse AML12 liver cells, and
inhibition of AR activity attenuates ethanol-induced hepatic
steatosis
Previous studies demonstrated that feeding C57BL/6
mice with Lieber-DeCarli ethanol diets induced fatty liver within
4–8 weeks (12). In order to
investigate whether AR was involved in the development of ethanol
diet-induced hepatosteatosis, the protein expression levels of
hepatic AR were analyzed in mice fed with the ethanol diet. As
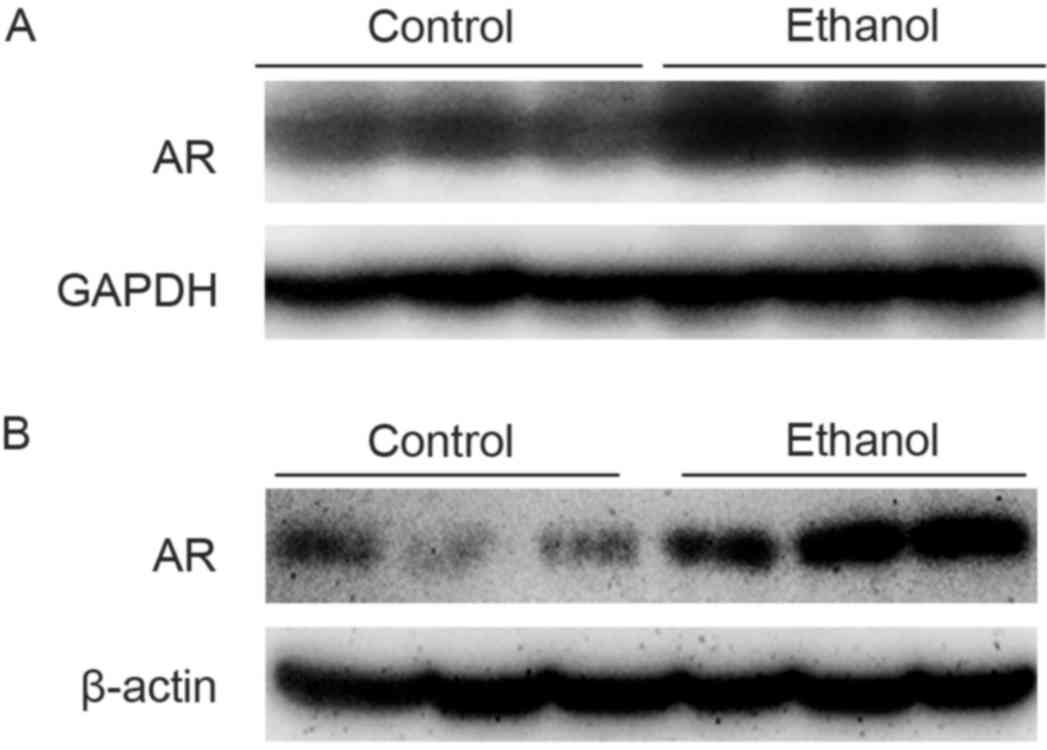
presented in Fig. 1A, hepatic AR
protein expression in C57BL/6 mice fed with the ethanol diet for 4
weeks was increased, compared with mice fed with the control diet.
In addition to the induction of AR expression in mouse liver
tissue, an increase in AR protein expression was observed in
hepatocytes exposed to 100 mM ethanol for 36 h (Fig. 1B).
