Introduction
Breast cancer is one of the most common malignant
cancers in women worldwide (1).
Its occurrence and development is a complicated process and can be
influenced by many factors, including abnormal expression of cell
surface receptors, abnormal activation of intracellular signal
transduction pathways and gene mutations (2–5). In
recent years, with significant progress in novel chemotherapy
regimens and proper combination of various therapeutic methods
including surgery, endocrine therapy, molecular targeted therapy,
chemotherapy and radiotherapy, the overall patient survival has
improved to some extent (6,7). As
a member of anthracyclines, Adriamycin (ADM) has been widely used
in different types of tumors due to its strong antitumor effects
(8–11). Especially in breast cancer, ADM has
become the cornerstone of many therapy regimens with very good
therapy outcomes (12,13). Unfortunately, drug resistance for
ADM, which usually occurs in most cases following a period of
treatment, restricts its further application and results in poor
long-term therapy outcomes (14).
Therefore, there is an urgent need for novel strategies to overcome
drug resistance that will lead to better prognosis for
patients.
Tumor necrosis factor receptor (TNFR) 2 is a member
of the TNFR family, and it is important in tumor progression and
prognosis, by regulating the malignant behavior of tumor cells via
stimulating AKT serine/threonine kinase 1 (AKT) or nuclear factor
(NF)-κB signaling pathways (15).
However, its role in ADM resistance of breast cancer has not been
reported. Aberrant stimulation of the phosphoinositide 3-kinase
(PI3K)/AKT signaling pathway, DNA damage repair and cancer stemness
are considered established events responsible for drug resistance
in many types of tumors (16–18).
But, whether TNFR2 could induce drug resistance through regulating
DNA repair or cancer stemness remains unknown.
In the present study, the role of TNFR2 in drug
resistance was explored from the perspective of its effect on the
DNA repair mechanism. The results demonstrated that TNFR2 induced
ADM resistance in breast cancer cells, by enhancing DNA damage
repair via regulating the DNA repair protein, poly(ADP-ribose)
polymerase (PARP). Furthermore, the AKT signaling pathway was
demonstrated to be required for TNFR2-induced PARP expression.
Materials and methods
Cell culture
Human breast cancer cell lines MCF-7 and MDA-MB-231
were purchased from the American Type Culture Collection (Manassas,
VA, USA). Both cell lines were cultured in minimum essential medium
(MEM) (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) supplemented with 10% fetal bovine serum (FBS) (Invitrogen;
Thermo Fisher Scientific, Inc.).
Cell transfection
Cells were plated in a 6-well plate at
4×105 cells per well. After 24 h, plasmid
pReceiver-M77-TNFR2 (410 ng/µl) (EX-A0254-M77; GeneCopoeia, Inc.,
Rockville, MD, USA) and control plasmid (320 ng/µl) were
transfected into MDA-MB-231 cells to upregulate TNFR2 expression;
plasmid psi-U6-GFP-TNFR2-sh (380 ng/µl) (RSH052309-CU6;
GeneCopoeia, Inc.) and control plasmid (440 ng/µl) were transfected
into MCF-7 cells to downregulate TNFR2 expression. All procedures
were performed using Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol and
empty vectors were used as a control. After 48 h, cells were
harvested and transfection efficiency was determined using western
blot analysis. Sequence silencing TNFR2,
5′-TTGACACCCTACAAGCCAGAA-3′; sequence as control plasmid,
5′-GTTCTGCGAACGTGTCACGT-3′.
Western blotting
Cells were washed twice in PBS and lysed in
radioimmunoprecipitation assay buffer (Beyotime Institute of
Biotechnology, Haimen, China) containing 1% protease inhibitor.
Protein concentration was measured by spectrophotometry (ND-1000;
Nano Drop Technologies; Thermo Fisher Scientific, Inc., Waltham,
MA, USA). Protein (200 µg) was separated by 10% SDS-PAGE and
transferred to a polyvinylidene fluoride membrane. Following
blocking in TBS/0.1% Tween-20 containing 5% non-fat dry milk for 1
h at room temperature, the membrane was incubated with primary
antibodies (listed in Table I) at
4°C overnight and then with horseradish peroxidase-conjugated
secondary antibody (ab97023/ab6802; 1:5,000; Epitomics; Abcam,
Cambridge, MA, USA) at room temperature for 1 h. Finally, signals
on the membrane were visualized by enhanced chemiluminescence
reagents (Pierce; Thermo Fisher Scientific, Inc.) and measured by
Image-Pro software (version 5.1; Media Cybernetics, Inc.,
Rockville, MA, USA).
 | Table I.Primary antibodies used in western
blot analyses. |
Table I.
Primary antibodies used in western
blot analyses.
| Protein | Cat. no. | Final dilution | Supplier |
|---|
| TNFR2 | ab8161 | 1:1,000 | Abcam, Cambridge, MA,
USA |
| pH2AX | ab22551 | 1:1,000 | Abcam, Cambridge, MA,
USA |
| PARP | 13371-1-AP | 1:1,000 | Wuhan Sanying
Biotechnology, Wuhan, China |
| MGMT | 17195-1-AP | 1:2,000 | Wuhan Sanying
Biotechnology, Wuhan, China |
| p-ERK1/2 | ab214362 | 1:1,000 | Abcam, Cambridge, MA,
USA |
| ERK1/2 | 9102 | 1:1,000 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| p-AKT | 13038 | 1:1,000 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| AKT | 4685 | 1:1,000 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| GAPDH | Ab181602 | 1:2,000 | Abcam, Cambridge, MA,
USA |
Drug resistance assay
Cells were plated in 96-well plates in triplicate in
MEM supplemented with 10% FBS at 8,000 cells per well. After 24 h,
the medium was replaced with MEM containing 0.02, 0.08, 0.32, 1.28,
5.12, or 20.48 µmol/l ADM for 48 h and then MTT was dissolved in
dimethyl sulfoxide (M1020-500T; Beijing Solarbio Science and
Technology Co., Ltd., Beijing, China) and MTT assay was performed
at 490 nm wavelength. The survival curves were constructed and the
half maximal inhibitory concentration (IC50) was calculated. The
experiment was repeated at least three times.