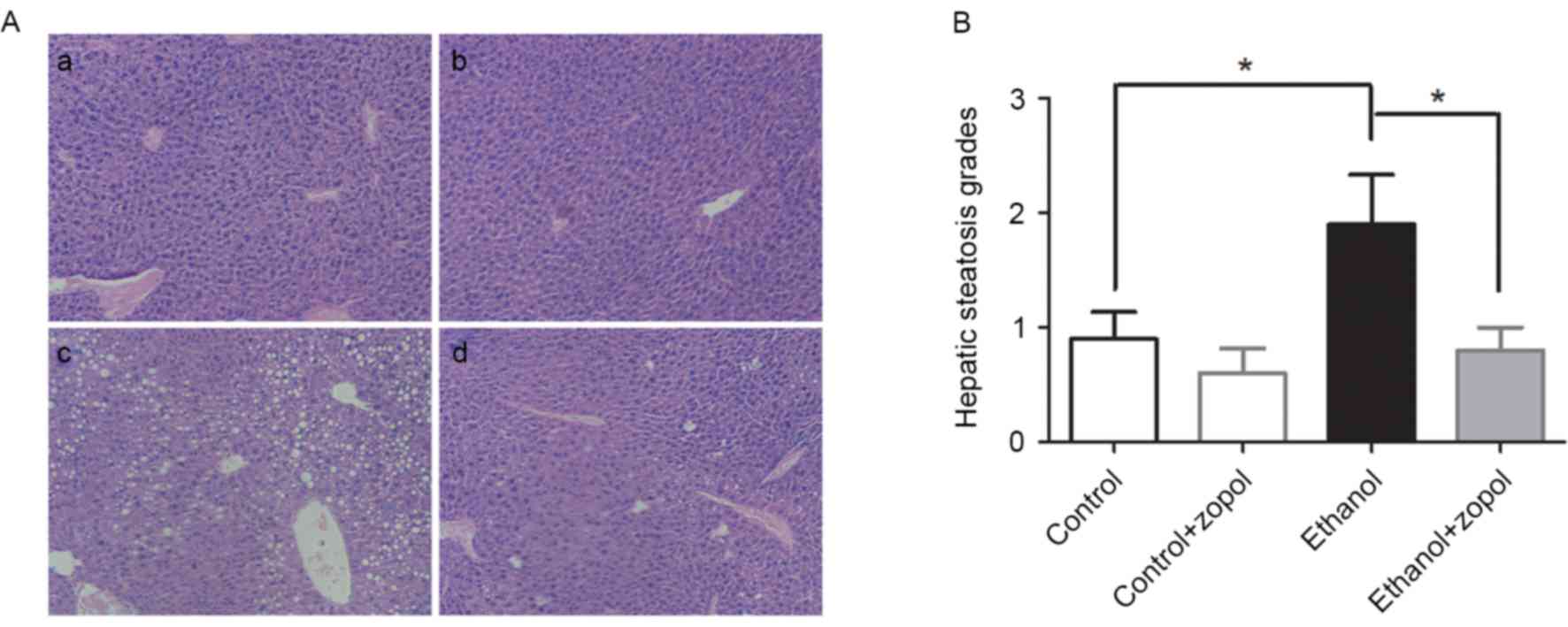
In order to further determine the role of AR in the
development of ethanol-induced hepatosteatosis, AR activity was
inhibited by treating mice with an AR-specific inhibitor, zopol. As
presented in Fig. 2A and B,
examination of H&E-stained sections demonstrated marked hepatic
steatosis in mice fed with the ethanol diet for 4 weeks, while mice
fed with the control diet exhibited low-grade steatosis. Following
inhibition of AR activity, hepatic steatosis in mice fed with the
ethanol diet was significantly attenuated (P<0.05).
AR inhibition decreases
ethanol-induced hepatic oxidative stress in vivo and in vitro
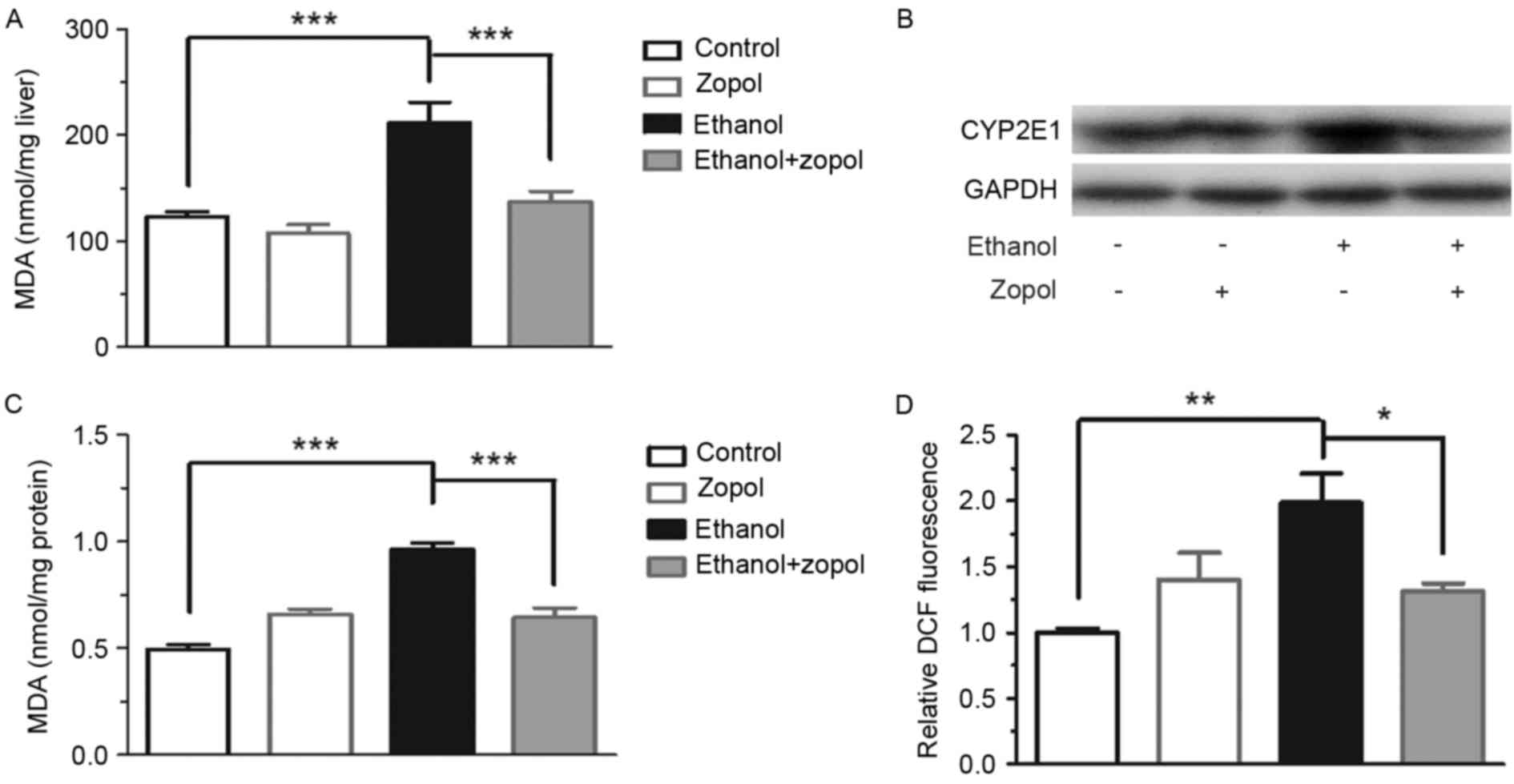
In order to clarify the mechanism through which AR
is associated with the development of ethanol-induced steatosis,
the effect of AR inhibition on hepatic lipoperoxide production was
investigated. As presented in Fig.
3A, consumption of the ethanol diet resulted in an increase in
hepatic TBARS levels, compared with the control diet, and the
increase was attenuated significantly by treating the ethanol
diet-fed mice with zopol (P<0.001). The protein expression of
CYP2E1, a principal mediator of lipid peroxidation (13,14),
was induced by ethanol, and AR inhibition significantly prevented
the induction of CYP2E1 (Fig. 3B).
In addition, the protective effect of AR inhibition on
ethanol-induced oxidative stress was analyzed in vitro. As
presented in Fig. 3C and D,
inhibition of AR resulted in a 33.0% decrease in MDA levels
(P<0.001) and a 33.9% decrease in ROS levels (P<0.05) in
ethanol-treated AML12 cells. However, CYP2E1 protein expression was
not detected in AML12 cells. The results of the present study
demonstrated that the induction of AR by ethanol may promote
hepatic oxidative stress in mice with alcoholic liver disease, and
that the increased hepatic oxidative stress is due in part to the
AR-mediated induction of CYP2E1.