Statistical analysis
Data were expressed as mean ± standard deviation.
SPSS 13.0 software (SPSS, Inc. Chicago, IL, USA) was used for
statistical analysis. IC50 was calculated by regression analysis.
Significance of differences between two groups was analyzed by
Student two-tailed t-test. Significance of differences between
multiple groups was analyzed by one-way analysis of variance
followed by Student-Newman-Keuls test. P<0.05 was considered to
indicate a statistically significant difference.
Results
TNFR2 expression levels are associated
with ADM resistance in breast cancer cells
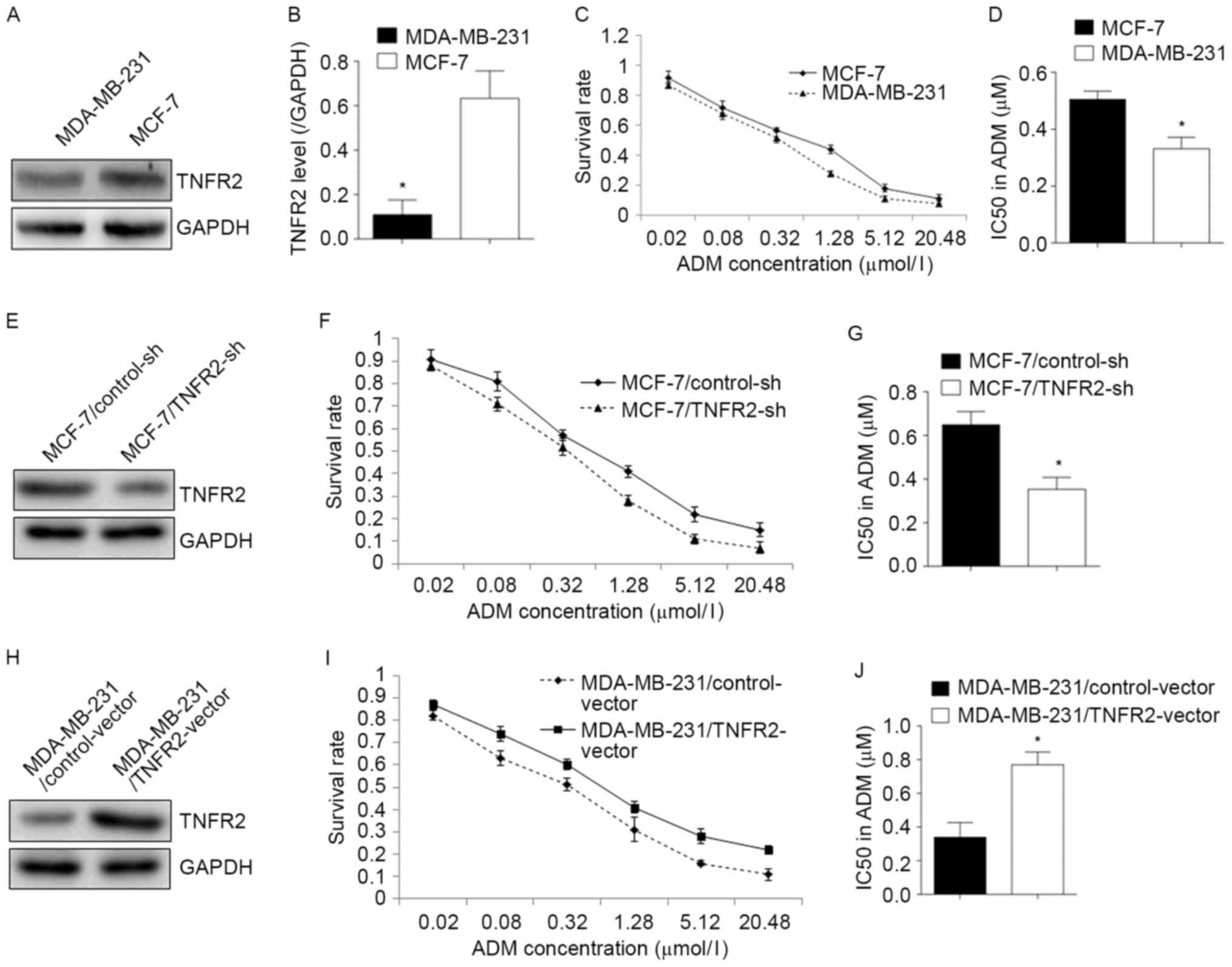
Firstly, the protein expression levels of TNFR2 were
detected in the breast cancer cell lines MCF-7 and MDA-MB-231. As
illustrated in Fig. 1A and B,
TNFR2 protein expression levels were significantly higher in MCF-7
cells compared with MDA-MB-231 cells, by ~3-fold. Of note, ADM
resistance of MCF-7 cells was also significantly higher compared
with MDA-MB-231 cells (Fig. 1C).
The IC50 was 0.505±0.028 and 0.331±0.039 µmol/l for MCF7 and
MDA-MB-231 cells respectively, which was a significant difference
(P<0.05; Fig. 1D). These
results suggested a potential correlation between TNFR2 expression
and AMD resistance in breast cancer cells. In order to further
explore this hypothesis, TNFR2 expression was silenced in MCF-7
cells by shRNA (Fig. 1E). The cell
survival rate of TNFR2-deficient MCF-7 cells declined significantly
following ADM treatment compared with control MCF-7 cells (Fig. 1F), with the IC50 decreasing from
0.649±0.06 µmol/l in the control cells to 0.353±0.054 µmol/l in the
TNFR2-deficient MCF-7 cells (P<0.05; Fig. 1G). By contrast, overexpressing
TNFR2 in MDA-MB-231 cells (Fig.