AR inhibition activates AMPK and
suppresses SREBP-1c mRNA expression
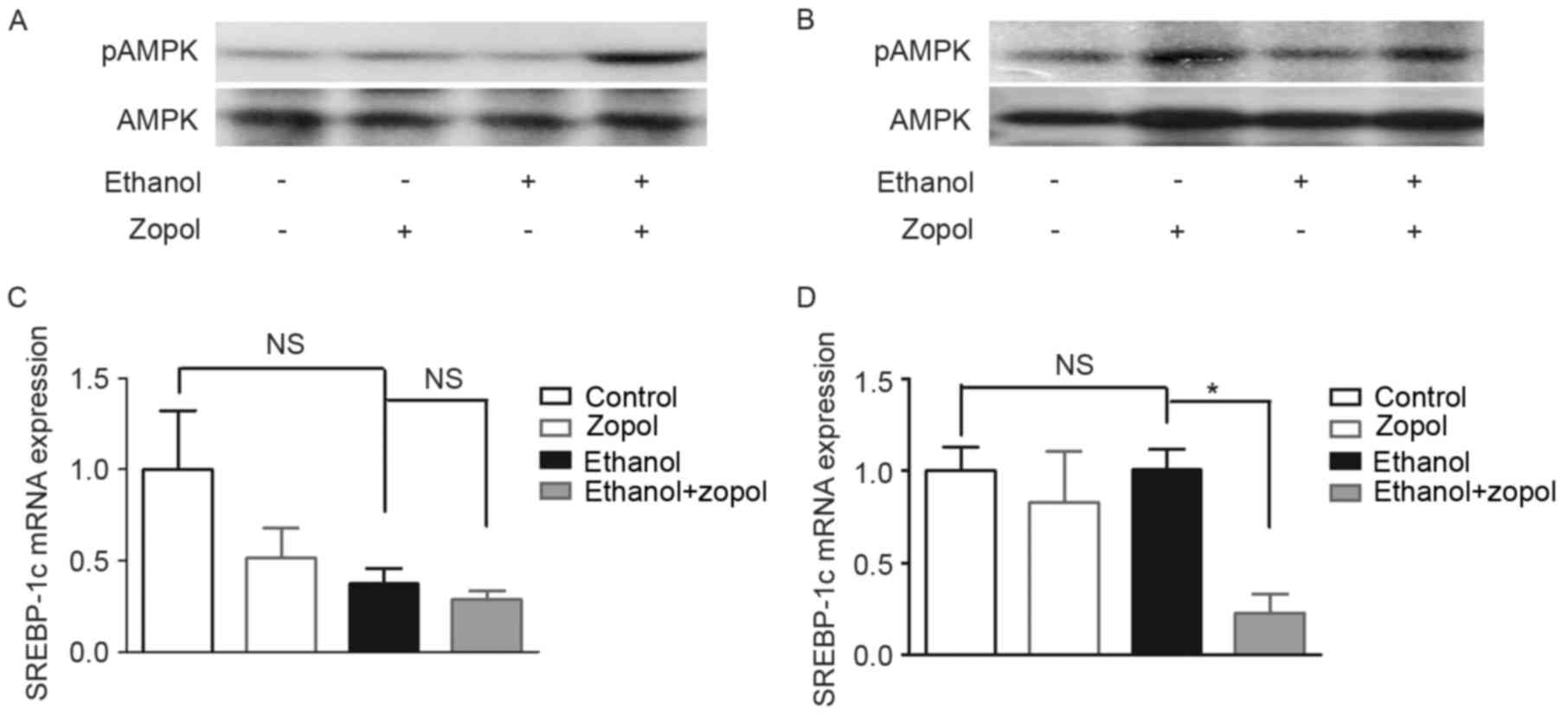
Previous studies demonstrated that AMPK inactivation
was associated with the development of ethanol-induced hepatic
steatosis (15,16). In order to clarify the mechanisms
whereby AR affects lipid accumulation in hepatocytes, the effect of
AR inhibition on AMPK activity was investigated. As presented in
Fig. 4A, phosphorylated AMPK
protein expression levels in the livers of ethanol-fed mice treated
with zopol were increased compared with ethanol-fed mice,
indicating that AR inhibition activated hepatic AMPK in ethanol-fed
mice. The activation on AMPK by the AR inhibitor was additionally
confirmed in ethanol-treated AML12 cells (Fig. 4B). The effect of AR inhibition on
the mRNA expression of SREBP-1c, a transcriptional factor regulated
by AMPK which controls the synthesis of fatty acids, was assessed
in the livers of ethanol-fed mice. As presented in Fig. 4C, AR inhibition did not
significantly decrease the mRNA expression levels of SREBP-1c in
ethanol-fed mice when compared with the ethanol-fed group. However,
AR inhibition significantly decreased the mRNA expression levels of
SREBP-1c in ethanol-treated AML12 cells (Fig. 4D).