1H) significantly increased the cell survival rate following
ADM treatment (Fig. 1I), with the
IC50 increasing from 0.339±0.087 µmol/l in the control cells to
0.769±0.075 µmol/l in the TNFR2-overexpressing cells (P<0.05;
Fig. 1J). These results
demonstrated that TNFR2 promoted ADM resistance in breast cancer
cells.
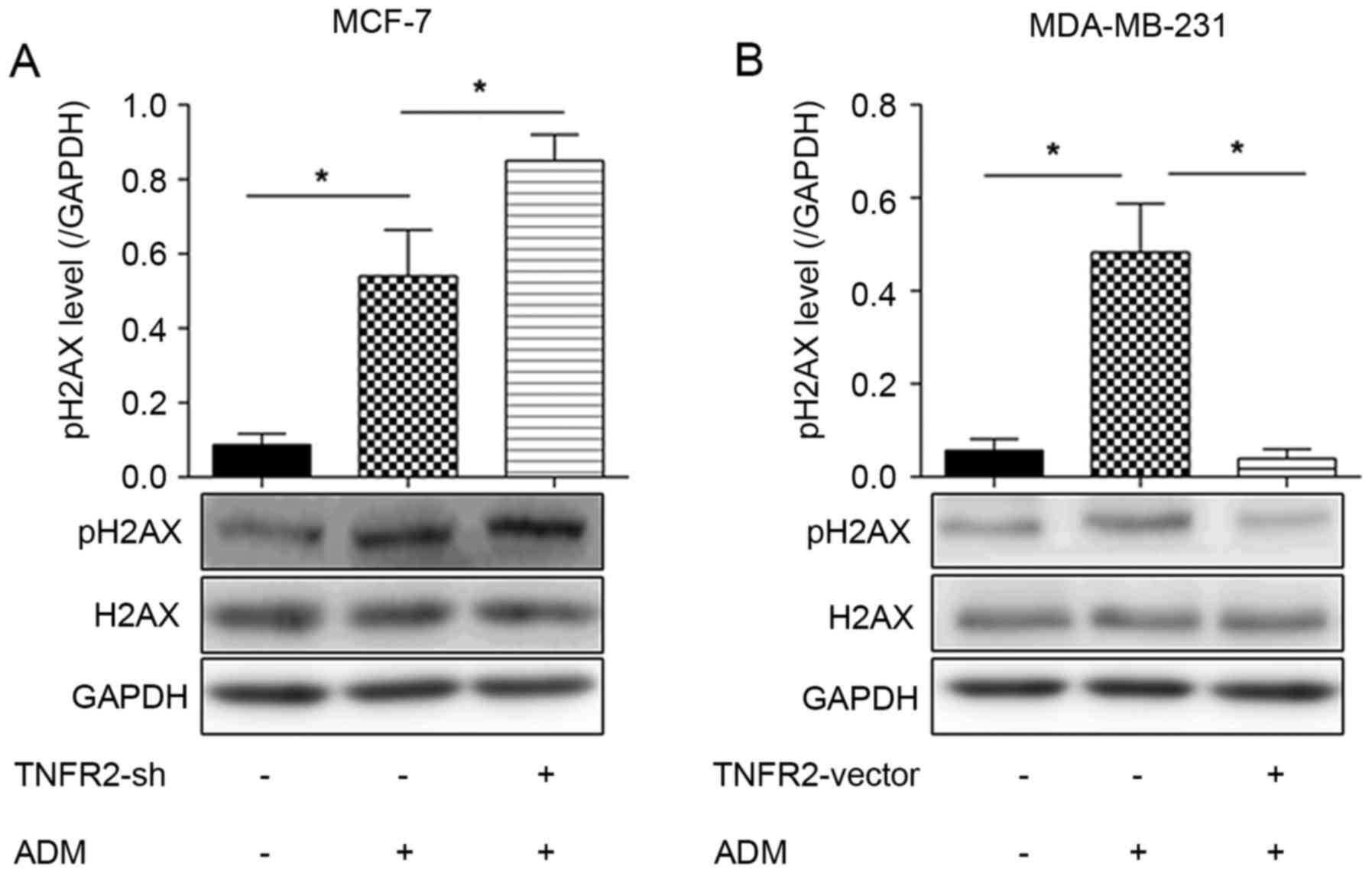
TNFR2 inhibits ADM-induced pH2AX
expression
As illustrated in Fig.
2A, phosphorylation of histone family 2A variant X (pH2AX),
which is indicative of DNA damage by double strand breakage,
increased by ~5-fold in ADM-treated MCF-7 cells compared with
untreated MCF-7 cells. No changes were observed in the levels of
total histone family 2A variant X (H2AX). When TNFR2 expression was
silenced in MCF-7 cells, pH2AX expression was further increased
(Fig. 2A). By contrast, TNFR2
overexpression in MDA-MB-231 cells resulted in a ~5-fold decrease
in pH2AX expression compared with control MDA-MB-231 cells treated
with ADM alone (Fig. 2B). These
results suggested that TNFR2 reduces the levels of DNA damage.
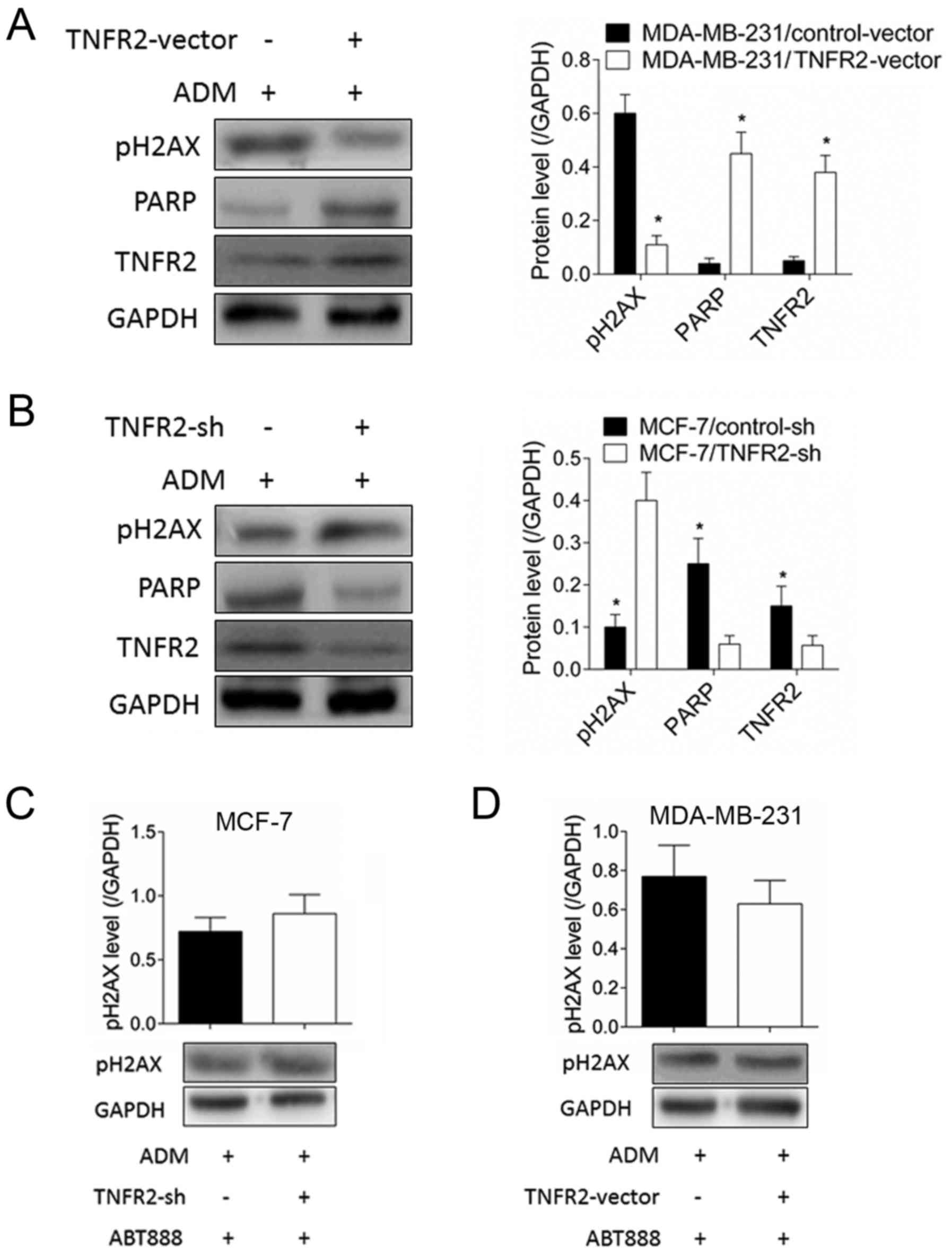
TNFR2 inhibits pH2AX expression
through regulation of PARP
As illustrated in Fig.
3A, TNFR2 overexpression significantly inhibited pH2AX
expression in MDA-MB-231 cells following ADM treatment, but
significantly increased PARP expression. By contrast, the pH2AX
increase induced by TNFR2 silencing in MCF-7 cells was accompanied
by PARP expression inhibition (Fig.
3B). No changes were observed to O6-methylguanine-DNA
methyltransferase (MGMT; data not shown). When the PARP inhibitor
ABT888 was used, TNFR2 silencing in MCF-7 cells did not result in
any significant changes of pH2AX expression following ADM treatment
(Fig. 3C). Similarly, pH2AX levels
in MDA-MB-231 cells were not affected by TNFR2 overexpression