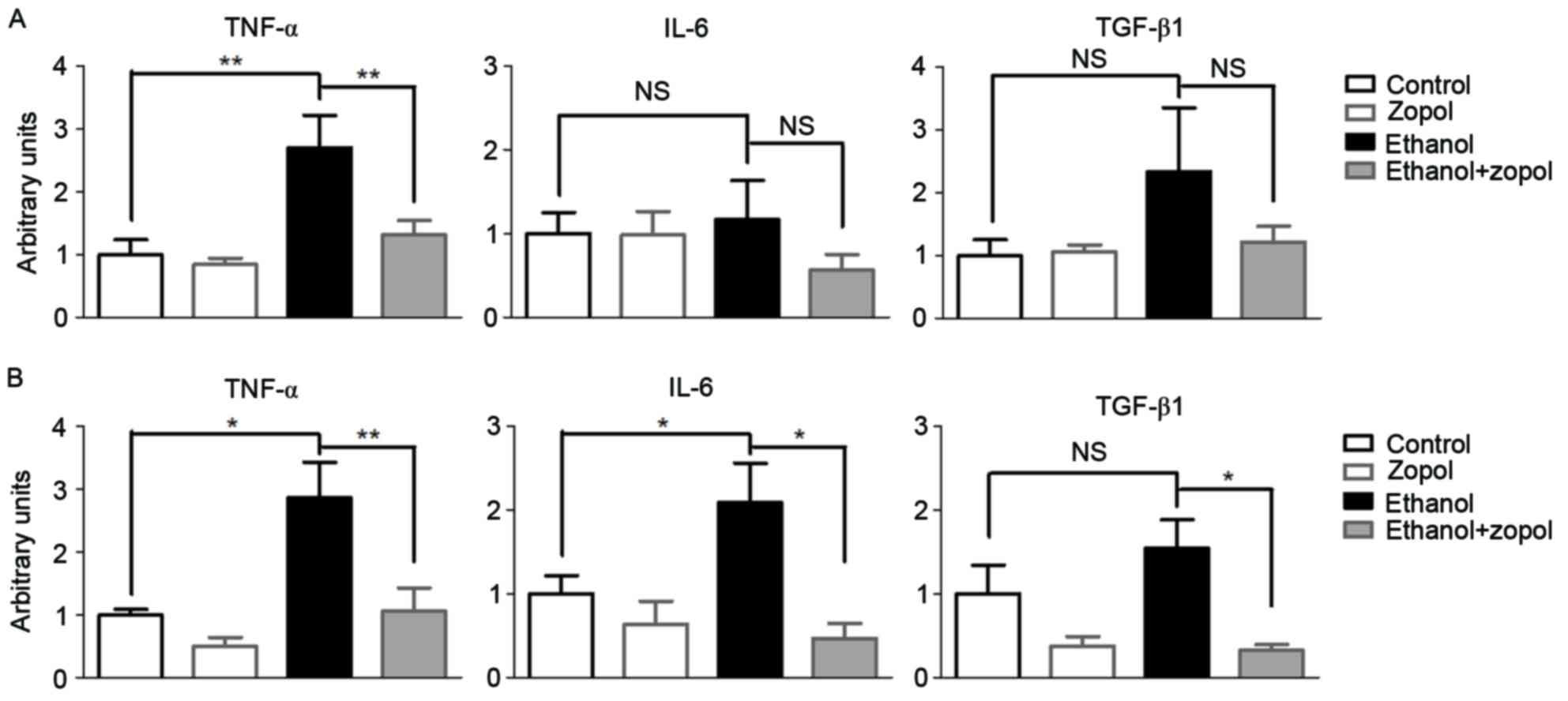
AR inhibition suppresses the mRNA
expression of certain inflammatory mediators in the livers of
ethanol diet-fed mice and in hepatocytes
In order to evaluate the effect of AR inhibition on
the development of ethanol-induced liver injuries, the expression
levels of certain proinflammatory cytokines were assessed,
including TNF-α, IL-6 and TGF-β1. Mice fed on the ethanol diet
exhibited a marked elevation of hepatic mRNA expression of TNF-α
compared with mice fed on the control diet (Fig. 5A). However, the ethanol-induced
TNF-α mRNA elevation was significantly attenuated by 51.4%
(P<0.01) in mice treated with zopol. Compared with TNF-α, the
effect of AR inhibition on IL-6 and TGF-β1 was less significant.
Consistent with the in vivo results, AR inhibition in
ethanol-treated AML12 cells resulted in a significant decrease in
the mRNA expression levels of TNF-α (P<0.01; Fig. 5B). AR inhibition in ethanol-treated
AML12 cells resulted in a significant decrease in the mRNA
expression levels of IL-6 (P<0.05) and TGF-β1 (P<0.05). The
results of the present study demonstrated that hepatic AR elevation
in mice fed on the ethanol diet may directly affect the production
of pro-inflammatory cytokines, particularly TNF-α, in the
liver.
Discussion
The induction of AR expression has been observed in
certain liver disease conditions, including alcoholic liver disease
and non-alcoholic fatty liver disease (6,8).
Inhibition of AR improves the development of hepatic steatosis and
liver inflammation in mice with non-alcoholic steatohepatitis
(7–9). However, the role of AR in the
development of ALD remains unclear. The results of the present
study demonstrated that hepatic AR protein was induced in ethanol
diet-induced hepatosteatosis in C57BL/6 mice, and in
ethanol-treated mouse AML12 liver cells. Pharmacological inhibition
of AR was conducted in the ethanol diet-fed C57BL/6 mice, to
examine the role of AR in the development of ethanol-induced
hepatosteatosis. It was demonstrated that AR inhibition improved
the development of hepatosteatosis. The results of the present
study indicated that AR may be involved in the development of
ALD.
The alcohol metabolite acetaldehyde has previously
been demonstrated to be one of the important causes of hepatic
triglyceride accumulation and hepatocellular injury in alcoholic
fatty livers (17). AR is able to
catalyze the reduction of a variety of aldehydes and carbonyls
(18,19). Therefore, AR has been postulated to
serve a cytoprotective function by rapidly detoxifying aldehydes.
AR inhibitors have been reported to prevent injuries in a variety
of rodent models, including ischemia/reperfusion-induced liver
(20), arterial balloon (21) and ischemic myocardial injuries
(22). In the present study, it
was demonstrated that AR exacerbated hepatocyte injury. As AR
possesses the capacity to rapidly detoxify aldehydes, it is notable
that AR inhibition prevented hepatocyte injury. The present study
demonstrated that AR inhibition prevented the induction of CYP2E1
in ethanol diet-fed C57BL/6 mice. CYP2E1 serves an important role
in the pathogenesis of ALD (13,14,23).
CYP2E1 is able to catalyze the reduction of alcohol to acetaldehyde
in the liver, resulting in the generation of ROS, including the
superoxide anion, hydrogen peroxide and the hydroxyl radical. The
generation of ROS may initiate lipid peroxidation in the liver
(13). The results of the present
study suggested that the elevation of AR resulted in the induction
of hepatic CYP2E1 expression in mice with ALD and, consequently,
exacerbated lipid peroxidation and oxidative stress, and affected
the development of ALD. Therefore, the prevention of the induction
of hepatic CYP2E1 in mice with ALD may be part of the mechanism via
which AR inhibition attenuates lipid peroxidation and ameliorates
ALD.