following ADM treatment in the presence of the PARP inhibitor
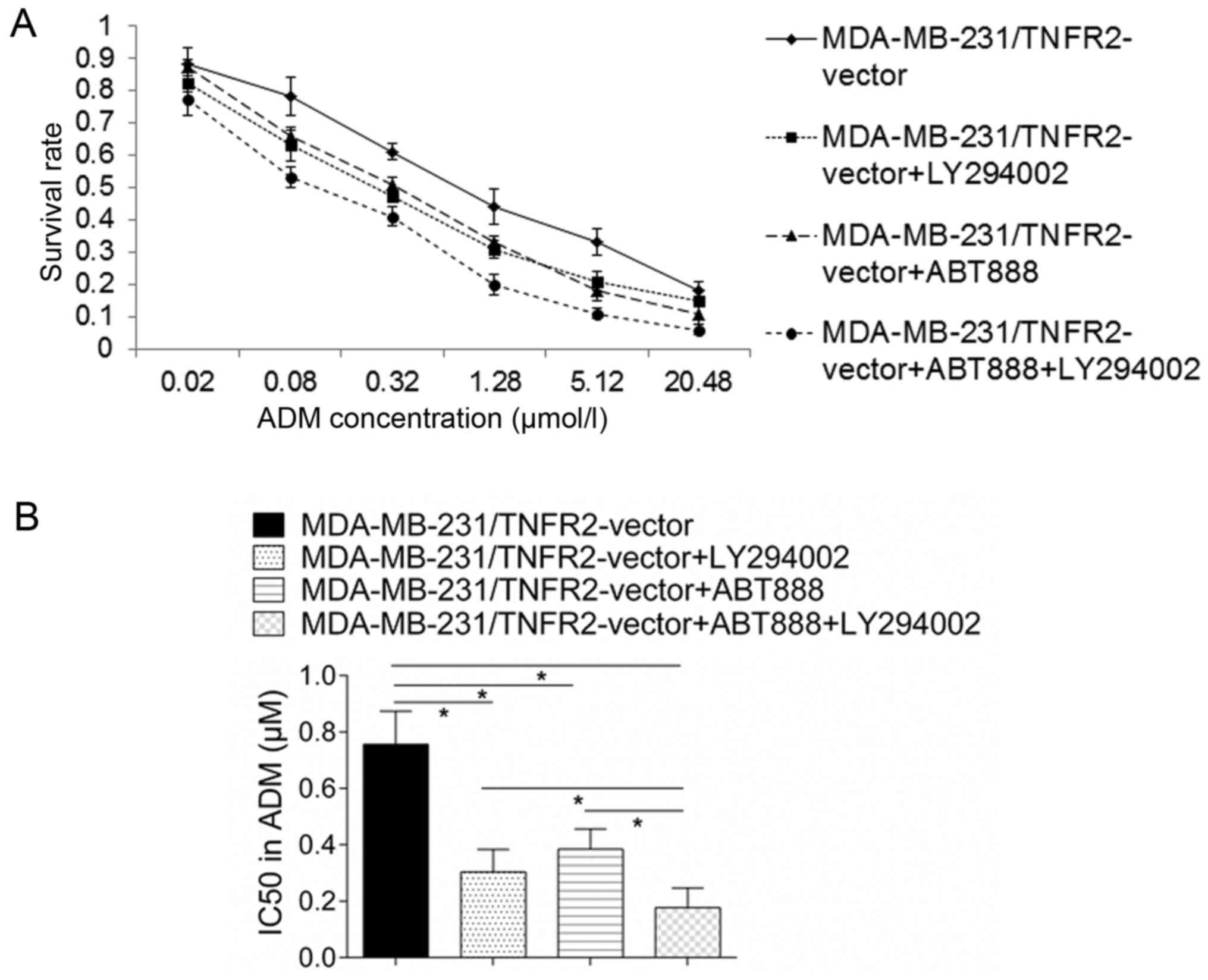
ABT888 (Fig. 3D). A drug
resistance assay demonstrated that the increase in survival rate
for MDA-MB-231 cells induced by TNFR2 overexpression declined
significantly following addition of ABT888 (Fig. 4A), with the IC50 declining from
0.756±0.117 to 0.384±0.071 µmol/l (P<0.05; Fig. 4B). These results suggested that
TNFR2 affected pH2AX expression partly by regulating PARP.
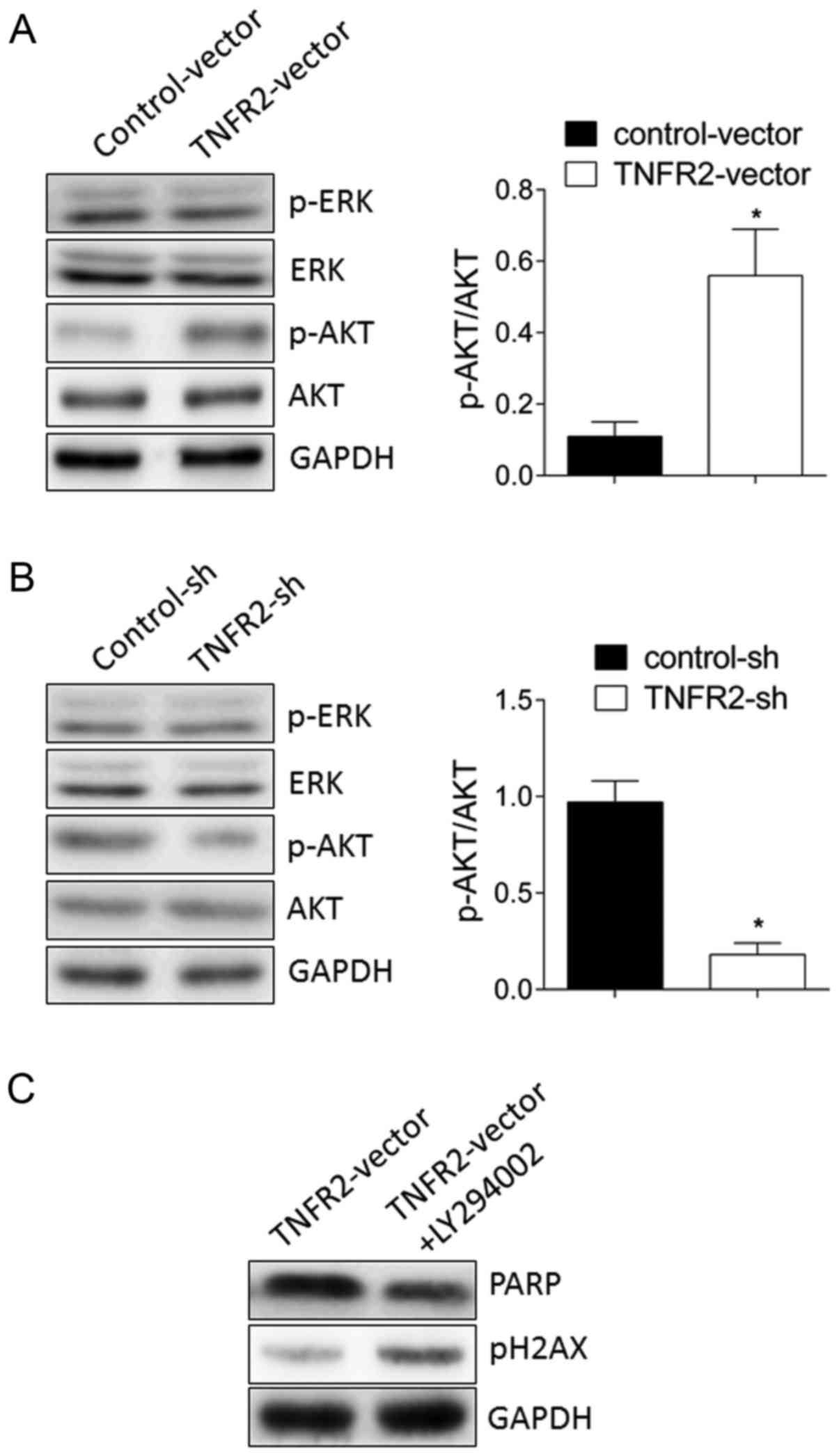
TNFR2 promotes PARP expression via AKT
signaling
To further study the potential molecular mechanism
responsible for PARP expression, AKT and extracellular
signal-regulated kinase (ERK) were examined as candidate signal
targets. As illustrated in Fig.