AMPK is an enzyme that serves a role in cellular
energy homeostasis. The effect of AMPK inhibition is suppression of
fatty acid oxidation and stimulation of lipogenesis (24). Ethanol has been reported to
suppress hepatic AMPK activity, and the inhibition of AMPK by
ethanol serves an important role in the development of
hepatosteatosis induced by chronic alcohol consumption (15,16).
In the present study, it was demonstrated that AR inhibition
activated AMPK in ethanol-treated AML12 liver cells and in the
livers of mice fed with ethanol. Therefore, activation of hepatic
AMPK may be part of the mechanism via which AR inhibition
ameliorates ethanol-induced hepatosteatosis.
It is well-known that chronic alcohol consumption
increases TNF-α production and leads to hepatosteatosis and liver
injury (2). Studies have
demonstrated that the expression of TNF-α is elevated in patients
with alcoholic liver diseases (25,26).
In addition, TNF-α has been observed to induce lipid accumulation,
and result in apoptosis and inflammation in liver cells (27). Therefore, the improvement of
steatosis and liver injury in alcoholic livers by treatment with an
AR inhibitor may be partially attributed to an inhibitory effect on
ethanol-induced TNF-α mRNA overexpression.
In conclusion, the present study demonstrated that
AR inhibition significantly ameliorated alcohol-induced hepatic
steatosis in mouse AML12 liver cells and in Kunming mice,
potentially by activating AMPK, decreasing CYP2E1-mediated
oxidative stress and suppressing the expression of pro-inflammatory
cytokines. These results indicate that AR inhibitors may have
potential as alternative therapeutic strategies for the treatment
of alcoholic fatty liver diseases.
Acknowledgements
The present study was supported in part by the
Training Program of Fujian Excellent Talents in University (grant
no. MJR201558) and the Science Planning Program of Longyan
University (grant no. LG2014012).
References
|
1
|
Ishii H, Kurose I and Kato S: Pathogenesis
of alcoholic liver disease with particular emphasis on oxidative
stress. J Gastroenterol Hepatol. 12:S272–S282. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kawaratani H, Tsujimoto T, Douhara A,
Takaya H, Moriya K, Namisaki T, Noguchi R, Yoshiji H, Fujimoto M
and Fukui H: The effect of inflammatory cytokines in alcoholic
liver disease. Mediators Inflamm. 2013:4951562013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hers HG: Aldose reductase. Biochim Biophys
Acta. 37:120–126. 1960.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nishimura-Yabe C: Aldose reductase in the
polyol pathway: A potential target for the therapeutic intervention
of diabetic complications. Nihon Yakurigaku Zasshi. 111:137–145.
1998.(In Japanese). View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brown KE, Broadhurst KA, Mathahs MM,
Kladney RD, Fimmel CJ, Srivastava SK and Brunt EM: Immunodetection
of aldose reductase in normal and diseased human liver. Histol
Histopathol. 20:429–436. 2005.PubMed/NCBI
|
|
6
|
O'Connor T, Ireland LS, Harrison DJ and
Hayes JD: Major differences exist in the function and
tissue-specific expression of human aflatoxin B1 aldehyde reductase
and the principal human aldo-keto reductase AKR1 family members.
Biochem J. 343:487–504. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qiu L, Lin J, Xu F, Gao Y, Zhang C, Liu Y,
Luo Y and Yang JY: Inhibition of aldose reductase activates hepatic
peroxisome proliferator-activated receptor-α and ameliorates
hepatosteatosis in diabetic db/db mice. Exp Diabetes Res.
2012:7897302012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Qiu L, Lin J, Ying M, Chen W, Yang J, Deng
T, Chen J, Shi D and Yang JY: Aldose reductase is involved in the
development of murine diet-induced nonalcoholic steatohepatitis.
PLoS One. 8:e735912013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen T, Shi D, Chen J, Yang Y, Qiu M, Wang
W and Qiu L: Inhibition of aldose reductase ameliorates
diet-induced nonalcoholic steatohepatitis in mice via modulating
the phosphorylation of hepatic peroxisome proliferator-activated
receptor α. Mol Med Rep. 11:303–308. 2015.PubMed/NCBI
|
|
10
|
Peng Z, Borea PA, Varani K, Wilder T, Yee
H, Chiriboga L, Blackburn MR, Azzena G, Resta G and Cronstein BN:
Adenosine signaling contributes to ethanol-induced fatty liver in
mice. J Clin Invest. 119:582–594. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lieber CS, DeCarli LM and Sorrell MF:
Experimental methods of ethanol administration. Hepatology.
10:501–510. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Leung TM and Nieto N: CYP2E1 and oxidant
stress in alcoholic and non-alcoholic fatty liver disease. J
Hepatol. 58:395–398. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lu Y and Cederbaum AI: CYP2E1 and
oxidative liver injury by alcohol. Free Radic Biol Med. 44:723–738.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
You M, Matsumoto M, Pacold CM, Cho WK and
Crabb DW: The role of AMP-activated protein kinase in the action of
ethanol in the liver. Gastroenterology. 127:1798–1808. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sid B, Verrax J and Calderon PB: Role of
AMPK activation in oxidative cell damage: Implications for
alcohol-induced liver disease. Biochem Pharmacol. 86:200–209. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Barry RE: Role of acetaldehyde in the
pathogenesis of alcoholic liver disease. Br J Addict. 83:1381–1386.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vander Jagt DL and Hunsaker LA: Substrate
specificity of reduced and oxidized forms of human aldose
reductase. Adv Exp Med Biol. 328:279–288. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vander Jagt DL, Kolb NS, Vander Jagt TJ,
Chino J, Martinez FJ, Hunsaker LA and Royer RE: Substrate
specificity of human aldose reductase: Identification of
4-hydroxynonenal as an endogenous substrate. Biochim Biophys Acta.
1249:117–126. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang C, Huang C, Tian Y and Li X: Polyol
pathway exacerbated ischemia/reperfusion-induced injury in
steatotic liver. Oxid Med Cell Longev. 2014:9636292014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ruef J, Liu SQ, Bode C, Tocchi M,
Srivastava S, Runge MS and Bhatnagar A: Involvement of aldose
reductase in vascular smooth muscle cell growth and lesion
formation after arterial injury. Arterioscler Thromb Vasc Biol.
20:1745–1752. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tracey WR, Magee WP, Ellery CA, MacAndrew
JT, Smith AH, Knight DR and Oates PJ: Aldose reductase inhibition
alone or combined with an adenosine A (3) agonist reduces ischemic
myocardial injury. Am J Physiol Heart Circ Physiol.
279:H1447–H1452. 2000.PubMed/NCBI
|
|
23
|
Morimoto M, Hagbjörk AL, Nanji AA,
Ingelman-Sundberg M, Lindros KO, Fu PC, Albano E and French SW:
Role of cytochrome P4502E1 in alcoholic liver disease pathogenesis.
Alcohol. 10:459–464. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Viollet B, Guigas B, Leclerc J, Hébrard S,
Lantier L, Mounier R, Andreelli F and Foretz M: AMP-activated
protein kinase in the regulation of hepatic energy metabolism: From
physiology to therapeutic perspectives. Acta Physiol (Oxf).
196:81–98. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
An L, Wang X and Cederbaum AI: Cytokines
in alcoholic liver disease. Arch Toxicol. 86:1337–1348. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
McClain CJ and Cohen DA: Increased tumor
necrosis factor production by monocytes in alcoholic hepatitis.
Hepatology. 9:349–351. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Endo M, Masaki T, Seike M and Yoshimatsu
H: TNF-alpha induces hepatic steatosis in mice by enhancing gene
expression of sterol regulatory element binding protein-1c
(SREBP-1c). Exp Biol Med (Maywood). 232:614–621. 2007.PubMed/NCBI
|