5A, TNFR2 overexpression in MDA-MB-231 cells significantly
stimulated phosphorylation of AKT, but no change ERK
phosphorylation was observed. By contrast, TNFR2 silencing in MCF-7
cells significantly inhibited phosphorylation of AKT, while again
no change was observed in ERK phosphorylation (Fig. 5B). To further confirm that AKT
activation mediated PARP expression, the AKT inhibitor LY294002 was
used. The results demonstrated that PARP upregulation induced by
TNFR2 overexpression in MDA-MB-231 cells was significantly
inhibited by addition of the AKT inhibitor LY294002 (Fig. 5C). A drug resistance assay
demonstrated that increase in survival rate of MDA-MB-231 cells
induced by TNFR2 overexpression declined significantly following
addition of LY294002 (Fig. 4A),
with the IC50 declining from 0.756±0.117 to 0.304±0.08 µmol/l
(P<0.05; Fig. 4B). Furthermore,
combination treatment of LY294002 and ABT888 inhibited the IC50 of
TNFR2-overexressing MDA-MB-231 cells from 0.756±0.117 to 0.176±0.07
µmol/l, with was significantly lower than either LY294002 or ABT888
treatment alone (Fig. 4B). These
results indicated that TNFR2 promoted PARP expression via AKT
signaling.
Discussion
Previous studies have reported two possible
mechanisms responsible for the antitumor effects of ADM: Inhibition
of DNA transcription and replication by intercalating between DNA
base pairs, and induction of DNA double strand breakage by
generating oxygen free radicals (19). Therefore, ADM resistance studies
may focus on the DNA damage repair mechanism.
TNFR2, which differs from TNFR1 mainly due to the
absence of death domain in its structure, promotes survival,
proliferation, migration and invasion in multiple types of cancer.
Tanimura et al (20)
reported that TNF-α promotes invasiveness of cholangiocarcinoma
cells via TNFR2. In addition, Yang et al (21) reported that progranulin promotes
proliferation and angiogenesis of colorectal cancer cells through
TNFR2. However, studies about the role of TNFR2 in drug resistance
are limited and remain controversial. Zhang et al (22) reported that apoptotic response of
colorectal cancer cells to 5-fluorouracil is mediated by induced
TNFR2, implying negative regulation of TNFR2 in drug resistance.
Sprowl et al (23) reported
that TNFR2 expression was upregulated in ADM resistant MCF-7 cells,
this suggested a possible correlation between TNFR2 and drug
resistance, but did not confirm the role of TNFR2 in drug
resistance, and the underlying mechanism remains to be elucidated.
In the present study, it was demonstrated that MCF-7 cells with
higher TNFR2 expression exhibited stronger ADM resistance than
MDA-MB-231 cells in which TNFR2 expression was significantly lower.
Furthermore, overexpression of TNFR2 in MDA-MB-231 cells enhanced
ADM resistance, while silencing of TNFR2 in MCF-7 cells weakened
ADM resistance. These results indicate that TNFR2 is important in
ADM resistance of breast cancer cells. The present findings are in
contrast to the findings of Zhang et al (22) for colorectal cancer cells. It is
possible that different types of tumors and different drugs may
involve different pharmacological mechanisms and pathways
regulating resistance.
The mechanism of ADM resistance is complicated and
TNFR2 effect on cell survival and proliferation may be partly
responsible for this. In addition, integrity of DNA is crucial for
cell survival (24). Chemotherapy
drugs kill tumor cells by destroying their DNA, but tumor cells can
repair DNA damage by activating the DNA damage repair mechanism,
resulting in drug resistance (25). To date, there are no reports on the
effect of TNFR2 on DNA damage repair. H2AX is a subtype of the core
histone 2A and it is localized to human chromosome 11q23 (26). Post-translational modification of
H2AX, such as phosphorylation, methylation and acetylation, usually
happens following DNA double strand breakage. Because H2AX is the
first substrate for phosphorylation following DNA double strand
breakage, phosphorylation of H2AX is routinely used as a marker of
DNA damage for recruitment of repair factors and chromosome
remodeling factors, thus maintaining genome stability (27). In the present study, pH2AX was
detected following ADM treatment, confirming the presence of DNA
damage induced by ADM. TNFR2 overexpression in MDA-MB-231 cells
significantly decreased pH2AX levels, while TNFR2 silencing in
MCF-7 cells significantly induced the levels of pH2AX, following
ADM challenge. These results suggested that TNFR2 affects the
levels of DNA damage induced by ADM in breast cancer cells.
MGMT and PARP are both important DNA repair
proteases (28,29). To test whether TNFR2 could repair
DNA damage by regulating DNA repair proteins, we examined the
expression levels of MGMT and PARP. The results demonstrated that
TNFR2 overexpression in MDA-MB-231 cells significantly upregulated
PARP expression, and TNFR2 silencing in MCF-7 cells significantly
inhibited PARP expression, following ADM challenge. No changes were
observed in MGMT expression (data not shown). pH2AX expression
levels exhibited opposite trends to PARP expression levels, when
TNFR2 was altered. In addition, when the PARP inhibitor ABT888 was
used, no significant change was observed in pH2AX levels in
TNFR2-overexpressing and control MDA-MB-231 cells following ADM
treatment. Similarly, no significant change was observed in pH2AX
levels in TNFR2-silenced and control MCF-7 cells following ADM
treatment. These results indicated that TNFR2 inhibited DNA damage
partly through PARP. However, other DNA damage repair proteins may
also be required and further studies will be needed to fully
explore the role of TNFR2 in DNA damage repair mechanisms.
AKT and ERK are important signaling pathways for
various cellular functions, including survival, proliferation, and
migration in multiple types of tumors (30–32).
Yang et al (21) have
reported that blocking TNFR2 significantly inhibited activation of
AKT signaling induced by progranulin, but no change was observed in
ERK phosphorylation. In the present study, TNFR2 overexpression was
also demonstrated to activate AKT signaling. In addition, the AKT
inhibitor LY294002 inhibited PARP expression. These results
suggested that TNFR2 promoted PARP expression via AKT signaling,
which is consistent to the study by Yang et al (21). A drug resistance assay demonstrated
that both ABT888 and LY294002 treatments alone enhanced the
sensitivity of MDA-MB-231 cells to ADM, and the combination
treatment had a synergistic effect, suggesting that a similar
combination may be beneficial for treatment of breast cancer. Of
course, further studies on the potential side effects and long-term
benefits for such a drug combination will be necessary for further
consideration of these results in the clinic.
In conclusion, the present study demonstrated a role
of TNFR2 in ADM resistance of breast cancer cells. This effect of
TNFR2 was partly mediated by the induction of the DNA damage repair
protease PARP via the AKT signaling pathway. The present results
may enrich our understanding regarding the role of TNFR2 in breast
cancer and in drug resistance and may provide novel therapy targets
for breast cancer treatment.
References
|
1
|
Raimondi S, Botteri E, Munzone E, Cipolla
C, Rotmensz N, DeCensi A and Gandini S: Use of beta-blockers,
angiotensin-converting enzyme inhibitors and angiotensin receptor
blockers and breast cancer survival: Systematic review and
meta-analysis. Int J Cancer. 139:212–219. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li M, Zhang J, Ouyang T, Li J, Wang T, Fan
Z, Fan T, Lin B and Xie Y: Incidence of BRCA1 somatic mutations and
response to neoadjuvant chemotherapy in Chinese women with
triple-negative breast cancer. Gene. 584:26–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wu Y, Yu DD, Yan DL, Hu Y, Chen D, Liu Y,
Zhang HD, Yu SR, Cao HX and Feng JF: Liver X receptor as a drug
target for the treatment of breast cancer. Anticancer Drugs.
27:373–382. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ugenskiene R, Myrzaliyeva D, Jankauskaite
R, Gedminaitė J, Jančiauskienė R, Šepetauskienė E and Juozaitytė E:
The contribution of SIPA1 and RRP1B germline polymorphisms to
breast cancer phenotype, lymph node status and survival in a group
of Lithuanian young breast cancer patients. Biomarkers. 21:363–370.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang ZY and Yin L: Estrogen receptor
alpha-36 (ER-α36): A new player in human breast cancer. Mol Cell
Endocrinol. 418:193–206. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sawant MA, Dasgupta A, Lavhale MS and
Sitasawad SL: Novel triterpenoid AECHL-1 induces apoptosis in
breast cancer cells by perturbing the mitochondria-endoplasmic
reticulum interactions and targeting diverse apoptotic pathways.
Biochim Biophys Acta. 1860:1056–1070. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Addetia K and DeCara JM: Cardiac
complications of HER2-targeted therapies in breast cancer. Curr
Treat Options Cardiovasc Med. 18:362016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Marques SC, Ranjbar B, Laursen MB,
Falgreen S, Bilgrau AE, Bødker JS, Jørgensen LK, Primo MN, Schmitz
A, Ettrup MS, et al: High miR-34a expression improves response to
doxorubicin in diffuse large B-cell lymphoma. Exp Hematol.
44:238–246.e2. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ni W, Chen B, Zhou G, Lu C, Xiao M, Guan
C, Zhang Y, He S, Shen A and Ni R: Overexpressed nuclear BAG-1 in
human hepatocellular carcinoma is associated with poor prognosis
and resistance to doxorubicin. J Cell Biochem. 114:2120–2130. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pelden S, Insawang T, Thuwajit C and
Thuwajit P: The trefoil factor 1 (TFF1) protein involved in
doxorubicin-induced apoptosis resistance is upregulated by estrogen
in breast cancer cells. Oncol Rep. 30:1518–1526. 2013.PubMed/NCBI
|
|
11
|
Kamba SA, Ismail M, Hussein-Al-Ali SH,
Ibrahim TA and Zakaria ZA: In vitro delivery and controlled release
of Doxorubicin for targeting osteosarcoma bone cancer. Molecules.
18:10580–10598. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cunha NL, Teixeira GM, Martins TD, Souza
AR, Oliveira PF, Símaro GV, Rezende KC, Gonçalves Ndos S, Souza DG,
Tavares DC, et al: (−)-Hinokinin induces G2/M arrest and
contributes to the antiproliferative effects of doxorubicin in
breast cancer cells. Planta Med. 82:530–538. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tarvirdipour S, Vasheghani-Farahani E,
Soleimani M and Bardania H: Functionalized magnetic
dextran-spermine nanocarriers for targeted delivery of doxorubicin
to breast cancer cells. Int J Pharm. 501:331–341. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hermawan A, Wagner E and Roidl A:
Consecutive salinomycin treatment reduces doxorubicin resistance of
breast tumor cells by diminishing drug efflux pump expression and
activity. Oncol Rep. 35:1732–1740. 2016.PubMed/NCBI
|
|
15
|
Zhong Z, Carroll KD, Policarpio D, Osborn
C, Gregory M, Bassi R, Jimenez X, Prewett M, Liebisch G, Persaud K,
et al: Anti-transforming growth factor beta receptor II antibody
has therapeutic efficacy against primary tumor growth and
metastasis through multieffects on cancer, stroma, and immune
cells. Clin Cancer Res. 16:1191–1205. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang J, Stevens MF and Bradshaw TD:
Temozolomide: Mechanisms of action, repair and resistance. Curr Mol
Pharmacol. 5:102–114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kang MK and Kang SK: Tumorigenesis of
chemotherapeutic drug-resistant cancer stem-like cells in brain
glioma. Stem Cells Dev. 16:837–847. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Du Y, Su T, Zhao L, Tan X, Chang W, Zhang
H and Cao G: Associations of polymorphisms in DNA repair genes and
MDR1 gene with chemotherapy response and survival of non-small cell
lung cancer. PLoS One. 9:e998432014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fornari F, Milazzo M, Chieco P, Negrini M,
Calin GA, Grazi GL, Pollutri D, Croce CM, Bolondi L and Gramantieri
L: MiR-199a-3p regulates mTOR and c-Met to influence the
doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res.
70:5184–5193. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tanimura Y, Kokuryo T, Tsunoda N, Yamazaki
Y, Oda K, Nimura Y, Mon N Naing, Huang P, Nakanuma Y, Chen MF, et
al: Tumor necrosis factor alpha promotes invasiveness of
cholangiocarcinoma cells via its receptor, TNFR2. Cancer Lett.
219:205–213. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang D, Wang LL, Dong TT, Shen YH, Guo XS,
Liu CY, Liu J, Zhang P, Li J and Sun YP: Progranulin promotes
colorectal cancer proliferation and angiogenesis through TNFR2/Akt
and ERK signaling pathways. Am J Cancer Res. 5:3085–3097.
2015.PubMed/NCBI
|
|
22
|
Zhang W, Ramdas L, Shen W, Song SW, Hu L
and Hamilton SR: Apoptotic response to 5-fluorouracil treatment is
mediated by reduced polyamines, non-autocrine Fas ligand and
induced tumor necrosis factor receptor 2. Cancer Biol Ther.
2:572–578. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sprowl JA, Reed K, Armstrong SR, Lanner C,
Guo B, Kalatskaya I, Stein L, Hembruff SL, Tam A and Parissenti AM:
Alterations in tumor necrosis factor signaling pathways are
associated with cytotoxicity and resistance to taxanes: A study in
isogenic resistant tumor cells. Breast Cancer Res. 14:R22012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dashzeveg N, Yogosawa S and Yoshida K:
Transcriptional induction of protein kinase C delta by p53 tumor
suppressor in the apoptotic response to DNA damage. Cancer Lett.
374:167–174. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Johnson NM, Lemmens BB and Tijsterman M: A
role for the malignant brain tumour (MBT) domain protein LIN-61 in
DNA double-strand break repair by homologous recombination. PLoS
Genet. 9:e10033392013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liubaviciute A, Krasko JA, Mlynska A,
Lagzdina J, Sužiedėlis K and Pašukonienė V: Evaluation of low-dose
proton beam radiation efficiency in MIA PaCa-2 pancreatic cancer
cell line vitality and H2AX formation. Medicina (Kaunas).
51:302–306. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jha HC, Upadhyay SK, Prasad AJM, Lu J, Cai
Q, Saha A and Robertson ES: H2AX phosphorylation is important for
LANA-mediated Kaposi's sarcoma-associated herpesvirus episome
persistence. J Virol. 87:5255–5269. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McGonigle S, Chen Z, Wu J, Chang P,
Kolber-Simonds D, Ackermann K, Twine NC, Shie JL, Miu JT, Huang KC,
et al: E7449: A dual inhibitor of PARP1/2 and tankyrase1/2 inhibits
growth of DNA repair deficient tumors and antagonizes Wnt
signaling. Oncotarget. 6:41307–41323. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bady P, Delorenzi M and Hegi ME:
Sensitivity analysis of the MGMT-STP27 model and impact of genetic
and epigenetic context to predict the MGMT methylation status in
gliomas and other tumors. J Mol Diagn. 18:350–361. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dahlmann M, Okhrimenko A, Marcinkowski P,
Osterland M, Herrmann P, Smith J, Heizmann CW, Schlag PM and Stein
U: RAGE mediates S100A4-induced cell motility via MAPK/ERK and
hypoxia signaling and is a prognostic biomarker for human
colorectal cancer metastasis. Oncotarget. 5:3220–3233. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li N, Cui J, Duan X, Chen H and Fan F:
Suppression of type I collagen expression by miR-29b via PI3K, Akt,
and Sp1 pathway in human Tenon's fibroblasts. Invest Ophthalmol Vis
Sci. 53:1670–1678. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Miao B and Degterev A: Targeting
phospshatidylinositol 3-kinase signaling with novel
phosphatidylinositol 3,4,5-triphosphate antagonists. Autophagy.
7:650–651. 2011. View Article : Google Scholar : PubMed/NCBI
|